
Abstract
The most paradigmatic examples of molecular evolution under positive selection involve genes related to the immune system. Recently, different chloroplastic factors have been shown to be important for plant defenses, among them, the α- and β-subunits of the ATP synthase. The β-subunit has been reported to interact with several viral proteins while both proteins have been implicated with sensitivity to tentoxin, a phytotoxin produced by the widespread fungus Alternaria alternata. Given the relation of both protein to virulence factors, we studied whether these proteins are evolving under positive selection. To this end, we used the dN/dS ratio to examine possible sites under positive selection in several Angiosperm clades. After examining 79 plant genera and 1232 species, we found three times more sites under pervasive diversifying selection in the N-terminal region of the β-subunit compared to the α-subunit, supporting previous results which identified this region as responsible for interacting with viral proteins. Moreover, we found the site 83 of β-subunit under positive selection in several plant genera, a site clearly related to the sensitivity to tentoxin according to biochemistry assays, which possibly reflects the selective pressure of the non-host specific tentoxin across various Angiosperm clades.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The contribution of positive selection to molecular evolution has been a long-standing question in evolutionary genetics. Within the most paradigmatic examples of molecular evolution under positive selection are the genes related to the immune system and antigen receptor evolution. Adaptation to pathogens often involves a constantly altering fitness landscape, setting up repeated adaptive steps by both host and pathogen. These cyclical host–pathogen interactions, also known as arms races (Daugherty and Malik 2012), exemplify a classic Red Queen genetic conflict, with both parties rapidly evolving to deviate from or restore the status quo.
Since they lack mobile defender cells and a somatic adaptive immune system, plants generally rely on the innate immunity of each cell and on systemic signals emanating from infection sites to confront pathogens, including bacteria, viruses, and fungi (Jones and Dangl 2006). In addition to its central role in energy production and redox homeostasis, the chloroplast also synthesizes major phytohormones and plays an active role in the defense response (Serrano et al. 2016). For example, the chloroplast is involved in the synthesis of defense-related molecules, such as the hormones salicylic acid, jasmonic acid, and abscisic acid, and secondary messengers, including calcium and ROS (Serrano et al. 2016). In turn, given its importance as a signaling center for immunity, chloroplasts become a valuable target for pathogenic effectors. These pathogenic effectors facilitate the proliferation of pathogens in the host and contribute to virulence by suppressing the plant immune response, by interfering with the host's physiology to promote nutrient acquisition, or by influencing the function of the organelles and gene expression (Kretschmer et al. 2020).
Among the most important chloroplastic targets of virulence factors are the α- and β-subunits of the catalytic region F1 of the ATP synthase complex, the primary function of which is the synthesis of ATP. Additionally, the β-subunit has also been reported to interact with several viral proteins (Seo et al. 2014; Tu et al. 2015; Gellért et al. 2018) while both proteins have been related to sensitivity to tentoxin (Steele et al., 1977; Avni et al. 1992; Groth and Pohl 2001; Tucker et al. 2001; Groth 2002), a phytotoxin produced by the wide-spread fungus Alternaria alternata (Meena et al. 2017). The β-subunit, for Arabidopsis thaliana and Nicotiana clevelandii, has been shown to interact with multiple viral proteins, among them: (i) the triple gene block protein (TGB1) of the Potexvirus Alternanthera mosaic virus (AltMV) (Seo et al. 2014); (ii) with the helper component-proteinase (HC-Pro) of Potato virus Y (PVY) (Tu et al. 2015); and (iii) the capsid protein of the Cucumber mosaic virus (Gellért et al. 2018). For the former viral protein, TGB1, a selective interaction between the β-subunit and TGB1 L88 variant has been shown to retards severe symptoms caused by the AltMV infection, suggesting an inducement of plant defense response (Seo et al. 2014). In contrast, other studies have found that interaction of this and other chloroplast factors with viral proteins could be prejudicial since it could arrest the ATP synthase complex F1 depleting the photosynthetic activity (Tu et al. 2015; Gellért et al. 2018). According to one study, the residues within the N-terminal region are those interacting with viral proteins (Tu et al. 2015). For the α-subunit, although it presents a highly similar protein structure to the β-subunit (both share the same protein domains (Leyva et al. 2003)), no evidence of virus interaction have been reported so far. In regards to the interaction with tentoxin, several biochemistry studies have indicated the site 83 of the β-subunit (β83) as key for conferring sensitivity to tentoxin in plant species, together with other sites of the α-subunit (e.g., Avni et al. 1992; Tucker et al. 2001; Groth 2002). Given the relation of the β-subunit with several viral proteins, its possible role in the immune systems of plants, and the interaction of the α- and β-subunit to tentoxin, we expected these proteins to be evolving under positive diversifying selection.
In order to assess the selective pressure on α- and β-subunit of chloroplast ATP synthase, we used the dN/dS ratio to examine possible sites under positive selection. The dN/dS ratio compares the rate of substitutions at non-synonymous sites (dN) to the rate of substitutions at synonymous sites (dS), which are presumed to be neutral. The dN/dS ratio indicates the evolutionary force acting on a set of homologous protein-coding sequences, with dN/dS = 1, < 1, and > 1 indicating neutral evolution, purifying selection, and positive diversifying selection, respectively (Yang and Nielsen 2001). This ratio is a stringent signal of positive Darwinian selection that has been used extensively in coding sequences of animal and plant species (e.g., D'Anatro et al. 2017; Qian et al. 2019). After interrogating 79 plant genera and 1232 species, we found three times more sites under pervasive diversifying selection in the N-terminal region of the β-subunit compared to the α-subunit, supporting previous results which identified this region as responsible for interacting with viral proteins. Moreover, we found the site β83 under positive selection in several and distant plant genera, which possibly reflects the selective pressure of the non-host specific tentoxin across various Angiosperm clades. Our results add to the understanding of the molecular evolution of the chloroplastic ATP synthase β-subunit and provide interesting candidate sites to further study the interaction of this protein with virulence factors.
Materials and Methods
Positive Selection Hypothesis on α- and β-Subunits
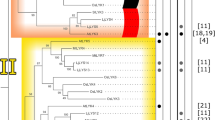
Our working hypothesis is that some residues of the chloroplast ATP synthase α-subunit (encoded by the chloroplastic atpA gene) and β-subunit (encoded by the chloroplastic atpB gene) are experiencing an arms race due to their interaction with virulence factors. To test our hypothesis, we worked with a large number of species belonging to different genera, widely covering the Angiosperms (Fig. 1).

Cladogram of Angiosperm genera analyzed in this work. Each genus presented at least seven species for the subsequent selection analysis. The number of codons under selection for atpB and atpA for each genus studied is also shown. Highlighted in violet are the seven genera that show the site β83 under positive selection. The site β83 is associated with the sensitivity to tentoxin according to previous studies. The cladogram was constructed based on the Angiosperm Phylogeny Group (2016)
The sites presumably interacting with virulence factors are expected to be evolving quickly, and thus, we expect sites under pervasive diversifying selection (i.e., constant positive selection pressure along time or across the entire phylogeny) in very closely related species (i.e., from the same genus). Since viral proteins have been described to interact only with the β-subunit, we expect more genera showing evidence of positive selection and a higher number of sites under selection per genus in the β-subunit compared to the α-subunit. For α- and β-subunit sites that interact with tentoxin, we also expect them to be under positive selection in close species since resistant and sensitive species have been described within genera (Durbin and Uchytil 1977).
Species Sequences, Alignments, and Phylogenies
The nucleotide sequences of the genes analyzed were obtained from the complete genomic sequence of chloroplasts from the NCBI database. After downloading the genome of chloroplasts, we selected only the Angiosperms (flowering plants) genera with seven or more species and extracted the atpA and atpB coding sequences (CDS) using custom python scripts. We required at least seven species per genus to achieve enough statistical power for the selection tests (see “dN/dS Selection Tests” section).
The analyzes performed looked for evidence of positive selection in both genes using two approaches. The first approach consisted of a genus-level analysis, while the second consisted of a global analysis using species from all the genera scanned in the first approach.
In the first approach, for each of the 79 genera obtained (Fig. 1, Table S1), we construct a multifasta file with the corresponding species and proceed to the CDS alignment. The CDS alignments were carried out with the software MACSE v2.03 (Ranwez et al. 2011) using the protein alignment as a template. The protein alignments were previously aligned using MUSCLE v3.8.31 (Edgar 2004a; 2004b). Each of the 79 CDS alignment for both genes was manually checked using AliView (Larsson 2014). Finally, for each genus alignment, we construct a maximum likelihood phylogeny with IQ-TREE v1.6.12 (Nguyen et al. 2015), using the best-fitted substitution model determined by ModelFinder (Kalyaanamoorthy et al. 2017) (Supplementary File 1). These phylogenies and alignments were used as input for the subsequent selection test (see “dN/dS Selection Tests” section).
For the global approach, we used 6 species per genus (totaling 474 species) to scan sites under episodic diversifying selection (see “dN/dS Selection Tests” section). The multiple sequence alignment was performed as previously described (Supplementary File 1). For this analysis, we did not use all the species due to computation constraints. Since this approach includes divergent genera, we checked for substitution saturation of the complete set of sequences (1232 species) before performing the phylogenetic analysis. The substitution saturation of a sequence decreases the phylogenetic signal, affects the phylogenetic analysis involving deep branches, and decreases the power of positive selection tests (Xia et al. 2003; Xia and Lemey 2009; Gharib and Robinson-Rechavi 2013). To test for substitution saturation, we employed the entropy-based index of substitution saturation approach implemented by the DAMBE7 software package (Xia 2018). Briefly, this approach tests if the observed entropy in the sequences (H) is significantly smaller than the entropy of ‘‘full substitution saturation’’ (HFSS). The ISS index is the ratio of observed entropy and the entropy of full substitution saturation (ISS = H/HFSS). If ISS is not significantly smaller than the critical ISS value (the value at which the sequences start to fail to recover the correct tree; ISS.C), one can conclude that the sequences have experienced severe substitution saturation (Xia and Lemey 2009). Finally, we constructed the phylogeny with IQ-TREE v1.6.12 (Nguyen et al. 2015) for atpB, using the best-fitted substitution model determined by ModelFinder (Kalyaanamoorthy et al. 2017).
dN/dS Selection Tests
Our main analysis consisted of scanning the atpA and atpB CDS for positive selection, using the dN/dS-based selection test, in a per genus approach. The selective footprint expected from the virulence factors (viral proteins and tentoxin) associated with the α- and β-subunit should be present in closely related species. The more typical phylogenetic selection approach, which analyzes jointly very divergent species, would also be sensitive to more broad selective processes and thus we mainly used it to further support the sites found in the main analysis and no to uncover additional sites (see below: MEME with 474 species including all the genera).
FUBAR (Fast Unconstrained Bayesian AppRoximation) (Murrell et al. 2013) was used to search for sites under pervasive diversifying selection in each genus. FUBAR takes a Bayesian approach to selection inference that uses a large number of predefined site classes spanning the range of negative, neutral, and positive selection regimes. This software is especially recommended for large analysis (involving numerous species—30–40 sequences) because it is much faster than the typical fixed or random effects likelihood approaches models (e.g., FEL, REL). We choose FUBAR over the other existing approaches because (i) some of our genera present up to 63 species; (ii) we ran multiple analyses (one per genus and gene); and (iii) because it presents more power than FEL (especially when the number of sequences is low, in the order of 7 to 10 sequences, and when positive selection is weak). Using only FUBAR, instead of using different tests for testing pervasive selection, allows for consistency and comparable results. Sites with a posterior probability (pp) equal or higher than 0.90 were considered under pervasive diversifying selection.
To evaluate the sites found with FUBAR, we also tested for episodic diversifying selection using MEME (Mixed Effects Model of Evolution) (Murrell et al. 2012). Briefly, MEME lets each site have a set of free parameters governing the strength of selection for two categories (dN + and dN-), and these parameters are shared for all branches at the site. Later, it detects sites where a proportion of lineages are evolving with dN/dS > 1. For this analysis, we worked with a subset of 474 (six species of each genus) of the 1232 species due to computational constraints. We tested for episodic selection because it is unexpected that the same sites across all the Angiosperm clades to be involved with virulence factors. For MEME, sites were considered under selection if p-values were equal or lower than 0.1. For atpA, the phylogenetic tree obtained for atpB was used as input.
FUBAR was run locally using the HyPhy software package (Kosakovsky Pond et al. 2005) v2.5.1, whereas MEME was run in the Datamonkey server (https://www.datamonkey.org/; Kosakovsky Pond and Frost 2005; Weaver et al. 2018). The results of the different selection tests were parsed using custom python scripts. All in house python scripts used for the analyses can be found on: https://github.com/fagire/atpB_selection_analysis.git.
Statistical Analyses
In order to get a better understanding of our results, we first checked for a correlation between the number of species per genus and the number of sites under selection, since each genus has a different number of species. We also tested for correlation between the number of selected codons per genus between the CDS of atpA and atpB. For both of these correlations, we used the non-parametric rank correlation Spearman’s Rho coefficient.
To evaluate if some sites were significantly over-represented (i.e., found in many genera) in the selection analyses, we calculated approximate p-values in the following manner. We made a script to answer which is the probability that one or more selected sites, of a total of X different selected sites, has Y or more selective instances, of a total of T selective instances (here “T selective instances” equals to the total selected sites -including the repeated ones-). In other words, the script, through an iterative process, distributes T selective instances across the X different selected sites to calculate the approximate p-values. Note that our p-values are conservative since many sites came from the same genus, and each genus presents only different selected sites. The correspondence between the selected sites in the different genera was established using the alignment of the 1232 species (Supplementary File 1).
Finally, since both proteins present a similar structure, we compared the distribution of dN/dS values obtained with the selection analyses of both genes using the non-parametric Kolmogorov–Smirnov test. Since distinct viruses are interacting with several residues of the β-subunit, it is expected that this protein shows higher dN/dS values, producing a longer tail in the distribution of dN/dS values. More in general, the comparison of the distribution of the dN/dS allows us to evaluate if both proteins are experimenting very different regimens of selection (e.g., pervasive, episodic, etc.).
Structural Prediction of Conserved and Variable Regions
We used the ConSurf server (https://consurf.tau.ac.il) (Ashkenazy et al. 2010, 2016; Celniker et al. 2013) to measure the degree of conservation at each aligned position of the β-subunit, especially for the sites identified to be under positive selection. Briefly, the software identifies conserved positions using multiple sequence alignment, calculates the evolutionary conservation rate using empirical Bayesian inference, and provides the evolutionary conservation profiles of the protein (Glaser et al. 2003; Landau et al. 2005). The ConSurf score ranges from 1 to 9, with 1 representing rapidly evolving (variable) sites and 9 representing slowly evolving (evolutionary conserved) sites. The chloroplast F1-ATP synthase's crystal structure complexed with the tentoxin (PDB ID: 1KMH) (Groth 2002) was used in this analysis. The multiple sequence alignment of all the sequences analyzed in this work (1232 species from 79 genera) was produced with MUSCLE and the phylogenetic tree with IQ-TREE. The conservation scores were calculated with a Bayesian method using the evolutionary substitution model cpREV for chloroplast proteins.
Results
Pervasive Diversifying Selection Per Genus
To assess the selective pressure on α- and β-subunit of chloroplast ATP synthase, we performed genus- and global-approaches using the dN/dS ratio to examine possible sites under positive selection. Of the total number of genera downloaded from the NCBI database, 79 presented at least seven or more species (Table S1), our minimum imposed to achieve enough statistical power for the selection tests.
In the first approach, codons under positive selection were sought, for both atpA and atpB, by performing a genus-analysis using FUBAR. For both genes studied, the genera which presented more codons under selection were, in general, those composed by a larger number of species (Spearman's Rho p-value < 0.01 for both genes). It is important to note that this fact did not bias our conclusion since, as the nucleotide sequences of each gene were obtained from the complete chloroplast genome, for each genus, both genes always have the same number of species.
atpB (which encodes the β-subunit of chloroplast ATP synthase) presented 26 genera (33%) with one or more codons under pervasive diversifying selection (Table 1, Table S2). The total number of codons under selection was 45 (Table 1, Table S2), but the number of unique codons was 36 (Table 1). In the case of atpA (which encodes the α-subunit of chloroplast ATP synthase), 23 genera (29%) showed evidence of pervasive diversifying selection, and the total number of codons under selection was 38 (Table S2). Of these, 33 were different or unique codons (Table 1). For both genes, Passiflora was the genus which exhibited more codons under selection (Fig. 1).
When comparing the number of codons under selection per genus, 19 genera presented more selected codons in atpB than in atpA, while 13 presented more selected codons in atpA than in atpB (Fig. 1, Table S2). Forty-seven genera presented exactly the same number of codons under selection (if any) between the two genes, of which only four presented evidence of selection (Fig. 1, Table S2). In general, a high correlation between genes was found across the genera regarding the number of selected codons (Spearman's Rho: 0.36; p-value < 0.001).
The genera which presented the larger differences in codons under selection between both genes were Pelargonium and Lathyrus (Fig. 1, Table S2), the former with four codons in atpB and none for atpA, and the latter with five codons in atpA and one for atpB.
Because both proteins have a very similar structure, it was interesting to know the distribution of the codons under selection throughout the coding sequence of both genes. The results showed that the residues under selection were preferentially concentrated on the N-terminal region and in the C-terminal region for both proteins (Fig. 2, Table S3A, and Table S3B). In the case of β-subunit, of the total sites identified under selection, 19 (of which 13 were unique sites) were located in the first 150 amino-acids of the protein (N-terminal region) and 15 (of which 14 were unique sites) in the last 150 amino-acids of the protein (C-terminal region) (Fig. 2a). However, the α-subunit presented a much higher frequency of amino-acids replacement in the C-terminal region respect to its N-terminal region, presenting only 5 sites (4 unique sites) under selection in the first 150 amino-acids and 24 (20 unique sites) in the last 150 amino-acids (Fig. 2b). Focusing on the N-terminal region, which in both proteins includes the N-terminal domain, the β-subunit presented three times more sites under selection than the α-subunit (13 vs. 4 unique sites, respectively). The average dN/dS for the sites under selection presented in the N-terminal region was close to 8 for both proteins (Table 1).

Distribution of sites under selection identified by FUBAR and MEME in the 79 Angiosperm genera studied. a Frequency of the sites detected under selection (identified by FUBAR) in the 79 genera for the β-subunit. b Frequency of the sites detected under selection (identified by FUBAR) in the 79 genera for the α-subunit. The shadowed zones highlight the main protein domains, according to the Pfam database. The violet arrowheads indicate the shared sites between the FUBAR and MEME analyzes. The inset of each panel shows the number of detected sites along the protein sequence by a disjoint sliding-windows analysis (bin size = 50 amino-acids). The protein position refers to the global alignment of the corresponding CDS
Afterward, we focused on which sites were independently detected under selection in more than one genus. In the alignment of atpB (β-subunit protein), we found that the codon 117 was independently detected under selection in 7 genera (Fig. 2a, Table S3A), which implies a significant overrepresentation of this codon (p-value ~ 0.001). Notably, the codon 117 in our global alignment corresponds to the β83 site related to tentoxin sensitivity in the spinach’s crystal structure (Groth and Pohl 2001; Groth 2002) (Fig. 3 and Supplementary File 2). These genera included Acer, Diplosthephium, Lonicera, Paeonia, Salix, Solanum, and Triodia. Other codons under selection, both in atpB and atpA, occurred in up to 2 or 3 genera (Fig. 2, Table S3A, and Table S3B), but this did not mean a significant overrepresentation (p-value > 0.1).

The variable β83 residue of the β-subunit is located in a highly conserved region of the protein. The evolutionary rates (ConSurf score) of the β-subunit residues were calculated with the ConSurf server (Ashkenazy et al. 2010, 2016; Celniker et al. 2013), using the multiple sequence alignment of all the species (from the 79 genera) analyzed in this work. Residue conservation is plotted onto the protein structure and is colored according to conservation scores ranging from 1 (cyan, variable site) to 9 (purple, high conservated site). The β83 site is indicated, as well as the tentoxin (TTX). In the inset is plotted the ribbon diagram of the structure of a single α-subunit (orange) and a single β-subunit (green), interacting with a molecule of tentoxin (black). The crystal structure of the chloroplast F1-ATP synthase complexed with the tentoxin (PDB ID: 1KMH) (Groth 2002) was used in this analysis. The site β83 of the crystal structure corresponds to the codon site 117 in our global alignment
Finally, we compared the distribution of the dN/dS value for both genes (Fig. S1, Table S3A, and Table S3B). We found that the distributions of the dN/dS values for each gene were statistically indistinguishable from one another (p-value > 0.05). The maximum values were 20 and 22 for atpA and atpB, respectively (Table S3A and Table S3B). For the alignment codon 117 of atpB, in particular, the dN/dS values ranged from 6 to 10 (Table 1, Table S3A).
Episodic Diversifying Selection Between the Species
To further check the sites obtained with FUBAR, we performed a global approach, with six species per genus, to find sites under episodic diversifying selection using MEME. According to the substitution saturation index obtained for the complete set of sequences, they were not saturated (Table S4A and Table S4B), which allowed us to continue with the subsequent analyses.
With MEME, 31 and 25 codons under episodic diversifying selection were found for atpB and atpA, respectively (Table S5A and Table S5B). For atpB, the codons with the highest number of branches (from 11 to 17) under selection were 36, 117, 343, and 527. In atpA, the codons with the highest number of branches (from 10 to 15) under selection were 84, 458, 469, and 490. For both genes, the distribution of selected codons across the CDS was similar to those found in the intra-genus analyses (Table S5A and Table S5B).
Eight codons were shared between FUBAR and MEME analyses for atpB. These codons were: 92, 117, 180, 343, 418, 419, 423, and 467 (Fig. 2a, violet arrowhead). The results for atpA were similar; eight shared codons between the analyses were found: 59, 310, 355, 357, 458, 461, 483, and 490 (Fig. 2b, violet arrowhead). The most interesting codon of those shared between FUBAR and MEME was the atpB codon 117, which corresponds to the β83 site of the crystal structure of spinach’s β-subunit (Supplementary File 2). This site was also identified independently under selection in seven of the genera analyzed.
As shown in Fig. 3, the amino acid residues along the β-subunit were highly conserved, presenting some variable sites preferentially located in the N-terminal and C-terminal regions of the protein. The interesting β83 site (codon 117 in our global alignment) showed a high variation across the sequences (ConSurf score = 1) but is located in a well-conserved region of the protein (ConSurf score = 9).
Discussion
In this work, we studied the α- and β-subunits of the ATP synthase (encoded by the chloroplastic atpA and atpB genes, respectively). These chloroplastic proteins are involved in host-virus and phytotoxin interactions. Since for many proteins that interact with pathogens an evolutionary arm race has been described, we studied if both proteins presented residues evolving at a fast rate compatible with that phenomena. For this end, we worked with 1232 species from 79 genera, representing all the biggest Angiosperm clades, and analyzed sites under selection within each genus.
β-Subunit Presented a Higher Proportion of Sites Under Selection in the N-Terminal Region than the α-Subunit
According to our site-to-site analyses of positive selection (FUBAR and MEME), both genes present widespread evidence of diversifying selection. Strong evidence of positive selection has already been found in other chloroplastic coding sequences along the plant phylogeny (e.g., Kapralov and Filatov 2007; Hermida-Carrera et al. 2017). In particular, for atpB, many authors have pointed out to specific substitutions to explain adaptation to various environments (Fan et al. 2018; Liu et al. 2018; Wu et al. 2018; Xie et al. 2018; Zhang et al. 2018), some of them with extreme characteristics (Zhang et al. 2018; Xie et al. 2018) in various species.
In line with our premise in relation to viral proteins, the β-subunit presented more genera with sites under selection and a higher number of sites under selection when compared to the α-subunit. However, these differences between both proteins were subtle. The main difference between these proteins is in the distribution of codons under selection along their respective CDS. Interestingly, the β-subunit presented a higher proportion of sites under selection in its N-terminal region, which includes the N-terminal domain, compared to the α-subunit. According to one study that addressed the interaction of the tobacco β-subunit with the HC-Pro of PVY, the β-subunit residues that interact with the virus belong to the N-terminal region (Tu et al. 2015). In particular, we found three times more sites under pervasive diversifying selection in the N-terminal region of β-subunit compared to the α-subunit. Besides the described interaction between the N-terminal domain with HC-Pro of PVY (Tu et al. 2015), it is plausible that this domain also interacts with other viral proteins, which would exert additional selective pressure on the domain. The sites found in this region (besides the codon 117/β83, which is known to be related to tentoxin, and will be discussed below) can be useful for future studies looking to characterize the interaction of β-subunit with virulence factors.
The Site β83 and Its Interaction with Tentoxin
The site β83 (codon 117 in our alignment) belong to the N-terminal domain of the β-subunit and, according to independent biochemistry essays, is related to plant resistant and sensitivity to tentoxin (Avni et al. 1992; Groth and Pohl 2001; Groth 2002), a phytotoxin produced by Alternaria alternata (Meena et al. 2017). This site was independently detected under selection in seven genera, showing a significant overrepresentation (p-value ~ 0.001). In turn, this site was also found under episodic diversifying selection using MEME, where 474 species were analyzed. Interestingly, this site presented a dN/dS ratio closer to those sites experimenting weak to intermediate positive selection (dN/dS ~ from 6 to 9) than to those experimenting very strong positive selection (dN/dS ~ 20). This could be due to functional constraints on this site. In fact, almost all the species analyzed (except for Cornus peruviana) presented one of these two amino-acids at this site: aspartic acid (D) or glutamic acid (E). These two amino-acids are exactly those that, according to biochemical analyses, are related to the sensitivity or resistance of plants to tentoxin. Plants carrying the β-subunit with the D variation are sensitive while those carrying the E are resistant to tentoxin (Avni et al. 1992; Groth and Pohl 2001; Groth 2002).
Tentoxin is a non-host specific toxin produced by the fungus Alternaria alternata, and has been recorded that the fungus could infest over 100 host species of plants (Meena et al. 2017). Although we found seven genera with positive selection signals for the codon 117, it is noteworthy that almost all genera carried both variations. For many of the genera used we were able to analyze only seven species (our imposed minimum) and, since the power of the selection test increases as the number of species, our finding is probably limited by the low number of species per genera available. For instance, if more relaxed posterior probabilities are considered, the codon 117 increases its presence significantly, up to 15 genera (9 genera if pp ≥ 0.85, 15 genera if pp ≥ 0.80), in comparison with the other sites under selection. As more chloroplast genomes become available, we think it will be possible to detect evidence of positive selection at this site in more genera. For example, our study did not incorporate the genus Nicotiana, a well-known genus to present sensitive and resistant species to tentoxin (Durbin and Uchytil 1977), because of the limited number of chloroplastic sequences available for this genus.
In regards to the genera that presented this site under selection, namely: Acer, Diplostephium, Lonicera, Paeonia, Salix, Solanum, and Triodia, they were not more closely related to each other in comparison with the remaining genera (Fig. 1). Five of the seven genera have been reported to be infected by Alternaria spp. (except for Diplostephium and Triodia) (Self and Zarko 1975; Akhtar et al. 2004; Oka et al. 2006; González-Díaz et al. 2011; Shao-hua et al. 2011; Hosseinnia and Mohammadi 2018; Mirzwa-Mróz et al. 2018; Wang et al. 2019), and for three of the seven Alternaria leaf spots, caused by Alternaria alternata, were described according to the Plantwise Knowledge Bank (https://www.plantwise.org/KnowledgeBank/). Of our seven genera, bioassays to explore sensitivity to tentoxin have been only carried out in Solanum species (Durbin and Uchytil 1977). The role of site β83 in promoting resistance has been described only for Nicotiana (Avni et al. 1992) and Spinacia oleracea (Groth 2002). In brief, our results not only uncovered new genera potentially affected by tentoxin but also pointed out that the same site across very divergent genera could be associated with tentoxin sensitivity.
The α-Subunit Sites and Tentoxin
As well as for the β-subunit, many sites were found under selection for the α-subunit in various species. However, significant differences can be found between the sites under selection in these two proteins. One, as described above, is the distribution of sites along the protein sequence. The β-subunit presented almost three times more sites under selection in the N-terminal region than the α-subunit, being the sites under selection of the α-subunit preferentially concentrated in the C-terminal region. Another difference is that the α-subunit did not present a significant shared site under selection across the distinct plant genera. This result could indicate that there is not an underlying process affecting the α-subunit in the genera studied, or that the response to such process is clade-specific.
For the α-subunit, six sites (α63, α65, α75, α237, α238, and α274) have been reported to interact, through hydrophobic contact, with tentoxin (Groth 2002). Other important residues controlling the binding of tentoxin in the α-subunit are sites α96 and α133, the latter being replaced by bulky hydrophobic or bulky basic residues in resistant species (Groth & Pohl, 2001). Another study highlighted the sites α129–α133 since substitutions in these positions altered the response to inhibition by tentoxin (Tucker et al. 2001). Additionally, tentoxin has been found to block the contact between the α297 and β83 sites (αβ interface), inhibiting the F1 complex (Groth 2002). However, none of these sites were found to be under positive selection in our analyses (neither in FUBAR nor in MEME). The actions of these sites in the sensitivity to tentoxin are subject to the variants present at the β83 site, which could reduce the individual importance of these sites in relation to tentoxin.
In conclusion, we found evidence of positive selection in the ATP synthase β-subunit consistent with what has been described for the interaction of this protein with virulence factors. Specifically, we found the site β83 under positive selection in various genera, a site related to the sensitivity to tentoxin, and a fast amino-acid replacement in the virus interacting N-terminal region. We think that our results add to the understanding of the molecular evolution of the β-subunit, as well as provide interesting candidate sites to study the interaction of this protein with virulence factors.
References
Akhtar KP, Saleem MY, Asghar M, Haq MA (2004) New report of Alternaria alternaria causing leaf blight of tomato in Pakistan. Plant Pathol 53:816
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44:W344–W350
Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38:529–533
Avni A, Anderson JD, Holland N, Rochaix JD, Gromet-Elhanan Z, Edelman M (1992) Tentoxin sensitivity of chloroplasts determined by Codon 83 of β subunit of proton-ATPase. Science (80-) 257:1245–1247
Celniker G, Nimrod G, Ashkenazy H, Glaser F, Martz E, Mayrose I, Pupko T, Ben-Tal N (2013) ConSurf: using evolutionary data to raise testable hypotheses about protein function. Isr J Chem 53:199–206
D’Anatro A, Giorello F, Feijoo M, Lessa EP (2017) Testing for the occurrence of selective episodes during the divergence of otophysan fishes: insights from mitogenomics. J Mol Evol 84:162–173
Daugherty MD, Malik HS (2012) Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet 46:677–700
Durbin RD, Uchytil TF (1977) A survey of plant insensitivity to tentoxin. Phytopathology 77:602
Edgar RC (2004a) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Edgar RC (2004b) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:1–19
Fan WB, Wu Y, Yang J, Shahzad K, Li ZH (2018) Comparative chloroplast genomics of dipsacales species: Insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front Plant Sci 9:1–13
Gellért Á, Pósa T, Fábián A, Szabó L, Bóka K, Forró B, Salánki K, Drahos L, Tóth E, Juhász A, Balázs E (2018) A single point mutation on the cucumber mosaic virus surface induces an unexpected and strong interaction with the F1 complex of the ATP synthase in Nicotiana clevelandii plants. Virus Res 251:47–55
Gharib WH, Robinson-Rechavi M (2013) The branch-site test of positive selection is surprisingly robust but lacks power under synonymous substitution saturation and variation in GC. Mol Biol Evol 30:1675–1686
Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N (2003) ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19:163–164
González-Díaz JG, García-Velasco R, Camacho-Cerón G, Nieto-Ángel D (2011) CANKER IN Salix bonplandiana KUNTH TWIGS CAUSED BY Alternaria tenuissima (KUNZE EX PERS.) WILTSHIRE. Agrociencia 45:75–86
Groth G (2002) Structure of spinach chloroplast F1-ATPase complexed with the phytopathogenic inhibitor tentoxin. Proc Natl Acad Sci USA 99:3464–3468
Groth G, Pohl E (2001) The structure of the chloroplast F1-ATPase at 3.2 Å resolution. J Biol Chem 276:1345–1352
Group TAP, Chase MW, Christenhusz MJM, Fay MF, Byng JW, Judd WS, Soltis DE, Mabberley DJ, Sennikov AN, Soltis PS, Stevens PF (2016) An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc 181:1–20
Hermida-Carrera C, Fares MA, Fernández Á, Gil-Pelegrín E, Kapralov MV, Mir A, Molins A, Peguero-Pina JJ, Rocha J, Sancho-Knapik D, Galmés J (2017) Positively selected amino acid replacements within the RuBisCO enzyme of oak trees are associated with ecological adaptations. PLoS ONE 12:1–21
Hosseinnia A, Mohammadi A (2018) Investigating the pathogenicity of Alternaria alternata on Lonicera japonica. Azarian J Agric 5:44–48
Jones JDG, Dangl JL (2006) The plant immune system. Nature 444:323–329
Kalyaanamoorthy S, Minh BQ, Wong TKF, Von HA, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Kapralov MV, Filatov DA (2007) Widespread positive selection in the photosynthetic Rubisco enzyme. BMC Evol Biol 7:1–10
Kosakovsky Pond SL, Frost SDW (2005) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533
Kosakovsky Pond SL, Frost SDW, Muse SV (2005) HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679
Kretschmer M, Damoo D, Djamei A, Kronstad J (2020) Chloroplasts and plant immunity: Where are the fungal effectors? Pathogens 9:1–16
Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 33:299–302
Larsson A (2014) AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30:3276–3278
Leyva JA, Bianchet MA, Amzel LM (2003) Understanding ATP synthesis: structure and mechanism of the F1-ATPase (review). Mol Membr Biol 20:27–33
Liu ML, Fan WB, Wang N, Bin DP, Zhang TT, Yue M, Li ZH (2018) Evolutionary analysis of plastid genomes of seven lonicera L. species: Implications for sequence divergence and phylogenetic relationships. Int J Mol Sci 19:1–17
Meena M, Gupta SK, Swapnil P, Zehra A, Dubey MK, Upadhyay RS (2017) Alternaria toxins: potential virulence factors and genes related to pathogenesis. Front Microbiol 8:1451
Mirzwa-Mróz E, Kukula W, Frydrych I, Wit M, Wakulińsk W (2018) First report of Alternaria black spot caused by Alternaria Alternata on blue honeysuckle in Poland. Plant Dis 102:820
Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K (2013) FUBAR: a fast, unconstrained bayesian AppRoximation for inferring selection. Mol Biol Evol 30:1196–1205
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL (2012) Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764
Nguyen LT, Schmidt HA, Von HA, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274
Oka K, Okubo A, Kodama M, Otani H (2006) Detoxification of α-tomatine by tomato pathogens Alternaria alternata tomato pathotype and Corynespora cassiicola and its role in infection. J Gen Plant Pathol 72:152–158
Qian J, Liu Y, Ma C, Chao N, Chen Q, Zhang Y, Luo Y, Cai D, Wu Y (2019) Positive selection of squalene synthase in Cucurbitaceae plants. Int J Genomics 2019:5913491
Ranwez V, Harispe S, Delsuc F, Douzery EJP (2011) MACSE: multiple alignment of coding sequences accounting for frameshifts and stop codons. PLoS ONE 6:e22594
Self RL, Zarko ML (1975) Control of Alternaria alternata on red maple (Acer palmatum). Proc SNA Res Conf Annu Rep South Nurserymen’s Assoc 20:57–58
Seo EY, Nam J, Kim HS, Park YH, Hong SM, Lakshman D, Bae H, Hammond J, Lim HS (2014) Selective Interaction between chloroplast β-ATPase and TGB1L88 retards severe symptoms caused by Alternanthera mosaic virus infection. Plant Pathol J 30:58–67
Serrano I, Audran C, Rivas S (2016) Chloroplasts at work during plant innate immunity. J Exp Bot 67:3845–3854
Shao-hua W, You-wei C, Zhi-ying L, Li-yuan Y, Shao-lan L (2011) Metabolites of the endophytic fungus Alternaria sp PR-14 of Paeonia delavayi. Nat Prod Res Dev 23:850
Steele JA, Uchytil TF, Durbin RD (1977) The binding of tentoxin to a tryptic digest of chloroplast coupling factor 1. Biochim Biophys Acta 459:347–350
Tu Y, Jin Y, Ma D, Li H, Zhang Z, Dong J, Wang T (2015) Interaction between PVY HC-Pro and the NtCF1 β-subunit reduces the amount of chloroplast ATP synthase in virus-infected tobacco. Sci Rep 5:1–14
Tucker WC, Du Z, Hein R, Gromet-Elhanan Z, Richter ML (2001) Role of the ATP synthase α-subunit in conferring sensitivity to tentoxin. Biochemistry 40:7542–7548
Wang JT, Ma ZH, Wang GK, Xu FQ, Chen L, Yu Y, Wang G, Liu JS (2019) Four meroterpenoids from Alternaria alternata isolated from Paeonia lactiflora. Phytochem Lett 31:1–4
Weaver S, Shank SD, Spielman SJ, Li M, Muse SV, Kosakovsky Pond SL (2018) Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol Biol Evol 35:773–777
Wu Y, Liu F, Yang DG, Li W, Zhou XJ, Pei XY, Liu YG, He KL, Zhang WS, Ren ZY, Zhou KH, Ma XF et al (2018) Comparative chloroplast genomics of Gossypium species: insights into repeat sequence variations and phylogeny. Front Plant Sci 9:1–14
Xia X (2018) DAMBE7: new and improved tools for data analysis in molecular biology and evolution. Mol Biol Evol 35:1550–1552
Xia X, Lemey P (2009) Assessing substitution saturation with DAMBE. In: Salemi M, Vandamme A-M, Lemey P (eds) The phylogenetic handbook: practical approach to phylogenetic analysis and hypothesis testing. Cambridge University Press, Cambridge, pp 615–630
Xia X, Xie Z, Salemi M, Chen L, Wang Y (2003) An index of substitution saturation and its application. Mol Phylogenet Evol 26:1–7
Xie DF, Yu Y, Deng YQ, Li J, Liu HY, Zhou SD, He XJ (2018) Comparative analysis of the chloroplast genomes of the chinese endemic genus urophysa and their contribution to chloroplast phylogeny and adaptive evolution. Int J Mol Sci 19:1–20
Yang Z, Nielsen R (2001) Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol 19:908–917
Zhang Z, An M, Miao J, Gu Z, Liu C, Zhong B (2018) The Antarctic sea ice alga Chlamydomonas sp. ICE-L provides insights into adaptive patterns of chloroplast evolution. BMC Plant Biol 18:1–12
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Handling Editor: David Alvarez-Ponce.
Electronic supplementary material
Below is the link to the electronic supplementary material.
239_2020_9968_MOESM3_ESM.fasta
Supplementary File 2. Alignment of atpB showing the correspondence of the codon 117 with the spinach’ β83 site, related to tentoxin sensitivity (FASTA 787 kb)
239_2020_9968_MOESM4_ESM.png
Figure S1. Distribution of dN/dS values for atpB and atpA. The density for atpB is represented in red while the density for atpA is displayed in blue (PNG 144 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Farias, J., Giorello, F.M. Positive Selection in the Chloroplastic ATP-Synthase β-Subunit and Its Relation to Virulence Factors. J Mol Evol 88, 703–713 (2020). https://doi.org/10.1007/s00239-020-09968-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-020-09968-8