
Abstract
Key enzymes play a vital role in plant growth and development. However, the evolutionary relationships between genes encoding key enzymes in the metabolic pathway of Tartary buckwheat flavonoids are poorly understood. Based on the published Tartary buckwheat genome sequence and related Tartary buckwheat transcriptome data, 48 key enzyme-encoding genes involved in flavonoid metabolism were screened from the Tartary buckwheat genome in this study; the chromosome localization, gene structure and promoter elements of these enzyme-encoding gene were also investigated. Gene structure analysis revealed relatively conserved 5′ exon sequences among the 48 genes, indicating that the structural diversity of key enzyme-encoding genes is low in Tartary buckwheat. Through promoter analysis, these key enzyme-encoding genes were found to contain a large number of light-response elements and hormone-response elements. In addition, some genes could bind MYB transcription factors, participating in the regulation of flavonoid biosynthesis. The transcription level of the 48 key enzyme-encoding gene varied greatly among tissues. In this study, we identified 48 key enzyme-encoding genes involved in flavonoid metabolic pathways, and elucidated the structure, evolution and tissue-specific expression patterns of these genes. These results lay a foundation for further understanding the functional characteristics and evolutionary relationships of key enzyme-encoding genes involved in the flavonoid metabolic pathway in Tartary buckwheat.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tartary buckwheat is a special grain plant that is rich in flavonoids such as rutin and has anti-diabetic and anti-inflammatory biological activity. Anthocyanins confer a desirable colour on Tartary buckwheat buds, and they have strong antioxidant activity. Therefore, Tartary buckwheat has become a nutritious and healthy crop favoured by consumers, and its flavonoid metabolism has also become a hot research topic. Flavonoids are secondary metabolites necessary for plant growth and possess multiple functions, including involvement in protection against biological and abiotic stresses, plant flower colour formation and stress resistance (Blount et al. 1992; Koes et al. 1994; Misra et al. 2010a, b). Flavonoids are products of the phenylalanine metabolic branch pathway. The phenylpropanoide metabolic pathway begins with phenylalanine, which is produced by coumaryl coenzyme A via catalysed by phenylalanine ammonia-lyase. In the presence of chalcone synthase, β-coumaryl CoA and 3-malonyl CoA are combined to produce chalcone (Dare et al. 2013), and this condensation reaction is a key step in the downstream flavonoid formation pathway (Holton and Cornish 1995).Chalcone is then used by the isoflavone reductase-like enzyme to form the isoflavone branch. The biosyntheses of flavonoids and anthocyanins occur downstream of the flavonoid metabolic pathway, and the final product by the corresponding enzymes is rutin and anthocyanin (Grotewold et al. 1991; Lepiniec et al. 2006) (Fig. 1).

Schematic diagram of the biosynthetic pathway of flavonoids in plants. PAL phenylalnine ammonia-lyase, C4H cinnamate-4-hydroxylase, 4CL 4-coumaryl coenzyme A, CHS chalcone synthase, CHI chalcone isomerase, F3H flavanone-3-hydroxylase, FLS flavonol synthase, F3′H flavonoid 3′-hydroxylase, F3′5′H flavonoid 3′,5′-hydroxylase, UFGT UDP flavonoid glycosyltransferases, DFR dihydroflavonol 4-reductase, ANS anthocyanidin synthase
Most of the enzyme-encoding genes involved in the flavonoid metabolic pathway of Tartary buckwheat have been cloned and characterized, including CHS, CHI, F3H, DFR, ANS and FLS (Park et al. 2004; Kobayashi et al. 1998; Laura et al. 2002; Yoshikazu et al. 1996). However, the overall structural characteristics of these enzyme-encoding genes and other features of in Tartary buckwheat are limited. Tartary buckwheat genome sequencing has provided a good opportunity for whole-genome level for research of the tissue, expression and evolutionary characteristics of enzyme-encoding genes and their homologues involved in the flavonoid metabolic pathway (Zhang et al. 2017). In this study, we describe in detail the exon–intron structure, protein motif composition, chromosome position, promoter composition and tissue expression specificity of 48 key enzyme-encoding genes. In addition, phylogenetic relationships between key enzyme-encoding genes involved in flavonoid metabolism in Arabidopsis, sugar beet, rice, soybean and grape were compared. In general, comprehensive mining of key enzyme-encoding genes in the flavonoid metabolic pathway is very important for understanding the tissue expression and evolutionary characteristics of enzyme-encoding genes, providing a theoretical basis for clarifying the regulation of the rutin and anthocyanin biosynthesis metabolic network in Tartary buckwheat.
Materials and Methods
Identification of Structural Genes Involved in the Flavonoid Metabolic Pathway
We downloaded buckwheat genomic data from an online source: http://www.mbkbase.org/Pinku1/. Enzyme-encoding genes with specific biological functions in the metabolic pathway of Tartary buckwheat flavonoids were used as seed sequences. The sequences of these genes were obtained by a two-step BLASTP search (Liu et al. 2019).
Sequence Analysis
The open reading frames of enzyme-encoding genes in the flavonoid metabolic pathway of Tartary buckwheat were predicted using the ORF Finder program (https://www.ncbi.nlm.nih.gov/orffinder/), and the ORFs were translated into amino acid sequences by with online software TBtools (Chen et al. 2020). Conserved structures of the translated amino acid sequences were identified by the SMART program (http://smart.embl-heidelberg.de/). Finally, the characteristic domain of the queried gene sequence was determined using the CDD website (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi), and the sequence length, molecular weight (MW) and isoelectric point (pI) of the enzymes were analysed via ExPASy (https://web.expasy.org/compute_pi/). Transmembrane domains (http://psort1.hgc.jp/form.html), signal peptides (http://www.cbs.dtu.dk/services/SignalP-3.0/) and subcellular localizations were also examined (https://www.genscript.com/psort.html).
Analysis of Enzyme-Encoding Gene Structure
To examine the structural characteristics of the genes encoding in the flavonoid metabolic pathway, we first analysed the structural exons-intron composition using software TBtools. Protein motifs were valuated using MEME software with the following optimized default parameters: motif width, 6–50 number, 25 (Bailey et al. 2009).
Chromosome Location, Gene Duplication and Evolutionary Analysis of Major Enzyme-Encoding Genes
With the help of the annotation file of the Tartary buckwheat genome, the distribution of genes encoding enzymes in the flavonoid metabolic pathway of Tartary buckwheat was analysed with TBtools. By referring to the Tartary buckwheat genomic data, we determined the length distribution of the chromosomes via Linux operating system commands. Circos was then employed to locate the 48 major enzyme-encoding genes on the chromosomes. We applied MCScanX (default parameters) to examine gene duplication. We also analysed homology of the major enzyme-encoding genes between Tartary buckwheat and five other plants species using Dual Synteny Plotter.
Analysis of Candidate Enzyme-Encoding Gene Promoters
Gene expression is related to structure and is regulated by the promoter. Therefore, we used the PlantCARE online database to analyse promoter elements of the candidate genes (Magali et al. 2002); cis-acting elements were visualized via the GSDS website (http://gsds.cbi.pku.edu.cn/).
Analysis of Gene Expression
According to transcriptomic data of different tissues (roots, stems, leaves and flowers) of Tartary buckwheat tested in our laboratory (unpublished), tissue-specific expression of genes encoding enzymes in the flavonoid metabolic pathway of Tartary buckwheat was assessed with TBtools. Gene expression is expressed as FPKM (reads per kilobase of exon per million mapped reads) values.
Identification of Tissue-Specific Expression of Enzyme-Encoding Genes via Real-Time PCR
Tartary buckwheat ‘Xiqiao No. 2′ was grown under field conditions at a farm at Baoxing, Ya'an, Sichuan. When the plants reached the full flowering stage, roots, stems, leaves and flowers were collected and treated with Sample Protector for RNA/DNA (Takara, China); the samples were then stored at – 80 °C until used for total RNA extraction and transcriptome sequencing. Total RNA from the four tissues was extracted using the RNAout kit (Tiandz, China) according to the manufacturer's procedure. First, the CDSs of structural genes were obtained from the Tartary buckwheat genomic database, and RT-qPCR primers were designed using the Primer3.0 online program (http://bioinfo.ut.ee/primer3/) (Table S1). Tissue-specific expression of the enzyme-encoding genes was verified by quantitative real-time PCR using the Tartary buckwheat histone H3 gene as a reference. The qPCR using SYBR Premix Ex Taq II (TaKaRa) was repeated at least three times, and the data were analysed by the 2−(∆∆Ct) method (Livak and Schmittgen 2001) to obtain related mRNA expression data.
Statistical Analysis
All analyses were performed at least three times. qRT-PCR result data were statistically evaluated using SPSS V20.0 (IBM, USA). Significant differences were confirmed with ANOVA tests and Fisher’s protected least-significant difference (LSD) tests at the 0.05 probability level.
Results
Identification of Flavonoid Metabolic Enzyme-Encoding genes in Tartary Buckwheat
A total of 48 candidate enzyme-encoding genes involved in the Tartary buckwheat flavonoid metabolic pathway were identified by means of a two-step BLAST comparison (Table S2). These 48 candidates included 10 CHS, 8 C4H and 7 IRL genes, which were the most numerous, followed by 5 each PAL and FLS genes. We also detected two 4CL, F3H, F3′H, CHI and DFR genes, and one ANS, ANR and F3′5′H gene (Table 1). The basic information of the 48 candidate enzyme-encoding genes is shown in detail in Table 1, including the predicted pI, MW, subcellular localization, transmembrane domain and signal peptide. We also translated the CDSs of the 48 candidate genes into protein sequences. Among them, the smallest protein is FtCHS5 with 64 aa; the largest protein is FtPAL1 with 722 aa. The MWs range from 7.30 to 78.38 kDa and the pIs from 4.86 (FtCHI1) to 10.38 (FtPAL5). Through subcellular mapping analysis, 19 of the encoded proteins are predicted to localize to the endoplasmic reticulum (membrane), 14 to the cytoplasm, 5 to the nucleus, 1 to the plasma membrane, 5 to microbodies (peroxisome), and the others to the mitochondrial matrix space. This diversity of subcellular localization indicates that these structural genes in Tartary buckwheat have a variety of biological functions. Based on transmembrane domain analysis of the 48 genes encoding key enzymes, only four of the proteins (FtCHS5, FtC4H5, FtF3′H2, FtANS) contain one transmembrane domain, whereas the others have no transmembrane domain. Finally, signal peptide analysis revealed signal peptides for only 12, with the other 36 having no signal peptide.
Chromosomal Location and Synteny Analysis of Major Enzyme-Encoding Genes
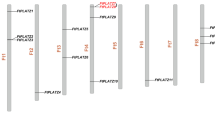
The 48 candidate genes are distributed on 8 chromosomes of Tartary buckwheat. As illustrated in the chromosome map, the 48 structural genes are mainly distributed on chromosomes Ft1, Ft2, Ft5, Ft7 and Ft8. It is worth noting that FtPAL3 and FtPAL5, FtCHS1 and FtCHS7, FtIRL2 and FtIRL7, FtIRL4 and FtIRL5, FtFLS1 and FtFLS2 are present in the form of tandem repeat genes. Some genes with the same function are distributed on different chromosomes, which have a positive effect on the evolution or expansion of a gene family (Fig. 2). To analyse duplication events, collinearity analysis of the 48 key enzyme-encoding genes was performed using MCScanX software. Four pairs of fragment duplication genes were found within the 48 major enzyme-encoding genes. These fragment duplication genes were the most common on chromosome 1, followed by chromosome 8; no fragment duplication gene pairs were found on chromosomes 4, 5, 6 or 7 (Fig. 3).

Distribution of structural genes involved in the flavonoid metabolic pathway among 8 chromosomes. The vertical bars represent the chromosomes of Tartary buckwheat. The number of chromosomes is on left of each chromosome. The scale on the left represents the chromosome length

Schematic representations of the interchromosomal relationships of the major enzyme-encoding genes of the Tartary buckwheat flavonoid metabolic pathway. The coloured lines indicate all the synteny blocks in the Tartary buckwheat genome, and the red lines indicate the major enzyme-encoding duplicate gene pairs (Color figure online)
To further explore evolutionary relationships of Tartary buckwheat genes encoding enzymes involved in flavonoid metabolism, we performed collinear relationship analysis with four dicotyledonous plants species (Arabidopsis thaliana, sugar beet, soybean and grape) and one monocotyledonous plant species (rice). A total of 30 key enzyme-encoding genes in the Tartary buckwheat flavonoid metabolic pathway displayed a syntenic relationship with those of soybean, followed by grape (19), beet (9), Arabidopsis (7) and rice (3). Tartary buckwheat chromosome 1 and chromosome 7 exhibit the most homologous gene pairs with those of the other species (Fig. 4).

Synteny analysis of major enzyme-encoding genes between Tartary buckwheat and five representative plant species. The grey line represents all the collinear blocks in the genome of Tartary buckwheat and other species, whereas the red lines highlight the syntenic major enzyme-encoding gene pairs (Color figure online)
Tartary buckwheat and soybean exhibits the most collinear blocks, which may indicate a crucial role for the buckwheat flavonoid-related enzyme gene family in the evolution Tartary buckwheat. In contrast, rice shares fewer than 5 colinear gene pairs, which may be associated with the phylogenetic relationship between these species.
Structure and Composition of Enzyme-Encoding Genes in the Flavonoid Metabolic Pathway
To analyse the gene structure of the 48 candidate genes in flavonoid metabolic pathway, the genomic DNA sequences of the genes were compared, and the composition of intron-exons was analysed. The coding sequences of the 48 genes are interrupted by introns, and 12 structural genes have two exons and one intron. The number of exons of other genes ranges from 3 to 6. It is worth noting that there is only one exon at the 3′ end of FtCHS5, with no corresponding intron (Fig. 5). We speculate that the incomplete FtCHS5 sequence may be due to the buckwheat genome annotation. Nevertheless, the exons and introns of highly homologous structural genes from the same family present the same distribution and have conserved characteristic regions.

Exon–intron structure of structural genes in the flavonoid metabolic pathway. The yellow blocks represent exons. The black line represents the intron. The green blocks represent upstream and downstream sequences (Color figure online)
Motif Composition of Structural Genes in the Flavonoid Metabolic Pathway
To evalute the characteristic regions, the protein motifs of the 48 candidate genes were analysed via the MEME website, and the resulting motifs were drawn using TBtools.
Based on this analysis, the proteins of the same family display a similar motif composition, and such similar motifs indicate protein structure conservation that the structure of the proteins is conserved within a particular family. To some extent, the catalytic function of these structural genes is determined by conserved domains. Five FtPAL genes contain motifs 15, 25, 17, 24, 14, 8 and 13. FtF3′H has six conserved motifs: 19, 18, 10, 7, 5 and 8. FtFLS and FtANS have two conserved motifs. FtDFR has only one motif, and FtANR contains motifs 17 and 14 (Fig. 6).

Composition of protein motifs of structural genes in the Tartary buckwheat flavonoid metabolic pathway (Color figure online)
Analysis of Upstream cis-Acting Elements
The basic building blocks of gene promoters are cis-regulatory DNA elements. In eukaryotes, clusters of cis-regulatory elements usually regulate gene expression by interacting with trans-acting factors, thus, altering metabolic flow through the flavonoid metabolic pathway. Plant cis-regulatory motifs are often reported as consensus sequences and are usually described by reporter gene expression analyses. Of note, we detected light-response elements in the promoter of each enzyme-encoding gene (Fig. 7). Hormone-response elements are also present. For example, abscisic acid (ABA) response elements (ABREs), gibberellin response elements, ethylene response elements (EREs), salicylic acid (SA) response elements, methyl jasmonate response elements, and auxin response elements are found in the promoters of the candidate genes. Among them, salicylic acid response elements are rarely reported in the study of enzyme-encoding genes but provide new avenues for future research. In addition, several CREs associated with abiotic stress responses were found in these candidate gene promoters (Fig. 7), including low-temperature-response elements in the promoters of 17. The promoters of 9 contain MYB-binding sites and participate in the regulation of flavonol biosynthesis pathway genes. The promoters of five structural genes contain MYB-binding sites participate in the response to light and drought. The promoters of 19 structural genes contain cis-acting elements involved in defence and stress responses. Finally, it is worth noting that the promoters of both FtCHS8 and FtCHI1 have a wound-response element, warranting further study. The details of the promoter elements are shown in Table 2.

Promoter elements of 48 structural genes. The black lines represent the length of the promoters, and modules of different colours represent various cis-acting elements. The length of the promoter is marked at the bottom of the figure (Color figure online)
Expression Pattern of Structural Genes in the Tartary Buckwheat Flavonoid Metabolic Pathway
Based on the Tartary buckwheat genome database and tissue-specific transcriptome data for Tartary buckwheat, we analysed the expression patterns of these candidate flavonoid metabolism genes in roots, stems, leaves and flowers. Our results showed significant differences in expression of 32 structural genes, with most exhibiting obvious tissue specificity. Compared with other tissues, FtCHS8 and FtC4H6 had the highest expression in roots and expression of Ft4CL2, FtPAL4 and FtPAL1 was higher in roots than in flowers and stems. FtANR and FtFLS2 were mainly expressed in roots and flowers. FtFLS1 was not expressed in roots but was significantly expressed in other tissues. FtCHS4 was specifically expressed in flowers and stems, suggesting that some of these key enzyme-coding genes have multiple functions during the growth and development of Tartary buckwheat. Only one gene (FtCHS1) was highly expressed in root, stem, leaf and flower tissues of Tartary buckwheat, and the expression level was stable. Furthermore, expression of five of the genes (FtFLS1, FtCHS4, FtC4H8, FtCHS1 and FtF3H2) was higher in the stems, leaves and flowers than in roots, indicating that these genes play an important role in the maturation of Tartary buckwheat (Fig. 8).

Expression of structural genes of the flavonoid metabolic pathway in the roots, stems, leaves and flowers of Tartary buckwheat. The red represents a positive correlation and the green represents a negative correlation. The colour bar on the top of the picture represents the log2 expression value (Color figure online)
To further study the tissue-specific expression of structural genes involved in the Tartary buckwheat flavonoid metabolic pathway, expression of 32 structural genes was measured by real-time quantitative PCR, and the results are depicted in Fig. 9.

Expression profiles of 32 candidate genes. The expression of 32 enzyme-encoding genes in the root (R), stem (S), leave (L) and flower (F) tissues was verified by qPCR. The lowest expression level of all genes in the four tissues is regarded as 1. The colour block above the qPCR data represents the HeatMap corresponding to the FPKM value of the gene. The colour bar on the bottom of the picture represents the log2 expression value. The error bars were obtained from three measurements (Color figure online)
Discussion
Tartary buckwheat, a small miscellaneous grain rich in flavonoids, has both nutritional value and health benefits, and regulation of the biosynthesis and metabolism of the components of this species has become a new research focus (Bankevich et al. 2012; Krkoskova and Mrazova 2005; Ohsako et al. 2002). Flavonoids are the most important biologically active components of Tartary buckwheat (Liu and Zhu 2007). For example, rutin accounts for 70–85% of the total flavonoid content and is mainly distributed in the leaves (3%) and grains (0.8–1.7%) of Tartary buckwheat (Fabjan et al. 2003). Flavonoids are secondary metabolites in plants (Misra et al. 2010a, b). These compounds are derived from the flavonoid synthesis pathway of the downstream branch of the flavonoid metabolic pathway and are jointly regulated by a variety of key enzymes and transcription factors (Tian et al. 2015). Therefore, the screening and identification of major genes encoding enzymes involved in the flavonoid metabolic pathway lay a foundation for increasing the flavonoid content, with important academic significance and application value.
It is greatly important that gene tandem and segmental replication can enrich protein function and promote the evolution and expansion of gene families. In plants, the expansion of gene families caused by tandem genes and fragment replication is ultimately very important for the evolution of functional diversity (Cannon et al. 2004; Lynch and Conery 2000; Otto and Yong 2002). Tandem repeats of genes are often caused by gene clusters (Mayerhofer et al. 1995). According to the results of the present study, it is worth noting that the CHS (FtPinG0000552100.01 and FtPinG0000551800.01) and FLS (FtPinG0006907100.01 and FtPinG0006907000.01) are repetitive. In Arabidopsis, AtFLS2-5 are tandemly duplicated genes distributed on chromosome 5 (Owens et al. 2008). Therefore, we speculate that the evolution of CHSs and FLSs is largely driven by such gene duplication events. Nonetheless, genes of the same family are also distributed on different chromosomes, and we speculate that this may be one of the reasons for expansion of a gene family.
Gene transcriptional regulation is a very intricate process that involves multiple proteins binding to cis-regulatory elements present in a sequence-specific way in the promoter region. In our research, we have found that many key enzyme-encoding genes contain light-response elements, including I-boxes (Donald and Cashmore 1990), G-boxes (Giuliano et al. 1988), GT1-motifs (Gao et al. 2004), GATA-motifs (Gerardo and Luis 1998) among others. These elements may be essential for light-mediated transcriptional activity. Under dark conditions, expression of FtPAL, Ft4CL, FtC4H, FtCHI, FtF3H, FtF3′H1 and FtFLS1 is significantly upregulated in Tartary buckwheat, and the content of rutin and anthocyanins in hairy roots increases significantly (Thwe et al. 2014). Overall, a large number of light-response elements in the promoters of key enzyme-encoding genes would promote their expression. In our study, 7 of the candidate gene promoters were found to contain motifs that control circadian rhythm (Table 2). In higher plants, expression of most genes is regulated in a circadian manner. Hormones also act as important key regulators of the process of plant growth and development, and elements responsive to ABA, auxin, JA and SA were found in the promoters of the candidate key enzyme-encoding genes. This indicates that hormones are vital for the regulation of genes encoding key enzymes in flavonoid metabolism. Liu et al. (2018) studied FtARF gene expression by spraying different concentrations of NAA solution at the bud stage, and the results revealed a positive response of FtARF to auxin during fruit development. Additionally, Li et al. (2013) applied exogenous ABA and SA to buckwheat buds, and found that both inhibited the transcription of FtFLS1; in contrast, ABA did not affect FtFLS2 transcription, and SA upregulated that of FtFLS2. According to our research, expression of key genes involved in flavonoid metabolism may be induced by abiotic stress and transcription factor binding to cis-regulatory elements present in the promoters.
Based on comparison the results of RNA-Seq and qPCR, the tissue-specific expression patterns of most of the genes were the same, except for a few, including Ft4CL1, Ft4CL2, FtC4H8 and FtFLS2 (Fig. 9). Our analysis may be that different detection methods lead to discrepancies in the detection system causing the above changes. Of the genes detected, FtC4Hs exhibited the highest expression in roots and stems, FtANS in roots, FtF3′H in stems and leaves, and FtF3H in stems and flowers. These findings are consistent with the view of Park et al. (2011). In addition, Li et al. (2012) cloned a PAL gene from Tartary buckwheat, and semiquantitative reverse transcription-PCR (RT-PCR) analysis showed that FtPAL was most highly expressed in the stem during the florescence period, Li et al. (2013) also identified the expression levels of FtFLS1 and FtFLS2 in the Tartary buckwheat cultivars 'Hokkai T10′ and 'Hokkai T8′ by quantitative real-time PCR, reporting that FtFLS1 expression levels in flowers were higher than in other tissues and that FtFLS2 showed the highest expression in 'Hokkai T10′ roots. Moreover, overexpression of FLS1 in Arabidopsis not only changed the colour of the seed coat (resulting in a light-brown colour), but also affected the accumulation of flavonoids (Nguyen et al. 2016). Jiang et al. (2020) also found that high expression of CHS enhanced the accumulation of flavonoids in Perilla frutescens, as based on transcriptome and metabolome analyses. Hence, we suggest that the tissue-specific expression of the above key enzyme-encoding genes may be due to the existence of tissue-specific promoters. The results of this research can be employed to further study the biosynthesis of flavonoids and metabolic engineering in Tartary buckwheat.
Conclusion
In our study, 48 major genes encoding enzymes in the metabolic pathway of Tartary buckwheat flavonoids were screened and identified. Through analysis exons and introns, conserved proteins motifs, cis-acting elements and evolutionary relationships, the results provide a basis for further examining the biological functions of these 48 genes. In addition, tissue-specific expression analysis of the genes revealed that some show broad expression but that the tissue-specific expression of other genes is not significant.
References
Bailey TL, Boden M, Buske FA, Martin F, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37(suppl2):W202–W208. https://doi.org/10.1093/nar/gkp335
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19(5):455–477. https://doi.org/10.1089/cmb.2012.0021
Blount JW, Dixon RA, Paiva NL (1992) Stress responses in alfalfa (Medicago sativa L.) XVI. Antifungal activity of medicarpin and its biosynthetic precursors; implications for the genetic manipulation of stress metabolites. Physiol Mol Plant Pathol 41(5):333–349. https://doi.org/10.1016/0885-5765(92)90020-V
Cannon SB, Mitra A, Baumgarten A, Young ND, May G (2004) The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol 4(1):796–815. https://doi.org/10.1186/1471-2229-4-10
Chen CJ, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R (2020) TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant 13(8):1194–1202. https://doi.org/10.1016/j.molp.2020.06.009
Dare AP, Tomes S, Jones M, McGhie TK, Hellens RP (2013) Phenotypic changes associated with RNA interference silencing of chalcone synthase in apple (Malus × domestica). Plant J 74(3):398–410. https://doi.org/10.1111/tpj.12140
Donald RGK, Cashmore AR (1990) Mutation of either G box or I box sequences profoundly affects expression from the Arabidopsis rbcS-1A promoter. EMBO J 9(6):1717–1726. https://doi.org/10.1002/j.1460-2075.1990.tb08295.x
Fabjan N, Rode J, Kosir IJ, Wang Z, Kreft I (2003) Tartary buckwheat (Fagopyrum tataricum Gaertn.) as a source of dietary rutin and quercitrin. J Agric Food Chem 51(22):6452–6455. https://doi.org/10.1021/jf034543e
Gao Y, Li JM, Strickland E, Hua SJ, Zhao SJ, Chen ZL, Qu LJ, Deng XW (2004) An Arabidopsis promoter microarray and its initial usage in the identification of HY5 binding targets in Vitro. Plant Mol Biol 54(5):683–699. https://doi.org/10.1023/B:PLAN.0000040898.86788.59
Gerardo AA, Luis HE (1998) Evolution of light-regulated plant promoters. Annu Rev Plant Physiol Plant Mol Biol 49:525–555. https://doi.org/10.1146/ANNUREV.ARPLANT.49.1.525
Giuliano G, Pichersky E, Malik VS, Timko MP, Scolnik PA, Cashmore AR (1988) An evolutionarily conserved protein binding sequence upstream of a plant light-regulated gene. Proc Natl Acad Sci USA 85(19):7089–7093. https://doi.org/10.1073/pnas.85.19.7089
Grotewold E, Athma P, Peterson T (1991) Alternatively spliced products of the maize P gene encode proteins with homology to the DNA-binding domain of myb-like transcription factors. Proc Natl Acad Sci USA 88(11):4587–4591. https://doi.org/10.2307/2357097
Holton TA, Cornish EC (1995) Genetics and biochemistry of anthocyanin biosynthesis. Plant Cell 7(7):1071–1083. https://doi.org/10.1105/tpc.7.7.1071
Jiang T, Guo KY, Liu LD, Tian W, Xie XL, Wen SQ, Wen CX (2020) Integrated transcriptomic and metabolomic data reveal the flavonoid biosynthesis metabolic pathway in Perilla frutescens (L.) leaves. Sci Rep 10(1):61–1403. https://doi.org/10.1038/s41598-020-73274-y
Park JS, Kim JB, Hahn BS, Kim HK, Ha SH, Kim JB, Kim YH (2004) EST analysis of genes involved in secondary metabolism in Camellia sinensis (tea), using suppression subtractive hybridization. Plant Sci 166(4):953–961. https://doi.org/10.1016/j.plantsci.2003.12.010
Kobayashi H, Oikawa Y, Koiwa H, Yamamura S (1998) Flower-specific gene expression directed by the promoter of a chalcone synthase gene from Gentiana triflora in Petunia hybrida. Plant Sci 131(2):173–180. https://doi.org/10.1016/S0168-9452(97)00236-7
Koes RE, Quattrocchio F, Mol JNM (1994) The flavonoid biosynthetic pathway in plants: function and evolution. BioEssays 16(2):123–132. https://doi.org/10.1002/bies.950160209
Krkoskova B, Mrazova Z (2005) Prophylactic components of buckwheat. Food Res Int 38(5):561–568. https://doi.org/10.1016/j.foodres.2004.11.009
Laura J, Kaisu MT, Anna Maria P, Riitta TRN, Sirpa KR, Anja H (2002) Expression of genes involved in anthocyanin biosynthesis in relation to anthocyanin, proanthocyanidin, and flavonol levels during bilberry fruit development. Plant Physiol 130(2):729–739. https://doi.org/10.1104/pp.006957
Lepiniec L, Debeaujon I, Routaboul JM, Baudry A, Pourcel L, Nesi N, Caboche M (2006) Genetics and biochemistry of seed flavonoids. Annu Rev Plant Biol 57:405–430. https://doi.org/10.1146/ANNUREV.ARPLANT.57.032905.105252
Li CL, Bai YC, Chen H, Zhao HX, Shao JR, Wu Q (2012) Cloning, characterization and functional analysis of a phenylalanine ammonia-lyase gene (FtPAL) from Fagopyrum tataricum Gaertn. Plant Mol Biol Rep 30(5):1172–1182. https://doi.org/10.1007/s11105-012-0431-9
Li X, Kim YB, Kim Y, Zhao S, Kim HH, Chung E, Jai-Heon L, Park SU (2013) Differential stress-response expression of two flavonol synthase genes and accumulation of flavonols in tartary buckwheat. J Plant Physiol 170(18):1630–1636. https://doi.org/10.1016/j.jplph.2013.06.010
Liu BG, Zhu YY (2007) Extraction of flavonoids from flavonoid-rich parts in tartary buckwheat and identification of the main flavonoids. J Food Eng 78(2):584–587. https://doi.org/10.1016/j.jfoodeng.2005.11.001
Liu MY, Fu QK, Ma ZT, Sun WJ, Huang L, Wu Q, Tang ZZ, Bu TL, Li CL, Chen H (2019) Genome-wide investigation of the MADS gene family and dehulling genes in tartary buckwheat (Fagopyrum tataricum). Planta 249(5):1301–1318. https://doi.org/10.1007/s00425-019-03089-3
Liu MY, Ma ZT, Wang AH, Zheng TR, Huang L, Sun WJ, Zhang YJ, Jin WQ, Zhan JY, Cai YT, Tang YJ, Wu Q, Tang ZZ, Bu TL, Li CL, Chen H (2018) Genome-wide investigation of the auxin response factor gene family in Tartary buckwheat (Fagopyrum tataricum). Int J Mol Sci 19(11):3526. https://doi.org/10.3390/ijms19113526
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290(5494):1151–1155. https://doi.org/10.1126/science.290.5494.1151
Magali L, Patrice D, Gert T, Kathleen M, Yves M, Yves VDP, Pierre R, Stephane R (2002) PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res 30(1):325–327. https://doi.org/10.1093/nar/30.1.325
Mayerhofer LE, Macario ECD, Macario AJL (1995) Conservation and variability in Archaea:protein antigens with tandem repeats encoded by a cluster of genes with common motifs in Methanosarcina mazei S-6. Gene 165(1):87–91. https://doi.org/10.1016/0378-1119(95)00524-A
Misra P, Pandey A, Tiwari M, Chandrashekar K, Sidhu OP, Asif MH, Charkrabarty D, Singh PK, Trivedi PK, Nath P (2010a) Modulation of transcriptome and metabolome of tobacco by Arabidopsis transcription factor, AtMYB12, leads to insect resistance. Plant Physiol 152(4):2258–2268. https://doi.org/10.1104/pp.109.150979
Nguyen NH, Kim JH, Kwon J, Jeong CY, Lee W, Lee D, Hong SW, Lee H (2016) Characterization of Arabidopsis thaliana FLAVONOL SYNTHASE 1 (FLS1) -overexpression plants in response to abiotic stress. Plant Physiol Biochem 103:133–142. https://doi.org/10.1016/j.plaphy.2016.03.010
Ohsako T, Yamane K, Ohnishi O (2002) Two new Fagopyrum (Polygonaceae) species, F. gracilipedoides and F. jinshaense from Yunnan, China. Genes Genet Syst 77(6):399–408. https://doi.org/10.1266/ggs.77.399
Otto SP, Yong P (2002) The evolution of gene duplicates. Adv Genet 46:451–483. https://doi.org/10.1016/s0065-2660(02)46017-8
Owens DK, Alerding AB, Crosby KC, Bandara AB, Winkel BSJ (2008) Functional analysis of a predicted flavonol synthase gene family in Arabidopsis. Plant Physiol 147(3):1046–1061. https://doi.org/10.1104/pp.108.117457
Park NI, Li X, Suzuki T, Kim SJ, Woo SH, Park CH, Park SU (2011) Differential expression of anthocyanin biosynthetic genes and anthocyanin accumulation in tartary buckwheat cultivars “Hokkai t8” and “Hokkai t10.” J Agric Food Chem 59(6):2356–2361. https://doi.org/10.1021/jf200020b
Misra P, Pandey A, Tiwari M, Chandrashekar K, Sidhu OP, Asif MH, Chakrabarty D, Singh PK, Trivedi PK, Nath P (2010b) Modulation of transcriptome and metabolome of tobacco by Arabidopsis transcription factor, AtMYB12, leads to insect resistance. Plant Physiol 152(4):2258–2268. https://doi.org/10.1104/pp.109.150979
Thwe AA, Kim YJ, Li X, Park YB, Kim HH, Kim SJ, Park SU (2014) Accumulation of phenylpropanoids and correlated gene expression in hairy roots of tartary buckwheat under light and dark conditions. Appl Biochem 174(7):2537–2547. https://doi.org/10.1007/s12010-014-1203-9
Tian J, Han ZY, Zhang J, Hu YJ, Song T, Yao Y (2015) The balance of expression of Dihydroflavonol 4-reductase and flavonol synthase regulates flavonoid biosynthesis and red foliage coloration in crabapples. Sci Rep 5(1):2258–2268. https://doi.org/10.1038/srep12228
Yoshikazu T, Keiko Y, Masako FM, Yuko F, Hiroyuki F, Toshihiko A, Takaaki K (1996) Molecular and biochemical characterization of three anthocyanin synthetic enzymes from Gentiana triflora. Plant Cell Physiol 37(5):711–716. https://doi.org/10.1093/oxfordjournals.pcp.a029004
Zhang LJ, Li XX, Ma B, Gao Q, Du HL, Han YH, Li Y, Cao YH, Qi M, Zhu YX, Lu HG, Ma MC, Liu LL, Zhou JP, Nan CH, Qin YJ, Wang J, Cui L, Liu HM, Liang CZ, Qiao ZJ (2017) The tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance. Mol Plant 10(9):1224–1237. https://doi.org/10.1016/j.molp.2017.08.013
Acknowledgements
This research was supported by the National Natural Science Foundation of China (31871698).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Handling editor: John Bracht.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yao, Y., Sun, L., Wu, W. et al. Genome-Wide Investigation of Major Enzyme-Encoding Genes in the Flavonoid Metabolic Pathway in Tartary Buckwheat (Fagopyrum tataricum). J Mol Evol 89, 269–286 (2021). https://doi.org/10.1007/s00239-021-10004-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-021-10004-6