
Abstract
Presenilin proteins make the catalytic component of γ-secretase, a multiprotein transmembrane protease, and are type II transmembrane proteins. Amyloid protein, Notch, and beta catenin are among more than 90 substrates of Presenilins. Mutations in Presenilins lead to defects in proteolytic cleavage of its substrate resulting in some of the most devastating pathological conditions including Alzheimer disease (AD), developmental disorders, and cancer. In addition to catalytic roles, Presenilin protein is also shown to be involved in many non-catalytic roles, i.e., calcium homeostasis, regulation of autophagy, and protein trafficking, etc. These proteolytic proteins are highly conserved and are present in almost all the major eukaryotic groups. Studies, performed on a wide variety of organisms ranging from human to unicellular dictyostelium, have shown the important catalytic and non-catalytic roles of Presenilins. In this study, we infer the evolutionary patterns and history of Presenilins as well as of other γ-secretase proteins. We show that Presenilins are the most ancient of the γ-secretase proteins and that Presenilins may have their origin in the last common ancestor (LCA) of Eukaryotes. We also demonstrate that Presenilin proteins generally lack diversifying selection during the course of their evolution. Through evolutionary trace analysis, we show that Presenilin protein sites that undergo mutations in Familial Alzheimer disease, are highly conserved in metazoans. Finally, we discuss the evolutionary, physiological, and pathological implications of our findings and propose that the evolutionary profile of Presenilins supports the loss of function hypothesis of AD pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
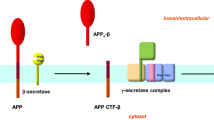
Presenilin proteins are known to perform important roles in a wide variety of catalytic and non-catalytic processes. For instance, Presenilins are the major catalytic component of γ-secretase, a multi-complex protease involved in the catalysis of many membrane proteins (De Strooper et al. 1997; Zhang et al. 2000). Presenilins perform this catalytic activity with the aid of three other γ-secretase proteins namely Nicastrin, APH-1 and PEN-2 (Goutte et al. 2002; Edbauer et al. 2003; Takasugi et al. 2003). Structurally, Presenilin is a multipass transmembrane protein with nine transmembrane domains (TMDs) (Laudon et al. 2005; Spasic et al. 2006). In humans, there are two Presenilin paralogs, Presenilin-1 and Presenilin-2, showing 67% sequence identity (Levy-Lahad et al. 1995). Both these Presenilin proteins are ubiquitously expressed in many human tissues including the brain, heart, kidney, and muscle (Lee et al. 1996). Inside the cells, Presenilin is abundantly found in the endoplasmic reticulum and Golgi bodies (Walter et al. 1996; De Strooper et al. 1997; Area-Gomez et al. 2009). An important aspect of Presenilin is that under normal physiological conditions, it undergoes endoproteolysis generating N and C terminal fragments (NTF and CTF, respectively) (Thinakaran et al. 1996; Brunkan et al. 2005) involving amino acids 292–299 (Podlisny et al. 1997; Fukumori et al. 2010). This endoproteolytic event is considered important for the γ-secretase activity. However, some Presenilin mutants with defective endoproteolysis still exhibit enzymatic activity (Jacobsen et al. 1999; Capell et al. 2000). There are more than 90 substrates of γ-secretase/Presenilin of which amyloid precursor protein (APP) and the Notch receptor are the predominant ones (Beel and Sanders 2008; McCarthy et al. 2009). It is the defect in Presenilin-based proteolysis of APP that leads to AD; the overproduction of neurotoxic Aβ42 peptide triggers inflammatory responses in brain leading to neuronal dysfunction and ultimately cell death (Duff et al. 1996; Jacobsen et al. 1999; Zhang et al. 2011). However, recently, the validity of the amyloid hypothesis as the sole explanation of AD has been questioned (Shen and Kelleher 2007; Kelleher and Shen 2017). The alternate explanation claims that mutations in Presenilin proteins cause loss of many important Presenilin functions (loss of function mutations) in the brain, which in turn leads to neurodegeneration and dementia, the hallmark features of AD (Sun et al. 2017).
In addition to acting as a major protease involved in the regulated intramembrane proteolysis, Presenilin is involved in many non-catalytic processes too, for instance, in calcium homeostasis (Ludtmann et al. 2014). Mutations in Presenilin are connected with defects in calcium signaling in both neuronal and non-neuronal cells, as well as with Familial Alzheimer disease (FAD) (LaFerla 2002; Supnet and Bezprozvanny 2010; Popugaeva and Bezprozvanny 2013). Presenilin is also involved in the regulation of autophagy in γ-secretase independent way (Bustos et al. 2017b). The mutations in Presenilin are known to be involved in the accumulation of immature autophagic vesicles (Bustos et al. 2017a). The defects in autophagy, due to Presenilin mutations, contribute towards the neurodegeneration in AD patients because of excessive neuronal death (Ohta et al. 2010; Nixon and Yang 2011; Menzies et al. 2015). Finally, Presenilin is also involved in the trafficking of proteins in both γ-secretase dependent (Brown et al. 2000; Cai et al. 2003; Wang et al. 2006b) as well as independent mechanism (Scheper et al. 2004; Suga et al. 2004).
Though it is known that Presenilins are present in a vast variety of eukaryotes, very little work has been done to explore the evolutionary history of this important protein. Various studies conducted in this direction are either limited to elucidate the functional aspects of Presenilins in the evolutionarily diverse panel of model organisms or are done before the advent of next-generation sequencing technology, with limited coverage (Brown et al. 2000; Murray et al. 2000; Ponting 2002; Wang et al. 2006a; Otto et al. 2016). In this regard, this is the first study to elucidate the phylogeny and evolutionary history of Presenilin. It also discusses the physiological implication of the evolutionary profile Presenilin exhibit. In our search, we confirmed through evolutionary analysis and conservation profile across various eukaryotic kingdoms that Presenilin is an ancient protein. We traced its possible presence in eukaryotic last common ancestor (LCA). We also confirmed the findings of Murray et al. (Murray et al. 2000) that Presenilins are very conserved proteins, characterized by a lack of diversifying evolution. We also observed that residues involved in FAD corresponded to conserved sites in Presenilins, which suggests that the evolutionary history of Presenilin does not support the “amyloid hypothesis” but supports the alternative “loss of function hypothesis” as an explanation of the etiology of Alzheimer disease.
Materials and Methods
Identification and Extraction of g-Secretase Proteins
We first made a list of 53 representative species with uniform representation from each clade of Animalia, Fungi, unicellular Eukaryotes, and Plantae, such that they were model organisms, and they had a well-annotated genome available (see suppl. mat. 1). For the collection of homologous protein sequences of the four γ-secretase proteins (Presenilin, Nicastrin, APHI and PEN-2) in each of these representative species, we deployed a three-step approach, whose details are given in suppl. mat. 1.
Phylogenetic Analysis
Phylogenetic analyses were performed using a Bayesian inference approach implemented in Bayesian Evolutionary Analysis Sampling Trees 2 (BEAST2) program version 2.6.0 (Bouckaert et al. 2019). Briefly, a total of 148 protein sequences (divided into 66, 25, 30, and 27 proteins for Presenilin, APH-1, Nicastrin, and PEN-2 gene families, respectively (see suppl. mat. 1), were collected from 53 different species ranging from primates to simplest of unicellular eukaryotes as described in the previous section. Then the amino acid sequences of each group were downloaded from the NCBI protein database using the identifiers (suppl. mat. 2). Syntenic information, based on the neighborhood of genes in the respective genome, was used to verify homologs in all the four gene families using GenFamClust version 1.0.3 (Walter et al. 1996; Ali et al. 2013, 2016) pipeline with default settings (further explanation and graphical display in suppl. mat. 3). Each of these four groups of homologs is aligned by using default settings in Clustal Omega version 1.2.1 (ClustalO) (Sievers and Higgins 2014a, b). Then the multiple sequence alignments were filtered/divvied using divvier version 1.0 (Ali et al. 2019) with default settings. Only the threshold for merging two columns was changed to 0.55 (using -thresh 0.55) instead of the default threshold of 0.81 (set for BAliBASE datasets—a conservative estimate). Snapshots of all four divvied alignments generated by using Jalview version 2.10.5 (Waterhouse et al. 2009) are shown in suppl. mat. 4.
The dated species tree, used to measure congruence between species tree and gene trees, was inferred from Timetree.org (Kumar et al. 2017) and is shown in suppl. mat. 5, where divergence times for only 30 species were found (the rest 9 species are marked as red in suppl. mat. 1 Table 1). Note that for some species, there is no clear bifurcation in the species tree and are represented by a 0 as the inter-node distance (see, e.g., distances between Zea mays, Brachypodium distachyon, and Oryza sativa). We did not check for incongruence between the gene tree and species tree for these multifurcations as well as for the missing species in the species tree.
The evolutionary history for Presenilin, APH-1, Nicastrin, and PEN-2 gene families were inferred by using BEAST 2 version 2.6.0 with default settings, and MCMC chain running for 10 million iterations, sampled after every 1000 iterations, to generate 10,001 samples for each gene family. Then VMCMC was used to assess burn-in and convergence of chain for each numeric parameter as well as for tree parameters. The convergence estimates by VMCMC from all parameters for all gene trees were less than 10%, so we removed 10% of samples (corresponding to 1000 samples) from each chain and the remaining 9000 samples were assessed for determining majority consensus tree for a gene family. All phylogenetic trees are drawn using forester in Archaeopteryx version 0.9920 beta (Han and Zmasek 2009).
For selection analysis, coding sequences of vertebrate Presenilin-1 and Presenilin-2 gene were extracted for nine vertebrates from the NCBI genome database (Fig. 2b and suppl. mat. 6). We looked for diversifying selection on each codon of Presenilins by making use of the Mixed-Effect Model of Episodic selection (MEME) employed at the Datamonkey server (Kosakovsky Pond and Frost 2005). MEME is a robust method that employs a mixed-effects maximum likelihood approach to test whether individual sites have been subjected to positive or diversifying selection. The method is very sensitive as it looks for episodic selection for each site too, while other existing methods look for pervasive selection (Murrell et al. 2012). The evolutionary distance between all possible pairs of vertebrate Presenilin paralogs was estimated by Tajima’s relative test employed in Mega6 software (Tajima 1993; Tamura et al. 2013). Each pair of paralogs was compared with the Presenilin protein sequence of Saccoglossus kowalevskii, which was chosen as an outgroup to other species.
Evolutionary Trace Analysis (ETA) and Homology Modeling
For determining the evolutionarily conserved residues, evolutionary trace analysis (ETA) was employed. ETA is a powerful technique that ranks the individual amino acids based on their conservation profile, i.e., lower the value of the rank, more conserved the residue is and vice versa (Lichtarge et al. 1996; Wilkins et al. 2012). The ETA results were incorporated for evolutionary structural analysis by employing pyETV and CHIMERA software tools (Pettersen et al. 2004; Lua and Lichtarge 2010). The heatmaps for the figurative representation of ETA results are generated by using the conditional formatting tool embedded in Microsoft excel (Meyer and Avery 2009). Secondary structure prediction was carried out by using SPLIT 4, an online server for the prediction of secondary structures of transmembrane proteins (Juretić et al. 2002). Homology modeling of Presenilin protein was done by using the human Presenilin-1 protein structure (pdb 5fn2) as a template. ClustalO tool, embedded in Mega6, was used to select homologous parts between 5fn2 and orthologous protein sequences. Modeller 9.20 software was used to generate the homology models keeping the number of models generated to 10 (Webb and Sali 2016). The models with the lowest DOPE score and best Ramachandran plot were selected for analysis. CHIMERA was used for visualizing and analyzing the models (Pettersen et al. 2004).
Results
Distribution and Extent of Duplication of g-Secretase Proteins Across Eukaryotes
For inferring the evolutionary history of metazoan Presenilins, we investigated the distribution of four γ-secretase proteins, Presenilins, APH-1, PEN-2, and Nicastrin, in 53 species representing all the major groups in eukaryotes and covering a time span of approximately one billion years. (Table 1 and suppl. mat. 1). The distribution of Presenilins among eukaryotes, in comparison with the other three gamma-secretase proteins, revealed two major findings. First, the four secretase proteins are present in all three multicellular divisions of eukaryotes (i.e., plants, animals, and fungi) though some major subgroups in unicellular eukaryotes do not contain homologs of APH-1, Nicastrin, and PEN-2 (for example, see for Rhizaria, Excavates and Rhodophytes in Table 1). Missing gene annotations for proteins/genome should be ruled out as a reason for the absence of homologs as we deployed tblastn and manually inspected the hit regions in the genome to address this issue specifically. Second, Presenilin is a nearly universal protein present in almost all major clades of eukaryotes. It is found in simple unicellular organisms, e.g., in Thalassiosira pseudonana as well as in complex organisms, e.g., in Mus musculus and all major subgroups of plants, e.g., in monocots and dicots (see suppl. mat. 1). Some exceptions to this nearly universal presence of Presenilins are members of Rhizaria clade, Nicotiana benthamiana (a eudicot), Tetrahymena thermophila (an alveolate), Polysphondylium pallidum (an amoebazoa), Naegleria gruberi (an excavate), and Saccharomyces from Ascomycotes group. Interestingly, we were unable to find any species/clade, in which Presenilin is absent but any of the other three γ-secretase protein is present (Table. 1).
This interesting distribution of γ-secretase proteins was intriguing and led us to explore the pattern of evolution of these proteins across the species tree as well. For this purpose, we inferred the pattern of evolution of protein homologs of APH-1A, APH-1B, Nicastrin, PEN-2, Presenilin-1, and Presenilin-2. The phylogenetic consensus tree, derived from the Bayesian inference method and divvied multiple sequence alignment for Presenilin proteins, is shown in Fig. 1. The corresponding phylogenetic trees for other γ-secretase proteins are shown in suppl. mat. 7–9. We compared for topological congruence between the dated species tree (shown in suppl. mat. 5) and the gene trees for all four γ-secretase proteins. The Bayesian consensus trees, inferred for all four γ-secretase proteins, are generally congruent with the dated species tree for the 30 species.

Bayesian phylogenetic tree of Presenilin proteins. The figure displays the evolutionary history of the Presenilin gene family through a rooted consensus phylogenetic tree of Presenilin proteins along with consensus values for each node on the tree. The tree was rooted in the most ancient eukaryotic species used in the analysis (Thalassiosira pseudonana). The proteins are named such that the first part before underscore is protein abbreviation (e.g., Pres for Presenilin) and the second part after the underscore is the species abbreviation (e.g., Hom-sap for Homo sapiens, see Suppl. Mat. 1 for all abbreviations). The protein’s names and branches are colored by the kingdom. Dark blue—unicellular Eukarya, dark green—Plantae, orange—Fungi, red and purple—Invertebrates and Vertebrates in Animalia, respectively. Consensus values below 0.6 are not shown. Ancient gene duplication events are marked with a golden star, representing gene duplications at the LCA of an important group of species (e.g., at the root of monocots and eudicots in Plantae in Presenilin). Other recent gene duplications, where a gene duplication event happened in the ancestor common to a few species, are marked with a silver star while gene duplication events within the species (termed “species-specific duplications” in literature) are marked with a silver circle. The topology of the tree is in general in agreement with the species tree (displayed in suppl. mat. 5) (Color figure online)
Another interesting observation is the extent of duplications of Presenilins and other γ-secretase genes in eukaryotes. Despite their presence in the same protein complex and functioning together, the four genes followed a different pattern of gene duplications in different species across eukaryotes. For example, in multicellular organisms, there is a probable ancient gene duplication event at the root of vertebrates for Presenilin and APH-1 but there is only one homolog of Nicastrin and PEN-2 in each of the vertebrates (see suppl. mat. 7, 8, and 9, in particular for events marked with a golden star in each tree). Presenilin had two ancient gene duplication events at the LCA of important subgroups (one at the root of vertebrates, and the other at the root of eudicots and monocots in plants—both complex organisms) as shown in Fig. 1. It is thus inferred that the recent gene duplication events leading to “species-specific duplications” (or recent paralogs—paralogs with roughly the same sequence), (Koonin 2005) are rare in APH-1, Nicastrin and PEN-2, but are abundant, in comparison, in Presenilin.
A simplistic way of comparing gene duplication rates is to count and compare the number of multiple paralogs in various species. In the case of γ-secretases, five different species from diverse groups across eukaryotes (Trichomonas vaginalis, Saprolegnia diclina, and Aphanomyces invadans from unicellular eukaryotes, Zea mays from plants, and Caenorhabditis elegans from Animalia) contain three paralogs of Presenilin, but only one species is containing three paralogs from the rest of three γ-secretase genes (APH-1 in Mus musculus), see suppl. mat. 1. Similarly, the number of species containing exactly two paralogs is highly unbalanced (17 in Presenilin vs. 6 for the other γ-secretase genes). Hence, there is a high gene duplication rate in Presenilin as compared to other γ-secretase genes.
Extent of Diversifying Selection
To investigate the extent of diversifying or positive selection in vertebrate Presenilin paralogs, namely Presenilin-1 and Presenilin-2, the most widely distributed γ-secretase proteins in eukaryotes, we employed MEME. In MEME analysis, we found only five codons in Presenilin-1 and two codons in Presenilin-2 that were under positive selection. Noticeably, a predominant majority of codons in both proteins not only exhibited p-values higher than the significance level of 0.05 but also higher than the very non-significant level of 0.5, e.g., 93% for Presenilin-1 and 90% for Presenilin-2 (Fig. 2 and suppl. mat. 11). Therefore, we inferred that overall, the vertebrate Presenilin proteins are evolving either under purifying selection or neutral selection but not under diversifying selection.

Selection analysis of Presenilin proteins in vertebrates. a Sliding window display of nonsynonymous and synonymous differences for each site (codon) in vertebrate Presenilins. The MEME method from the datamonkey server was employed for this analysis. The position of each codon is given on X-axis. The horizontal red bar in the graph refers to a p-value of 0.05, which is taken as the significance level for positive selection. The Y-axis is broken to accommodate the whole data for clarity. b Representation of MEME results on branches of Presenilin-1 and Presenilin-2 tree. Branches with red color represent positive selection. Numbers on the positively selected branches indicate the codon number which is undergoing positive selection (Color figure online)
Finally, we employed Tajima’s relative rate test to find out if there is any significant difference between rates of evolution of two Presenilin proteins. In this test, we compared the extent of substitutions in both vertebrate Presenilin paralogs by taking the protein sequence of a third species as an outgroup in which the respective protein exists as an unduplicated singleton, e.g., the invertebrate Saccoglossus kowalevskii. The p-values from Tajima’s test showed that there is no significant difference in evolutionary rate between two paralogs (Suppl. Mat. 12).
Evolutionary Traces in Metazoan Presenilins
From ETA, we identified 103 residues in Presenilins which were conserved (with the rank value of 1) throughout the metazoans, from primates to choanoflagellates (Fig. 3 and Supplementary file 10). We also looked for the location of these highly ranked residues along the whole length of Presenilin protein. Interestingly, the majority of highly ranked residues were present in the transmembrane part of the protein (Fig. 3b, c). In order to see the biological importance of these traces, we made use of the site-directed mutagenesis data for Presenilin-1 available on Uniprot (Pundir et al. 2017) (Fig. 3d). These mutations lead to a wide range of phenotypic effects, i.e., loss of interaction with Glial Fibrillary Acidic Protein (GFAP) (Φ), increased protease activity (Ψ), altered γ-secretase specificity (α), reduced Noch processing (Ω), abolished protease activity (β), abolished caspase cleavage (∆), and abolished protein kinase A (PKA) signaling (Π). As was expected, a considerable number of mutating sites (60%) were highly conserved among the metazoans as depicted by their very low rank values of 1 or 2.

taken from the work of Zhang et al. (2013). c Bar graph showing the frequency of rank values/conservation score (with 1 being for the most conserved and 10 being the least conserved residue) with their corresponding rank values/conservation scores in the transmembrane and non-transmembrane portion of the Presenilin-1 protein. d List of site-directed mutations (taken from UniProt database) which have been carried out in the Presenilin-1 gene. Column-1 represents the position of residues; column-2 shows the heatmap, depicting conservation profile of mutated sites; column-3 represents substituted residues on the left side of the arrow and replacement residues on the right side of the arrow with more than one replacement residues giving rise to a specific phenotype are separated by a comma, and finally, the phenotypic effects due to these mutations are given in columns 4 where Φ-Loss of interaction with GFAP, Ω-abolished protease activity, Ψ-increased protease activity, ∆-abolished caspase cleavage, α-alters γ-secretase specificity, Π-abolished PKA signaling, and β-reduced notch processing (Color figure online)
Evolutionary traces analysis of Presenilin-1 protein. a A heatmap of evolutionary traces found in metazoan Presenilin. The color shades from dark brown to white correspond to the most conserved to the least conserved residues. The ETA is carried out by using 15 species, covering major metazoan groups from primates to choanoflagellates. b A Schematic heatmap-based representation of Presenilin-1 protein across the cell membrane. The color shades correspond to the same conservation profile as mentioned in a. The scheme of arrangement of Presenilin across the membrane is
Homology Modeling and Molecular Docking
In addition to the simple sequence-based analysis, we also performed structure-based bioinformatics analysis. Taking the 3D structure of human Presenilin protein (5FN2), we made homology models of Presenilin protein from fish and sponge (metazoans) and from dictyostelium (unicellular eukaryote) to see the effect of sequence divergence on Presenilin structure during the course of eukaryotic evolution (spanning about 1 billion years). Both secondary and tertiary structures of Presenilins in these species (Fig. 4a, b) showed an extreme level of conservation as depicted by the number of TMDs, the presence of catalytic aspartates along with the surrounding catalytic cavity, the distance of about 4 Å between the two catalytic aspartates present in the catalytic cavity in membrane 6 and 7, and columbic surface coloring depicting the electrostatic overview of these orthologous proteins.

Homology modeling of Presenilin protein. a A representation of the secondary structure predicted for a diverse range of species, ranging from vertebrates to non-vertebrates and protists. Red circles in the TMD6 and TMD7 represent the catalytic aspartate residues. b Upper panel: Human Presenilin protein structure along with its homologous models from three different species. The Brown highlighted portion represents the presence of two catalytic aspartates. Middle panel: Distance between the catalytic aspartates in transmembrane segments 6 and 7. Lower panel: Depiction of electrostatic charge distribution on Presenilin orthologs models by columbic surface coloring. The electrostatic potential is represented by coulombic surface coloring in the range of values-10 (red), 0 (white), and 10 (blue) kcal/mol. Preservation of electrostatic profile from human to unicellular eukaryotes can clearly be seen (Color figure online)
Alzheimer Disease and Presenilin Evolution
We used the Uniprot database for natural variants (Pundir et al. 2017) in human Presenilin-1 protein causing familial Alzheimer disease (FAD) to elucidate the relationship between the evolutionary profile of Presenilin protein and Alzheimer disease. When ETA was performed on these AD/FAD-causing sites, a predominant number of sites exhibited a significantly high level of conservation (70%, with a z-score of 5.52 and p-value < 0.00001 at 0.5 significance level) throughout metazoans from human to choanoflagellates (Fig. 5a, b). The majority of rank 1 residues lie in the alpha helix as was observed during the structural analysis in which ETA-based ranked residues were mapped on existing human Presenilin-1 structure (Fig. 5c). Similarly, the surface view of the structure (Fig. 5d) revealed the predominant presence of these conserved sites in the hydrophobic region of the protein, which is consistent with our analysis (Fig. 3b) that a big majority of these residues lie in the transmembrane part of the protein.

Evolution of Presenilin and Alzheimer's disease. a List of Presenilin-1 mutations leading to AD along with the conservation profile of each mutated site in the form of a heatmap. Column-1 represents the position of each residue; column-2 represents heatmap in the form of color shades from dark brown to white corresponding to most conserved to least conserved residues; column-3 represents substituted residues and column-4 is showing replacement residues. b Pie graph showing the extent of conservation (obtained from multiple sequence alignment of Presenilin proteins in different eukaryotic species, ranging from primates to unicellular organisms) of evolutionary traces shown in 5A. c and d The ETA-based structural analysis of the Presenilin protein by using the Cryo-EM structure of human Presenilin-1 (5NF2) as a template. Both ribbon (c) and surface (d) representations are shown. In surface representation, the dodger blue color refers to the most hydrophilic and orange-red to the most hydrophobic residues while the white color refers to neutral residues. The highly conserved residues (traces) susceptible to AD are indicated by black colored one letter name. ETA-based ranks are generated by comparing protein sequences of 15 species, covering major metazoan groups from primates to choanoflagellates (Color figure online)
Discussion
γ-Secretase is a protein complex which along with β-secretase is involved in the successive proteolysis of the amyloid precursor protein. This produces amyloid β-protein whose abnormal deposition in the brain leads to AD. Of the four proteins making γ-secretase complex, it is the catalytic role of Presenilin which is most prominently involved in AD pathogenesis. Since Presenilin performs a significant role in AD as well as other catalytic and non-catalytic roles in a wide range of evolutionarily diverse eukaryotes, it is important to elucidate the evolutionary history of Presenilin and its implication in the functions it performs.
A protein blastp-based search, reaffirmed by protein blastn search on genomic regions and subsequent syntenic analysis (for plant and vertebrate homologs only) for all the γ-secretase proteins, showed that these proteins are widely distributed in all the eukaryotic lineages (see Table 1 and suppl. mat. 1). However, Presenilin showed the widest distribution among the four proteins, reflecting its vital role. Among all the 64 species included in our sample for protein evolution study, only a few species, namely Bigelowiella natans, Reticulomyxa filose, Polysphondylium pallidum, Nicotiana benthamiana, Naegleria gruberi (an Excavate), Tetrahymena thermophila, Saccharomyces cerevisiae, and Schizosaccharomyces pombe (Ascomycotes), lacked Presenilin gene in their genome. The absence of some genes in parasitic species, e.g., in Naegleria gruberi, is understandable as parasitic organisms tend to have reduced genome because of repeated gene loss during evolution (Keeling and Slamovits 2005; Sakharkar and Chow 2005; Slamovits 2013). Similarly, the absence of Presenilin in Saccharomyces cerevisae and Schizosaccharomyces pombe, model organisms representing Ascomycotes (a symbiont Fungi), may be explained as well through similar mechanisms (Espagne et al. 2008; Fan et al. 2015). But the absence of Presenilin in Tetrahymena thermophila, a free-living ciliate, is surprising. However, it is worth noting that Ciliates also seem to lack other important substrates of Presenilin, e.g., amyloid precursor protein and Notch (data not shown). This reflects the possibility of the coevolution of Presenilin with its substrates, though more work is needed to draw a conclusion on this aspect of Presenilin evolution. Interestingly, we did not find a single species in which any of the other three γ-secretase proteins was present when Presenilin was absent. This leads to the hypothesis that Presenilin plays a vital role in the gamma-secretase function, and perhaps, γ-secretase activity was assumed solely by Presenilins in the past. This hints towards a possible functional dependence of other γ-secretases on Presenilins or possibly a translational regulation making it possible for the survival of species without these proteins. However, further investigation is required to comprehensively identify the role of Presenilin in unicellular species, where it is the only γ-secretase protein. This investigation might also explain if Presenilin alone is responsible for some rudimentary γ-secretase activity or other transcriptional and post-translational modifications are responsible for the survival of these organisms in the absence of other three γ-secretase proteins.
The phylogenetic gene trees, inferred in Fig. 1, for Presenilin and in suppl. mat. 7–9 for the other three γ-secretase genes have little incongruence with the species tree in suppl. mat. 5 and therefore, in addition to tblastn and syntenic evidence (for homologs in plants and vertebrates only), give more confidence in inferring true homologs for the γ-secretase families. The small incongruence between the species tree and the gene tree can be caused by three main biological processes, namely lineage sorting, gene duplication and loss, and reticulate evolution (Maddison 1997; Degnan and Rosenberg 2009), and general lack of incongruence between gene trees and the species tree supports the absence of these events in the γ-secretase genes.
The Presenilin phylogeny also shows some interesting gene duplications in the LCA of important clades. There were at least two ancient duplication events (represented with a golden star) at the root of important speciation events—one in vertebrate and one at the root of monocots and dicots. Additionally, there is a flow of recent gene duplications (represented with a silver circle), giving rise to several “species-specific duplications” across various eukaryotic species. This is well established that gene duplication may lead to conservation of the function of a gene so that if one of the paralogs malfunctions, the other paralog compensates for it, thus providing higher resilience to the cell in such conditions (Ohno 1970; Gu et al. 2003; DeLuna et al. 2008). The same duplication rates are not observed for other γ-secretase proteins.
It is interesting to note that despite high conservation in metazoans, the amyloid-beta protein is not present in unicellular organisms (Tharp and Sarkar 2013). On the other hand, Notch proteins are present throughout the eukaryotes (unicellular eukaryotes, Planta, Animalia and Fungi) in the same way as Presenilin. Therefore, it can be hypothesized that the main proteolytic function of Presenilins in eukaryotic LCA was the catalysis of Notch-related proteins while the catalysis of amyloid-beta is rather a metazoan innovation. Likewise, incorporation of Presenilin to form γ-secretase complex probably is also a metazoan innovation and contributes towards the diversity of its proteolytic activity to fulfill the requirements of a complex multicellular organization. The independent evolution of these two catalytic activities of Presenilin is also reflected by the fact that mutations in Presenilin protein that affect amyloid-beta proteolysis do not primarily affect the proteolysis of Notch by Presenilin (Capell et al. 2000; Kulic et al. 2000). The conservation of the presence and location of Asp-257 and Asp-385 throughout the eukaryotic life, from human to hydra to almost all the unicellular eukaryotes included in this analysis, signifies the pivotal role of Presenilins as aspartate protease. The preservation of catalytic residues in species in which Presenilin is the only representative protein of γ-secretase indicates that ancestral Presenilin in eukaryotic LCA could exhibit its proteolytic activity without getting incorporated into a larger complex. Therefore, the presence of Presenilin in these unicellular organisms indicates that the non-catalytic functions of Presenilin, e.g., protein trafficking, calcium homeostasis, and apoptosis, and so on (Capell et al. 2000; Otto et al. 2016), performed independently of its γ-secretase activity, originated before the origin of catalytic activity. However, with the advent of premetazoans and metazoans, this proteolytic activity would have been reinforced due to the presence of all the proteins required for making multiprotein secretase complex.
These findings lead to the hypothesis that a protein present in a diverse group of organisms and performing a variety of important functions (of both catalytic as well as non-catalytic nature) must also be evolving under some considerable functional constraints, either evolving under neutral or purifying selection but very little diversifying selection. This is positively observed in our effort to search for diversifying evolution in Presenilins. The MEME analysis in this direction helped us to estimate the extent of positive selection for each site along the whole length of Presenilin proteins. Of the five and two positively selected codons for Presenilin-1 and Presenilin-2, respectively, none corresponded to the highly ranked residues we got from ETA. These positively selected codons are located in the non-transmembrane part of the protein which is understandable as our ETA analysis clearly showed that the TM region is more conserved than the non-TM region (Fig. 3b, c). A low number of codons under positive selection as well as much higher p-values of the predominant majority of the codons (as seen clearly in Fig. 2 and suppl. mat. 11) indicate that vertebrate Presenilin proteins are generally not evolving under diversifying selection. This conclusion is in line with the general understanding that older genes (especially those present in a wide range of organisms) and genes necessary for the survival of an organism evolve slowly than younger genes (Wilson et al. 1977; Hirsh and Fraser 2001; Domazet-Loso and Tautz 2003; Albà and Castresana 2005; Wolf et al. 2009). Likewise, the Tajima’s relative rate test showed that even duplication of vertebrate Presenilin did not affect the rate of evolution of Presenilins, as both Presenilin paralogs continued to evolve with almost the same evolutionary rate when compared with an outgroup.
The study of protein topology of Presenilin protein has revealed important aspects of its structure–function relationship (Bai et al. 2015). For instance, several studies have shown the importance of transmembrane portions of Presenilin involved in the catalytic activity of γ-secretase (Sato et al. 2006, 2008). Similarly, studies based upon Substituted Cysteine Accessibility Method (SCAM) and Nuclear Magnetic Resonance (NMR) have shown the importance of TMD1, TMD5, TMD6, TMD7, and TMD9 in the catalysis of transmembrane proteins (Tolia et al. 2006, 2008; Watanabe et al. 2010). Hence, we also studied the topology of Presenilin protein and investigated if there exists any selective constraint on the TMDs of Presenilin in comparison with the cytosolic or luminal portions. As expected, the ETA showed that a significant majority (65%) of the highly conserved amino acids (with ETA rank value of 1) were present in the transmembrane region of the protein (see Fig. 3). This conservation of the transmembrane portion of Presenilin reflects the functional constraint TMDs exhibited by providing a hydrophilic catalytic pore in the hydrophobic milieu for efficient catalysis of the membrane proteins. The conservation analysis of site-directed mutagenesis also indicates the physiological importance of highly ranked residues obtained from ETA (see Fig. 4). For instance, mutagenesis of D-257 and D-385, two highly ranked residues, leads to disruption in the proteolytic activity of Presenilins. This phenotypic effect reflects the functional constraint on these residues that lead to the conservation of these residues for more than one billion years, from the simplest unicellular protists to highly complex organisms, e.g., primates. Similarly, the mutations in highly ranked residues P-433, A-434, and L-435 lead to disruption in protein kinase A (PKA) signaling, which affects the phosphorylation of β-catenin (Kang et al. 2002). It will be interesting to study the phenotypic changes resulting from the same mutation in other organisms, especially in invertebrates and protists.
Intrigued by the evolutionary history of Presenilin, we looked at the conservation profile of amino acid sites in Presenilin whose mutation causes FAD. This evolutionary analysis of FAD-causing mutations gave some interesting results (see Fig. 5). For instance, as was expected from structure–function evolutionary analysis, the majority of these conserved FAD-causing sites were located in the alpha helical transmembrane part of Presenilin. In addition, many of these mutations seem to be exposed to a hydrophilic aqueous environment as depicted in the surface view of the structure analysis.
Interestingly, a significant majority of amino acid sites susceptible to FAD were highly conserved among metazoans from humans to choanoflagellates (~ 70%). The genes/loci that are involved in old age diseases usually tend to evolve under higher functional constraint. It reflects that the sites, whose mutation leads to FAD, might be involved in performing important functions (Kirkwood and Austad 2000; Drenos and Kirkwood 2010). These functions may involve eliciting those signaling cascades and networks which control physiological manifestations as complex as learning and memory in higher vertebrates or other important neuro-physiological functions in lower vertebrates and invertebrates. However, more investigations are required to elucidate the function of these conserved residues in invertebrates and protists, where neurological structures are either relatively simple or are absent.
It is known that genes involved in a disease evolve under purifying selection due to dominant mutations (Blekhman et al. 2008). Moreover, it has recently been proposed that only the amyloid hypothesis cannot account for the occurrence and progression of AD (Watanabe and Shen 2017). According to this new hypothesis, mutations in Presenilin leading to AD/FAD should rather be considered as loss of function dominant-negative mutations rather than gain of function mutations, as a significant number of these mutations severely impair the functioning of γ-secretase instead of only increasing Aβ42 production (Kelleher and Shen 2017; Sun et al. 2017; Watanabe and Shen 2017). Our findings, i.e., the ancient origin of Presenilins, their evolution under negligible positive selection, and strong conservation of the amino acid sites involved in AD pathogenesis also favor this new hypothesis. Also, our findings indicate the importance of exploring amyloid-beta as well as other candidates to determine the role of Presenilin in AD etiology. More experimental work on a broad spectrum of model organisms will not only contribute to a better understanding of the mechanistic details of AD pathogenicity but will also help in formulating better therapeutic strategies for this devastating disease.
Conclusion
Presenilins are type II membrane proteins involved in the proteolysis of many important proteins. Of the four γ-secretase proteins, Presenilins are the most primitive ones. Their primitive origin supports the hypothesis that they may be present in eukaryotic LCA and maybe the only representative of γ-secretase activity in eukaryotic LCA, performing both catalytic and non-catalytic roles. Presenilins are very conserved proteins, exhibiting very little diversifying evolution which hints towards the functional constraint posed by the physiological vitality of these proteins. Remarkably, the Presenilin residues susceptible to AD show significantly high conservation in metazoans manifesting the involvement of these residues in important physiological processes. The mutation in such functionally constrained residues would eventually lead to AD-like complex and multifaceted disorder. Based upon their evolutionary profile we can predict that involvement of Presenilin in AD pathogenicity cannot be ascribed to a single cause like amyloid-beta production, but other factors should also be explored for more effective therapeutic solutions.
References
Albà MM, Castresana J (2005) Inverse relationship between evolutionary rate and age of mammalian genes. Mol Biol Evol 22:598–606. https://doi.org/10.1093/molbev/msi045
Ali R, Muhammad S, Khan M, Arvestad L (2013) Quantitative synteny scoring improves homology inference and partitioning of gene families. BMC Bioinform. https://doi.org/10.1186/1471-2105-14-S15-S12
Ali RH, Muhammad SA, Arvestad L (2016) GenFamClust: an accurate, synteny-aware and reliable homology inference algorithm. BMC Evol Biol. https://doi.org/10.1186/s12862-016-0684-2
Ali RH, Bogusz M, Whelan S, Tamura K (2019) Identifying clusters of high confidence homologies in multiple sequence alignments. Mol Biol Evol. https://doi.org/10.1093/molbev/msz142
Altschul SF, Madden TL, Schäffer AA et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Area-Gomez E, de Groof AJC, Boldogh I et al (2009) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175:1810–1816. https://doi.org/10.2353/ajpath.2009.090219
Bai XC, Yan C, Yang G et al (2015) An atomic structure of human gamma-secretase. Nature 525:212–217. https://doi.org/10.1038/nature14892
Beel AJ, Sanders CR (2008) Substrate specificity of gamma-secretase and other intramembrane proteases. Cell Mol Life Sci 65:1311–1334. https://doi.org/10.1007/s00018-008-7462-2
Blekhman R, Man O, Herrmann L et al (2008) Natural selection on genes that underlie human disease susceptibility. Curr Biol 18:883–889. https://doi.org/10.1016/j.cub.2008.04.074
Bouckaert R, Vaughan TG, Barido-Sottani J et al (2019) BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1006650
Brown MS, Ye J, Rawson RB, Goldstein JL (2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100:391–398. https://doi.org/10.1016/S0092-8674(00)80675-3
Brunkan AL, Martinez M, Walker ES, Goate AM (2005) Presenilin endoproteolysis is an intramolecular cleavage. Mol Cell Neurosci 29:65–73. https://doi.org/10.1016/j.mcn.2004.12.012
Bustos V, Pulina MV, Bispo A et al (2017a) Phosphorylated Presenilin 1 decreases β-amyloid by facilitating autophagosome-lysosome fusion. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1705240114
Bustos V, Pulina MV, Kelahmetoglu Y et al (2017b) Bidirectional regulation of Aβ levels by Presenilin 1. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1705235114
Cai D, Leem JY, Greenfield JP et al (2003) Presenilin-1 regulates intracellular trafficking and cell surface delivery of beta-amyloid precursor protein. J Biol Chem 278:3446–3454. https://doi.org/10.1074/jbc.M209065200
Capell A, Steiner H, Romig H et al (2000) Presenilin-1 differentially facilitates endoproteolysis of the beta-amyloid precursor protein and Notch. Nat Cell Biol 2:205–211. https://doi.org/10.1038/35008626
De Strooper B, Beullens M, Contreras B et al (1997) Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer’s disease-associated presenilins. J Biol Chem 272:3590–3598. https://doi.org/10.1074/jbc.272.6.3590
Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol 24:332–340
DeLuna A, Vetsigian K, Shoresh N et al (2008) Exposing the fitness contribution of duplicated genes. Nat Genet. https://doi.org/10.1038/ng.123
Domazet-Loso T, Tautz D (2003) An evolutionary analysis of orphan genes in Drosophila. Genome Res 13:2213–2219. https://doi.org/10.1101/gr.1311003
Drenos F, Kirkwood TBL (2010) Selection on alleles affecting human longevity and late-life disease: the example of apolipoprotein E. PLoS ONE. https://doi.org/10.1371/journal.pone.0010022
Duff K, Eckman C, Zehr C et al (1996) Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature 383:710–713. https://doi.org/10.1038/383710a0
Edbauer D, Winkler E, Regula JT et al (2003) Reconstitution of gamma-secretase activity. Nat Cell Biol 5:486–488. https://doi.org/10.1038/ncb960
Espagne E, Lespinet O, Malagnac F et al (2008) The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol. https://doi.org/10.1186/gb-2008-9-5-r77
Fan HW, Noda H, Xie HQ et al (2015) Genomic analysis of an ascomycete fungus from the rice planthopper reveals how it adapts to an endosymbiotic lifestyle. Genome Biol Evol. https://doi.org/10.1093/gbe/evv169
Fukumori A, Fluhrer R, Steiner H, Haass C (2010) Three-amino acid spacing of presenilin endoproteolysis suggests a general stepwise cleavage of -secretase-mediated intramembrane proteolysis. J Neurosci 30:7853–7862. https://doi.org/10.1523/JNEUROSCI.1443-10.2010
Goutte C, Tsunozaki M, Hale VA, Priess JR (2002) APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA 99:775–779. https://doi.org/10.1073/pnas.022523499
Gu Z, Steinmetz LM, Gu X et al (2003) Role of duplicate genes in genetic robustness against null mutations. Nature. https://doi.org/10.1038/nature01198
Han MV, Zmasek CM (2009) PhyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinform 10:356. https://doi.org/10.1186/1471-2105-10-356
Hirsh AE, Fraser HB (2001) Protein dispensability and rate of evolution. Nature 411:1046–1049. https://doi.org/10.1038/35082561
Jacobsen H, Reinhardt D, Brockhaus M et al (1999) The influence of endoproteolytic processing of familial Alzheimer’s disease presenilin 2 on Aβ42 amyloid peptide formation. J Biol Chem 274:35233–35239. https://doi.org/10.1074/jbc.274.49.35233
Juretić D, Zoranić L, Zucić D (2002) Basic charge clusters and predictions of membrane protein topology. J Chem Inf Comput Sci. https://doi.org/10.1021/ci010263s
Kang DE, Soriano S, Xia X et al (2002) Presenilin couples the paired phosphorylation of β-catenin independent of axin: Implications for β-catenin activation in tumorigenesis. Cell. https://doi.org/10.1016/S0092-8674(02)00970-4
Keeling PJ, Slamovits CH (2005) Causes and effects of nuclear genome reduction. Curr Opin Genet Dev 15:601
Kelleher RJ, Shen J (2017) Presenilin-1 mutations and Alzheimer’s disease. Proc Natl Acad Sci USA 114:629–631. https://doi.org/10.1073/pnas.1619574114
Kirkwood TBL, Austad SN (2000) Why do we age? Nature 408:233–238. https://doi.org/10.1038/35041682
Koonin EV (2005) Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. https://doi.org/10.1146/annurev.genet.39.073003.114725
Kosakovsky Pond SL, Frost SDW (2005) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533. https://doi.org/10.1093/bioinformatics/bti320
Kulic L, Walter J, Multhaup G et al (2000) Separation of presenilin function in amyloid beta-peptide generation and endoproteolysis of Notch. Proc Natl Acad Sci USA 97:5913–5918. https://doi.org/10.1073/pnas.100049897
Kumar S, Stecher G, Suleski M, Hedges SB (2017) TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol. https://doi.org/10.1093/molbev/msx116
LaFerla FM (2002) Calcium dyshomeostasis and intracellular signalling in alzheimer’s disease. Nat Rev Neurosci 3:862–872. https://doi.org/10.1038/nrn960
Laudon H, Hansson EM, Melén K et al (2005) A nine-transmembrane domain topology for presenilin 1. J Biol Chem 280:35352–35360. https://doi.org/10.1074/jbc.M507217200
Lee MK, Slunt HH, Martin LJ et al (1996) Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J Neurosci 16:7513–7525
Levy-Lahad E, Wasco W, Poorkaj P et al (1995) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269:973–977
Lichtarge O, Bourne HR, Cohen FE (1996) An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol 257:342–358. https://doi.org/10.1006/jmbi.1996.0167
Lua RC, Lichtarge O (2010) PyETV: a PyMOL evolutionary trace viewer to analyze functional site predictions in protein complexes. Bioinformatics 26:2981–2982. https://doi.org/10.1093/bioinformatics/btq566
Ludtmann MHR, Otto GP, Schilde C et al (2014) An ancestral non-proteolytic role for presenilin proteins in multicellular development of the social amoeba dictyostelium discoideum. J Cell Sci. https://doi.org/10.1242/jcs.140939
Maddison WP (1997) Gene trees in species trees. Syst Biol. https://doi.org/10.1093/sysbio/46.3.523
Marchler-Bauer A, Bo Y, Han L et al (2016) CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw1129
McCarthy JV, Twomey C, Wujek P (2009) Presenilin-dependent regulated intramembrane proteolysis and γ-secretase activity. Cell Mol Life Sci 66:1534–1555
Menzies FM, Fleming A, Rubinsztein DC (2015) Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 16:345–357. https://doi.org/10.1038/nrn3961
Meyer DZ, Avery LM (2009) Excel as a qualitative data analysis tool. Field Methods 21:91–112. https://doi.org/10.1177/1525822X08323985
Murray MM, Jones CJ, Clark RF (2000) The presenilin gene is very highly conserved throughout evolution. Neurobiol Aging 21:48. https://doi.org/10.1016/S0197-4580(00)82893-6
Murrell B, Wertheim JO, Moola S et al (2012) Detecting individual sites subject to episodic diversifying selection. PLoS Genet. https://doi.org/10.1371/journal.pgen.1002764
Nixon RA, Yang DS (2011) Autophagy failure in Alzheimer’s disease-locating the primary defect. Neurobiol Dis 43:38–45
Ohno S (1970) Evolution by gene duplication. Springer, Berlin
Ohta K, Mizuno A, Ueda M et al (2010) Autophagy impairment stimulates PS1 expression and γ-secretase activity. Autophagy 6:345–352
Otto GP, Sharma D, Williams RSB (2016) Non-catalytic roles of presenilin throughout evolution. J Alzheimer’s Dis 52:1177–1187
Pettersen EF, Goddard TD, Huang CC et al (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084
Podlisny MB, Citron M, Amarante P et al (1997) Presenilin proteins undergo heterogeneous endoproteolysis between Thr291and Ala299and occur as stable N- and C-terminal fragments in normal and alzheimer brain tissue. Neurobiol Dis 3:325–337. https://doi.org/10.1006/nbdi.1997.0129
Ponting CP (2002) Identification of a novel family of presenilin homologues. Hum Mol Genet. https://doi.org/10.1093/hmg/11.9.1037
Popugaeva E, Bezprozvanny I (2013) Role of endoplasmic reticulum Ca2+ signaling in the pathogenesis of Alzheimer disease. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2013.00029
Pundir S, Martin MJ, O’Donovan C (2017) UniProt protein knowledgebase. In: Wu CH, Arighi CN, Ross KE (eds) Methods in molecular biology. Humana Press, New York, pp 41–55
Sakharkar KR, Chow VTK (2005) Strategies for genome reduction in microbial genomes. Genome Inform 16:69–75
Sato C, Morohashi Y, Tomita T, Iwatsubo T (2006) Structure of the catalytic pore of gamma-secretase probed by the accessibility of substituted cysteines. J Neurosci 26:12081–12088. https://doi.org/10.1523/JNEUROSCI.3614-06.2006
Sato C, Takagi S, Tomita T, Iwatsubo T (2008) The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the gamma-secretase. J Neurosci 28:6264–6271. https://doi.org/10.1523/JNEUROSCI.1163-08.2008
Scheper W, Zwart R, Baas F (2004) Rab6 membrane association is dependent of Presenilin 1 and cellular phosphorylation events. Mol Brain Res 122:17–23. https://doi.org/10.1016/j.molbrainres.2003.11.013
Shen J, Kelleher RJ (2007) The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci USA 104:403–409. https://doi.org/10.1073/pnas.0608332104
Sievers F, Higgins DG (2014a) Clustal omega. Curr Protoc Bioinform. https://doi.org/10.1002/0471250953.bi0313s48
Sievers F, Higgins DG (2014b) Clustal omega, accurate alignment of very large numbers of sequences. Methods Mol Biol. https://doi.org/10.1007/978-1-62703-646-7_6
Slamovits CH (2013) Extreme genome reduction in microbial parasites
Spasic D, Tolia A, Dillen K et al (2006) Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J Biol Chem 281:26569–26577. https://doi.org/10.1074/jbc.M600592200
Suga K, Tomiyama T, Mori H, Akagawa K (2004) Syntaxin 5 interacts with presenilin holoproteins, but not with their N- or C-terminal fragments, and affects beta-amyloid peptide production. Biochem J 381:619–628. https://doi.org/10.1042/BJ20040618
Sun L, Zhou R, Yang G, Shi Y (2017) Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc Natl Acad Sci USA 114:E476–E485. https://doi.org/10.1073/pnas.1618657114
Supnet C, Bezprozvanny I (2010) The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 47:183–189
Tajima F (1993) Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 135:599–607
Takasugi N, Tomita T, Hayashi I et al (2003) The role of presenilin cofactors in the γ-secretase complex. Nature 422:438–441. https://doi.org/10.1038/nature01506
Tamura K, Stecher G, Peterson D et al (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. https://doi.org/10.1093/molbev/mst197
Tharp WG, Sarkar IN (2013) Origins of amyloid-β. BMC Genomics. https://doi.org/10.1186/1471-2164-14-290
Thinakaran G, Borchelt DR, Lee MK et al (1996) Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17:181–190. https://doi.org/10.1016/S0896-6273(00)80291-3
Tolia A, Chávez-Gutiérrez L, De Strooper B (2006) Contribution of presenilin transmembrane domains 6 and 7 to a water-containing cavity in the ??-secretase complex. J Biol Chem 281:27633–27642. https://doi.org/10.1074/jbc.M604997200
Tolia A, Horré K, De Strooper B (2008) Transmembrane domain 9 of presenilin determines the dynamic conformation of the catalytic site of γ-secretase. J Biol Chem 283:19793–19803. https://doi.org/10.1074/jbc.M802461200
Walter J, Capell A, Grünberg J et al (1996) The Alzheimer’s disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Mol Med 2:673–691
Wang J, Beher D, Nyborg AC et al (2006a) C-terminal PAL motif of presenilin and presenilin homologues required for normal active site conformation. J Neurochem 96:218–227. https://doi.org/10.1111/j.1471-4159.2005.03548.x
Wang R, Tang P, Wang P et al (2006b) Regulation of tyrosinase trafficking and processing by presenilins: partial loss of function by familial Alzheimer’s disease mutation. Proc Natl Acad Sci USA 103:353–358. https://doi.org/10.1073/pnas.0509822102
Watanabe H, Shen J (2017) Dominant negative mechanism of Presenilin-1 mutations in FAD. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1717180114
Watanabe N, Takagi S, Tominaga A et al (2010) Functional analysis of the transmembrane domains of presenilin 1: Participation of transmembrane domains 2 and 6 in the formation of initial substrate-binding site of γ-secretase. J Biol Chem 285:19738–19746. https://doi.org/10.1074/jbc.M110.101287
Waterhouse AM, Procter JB, Martin DMA et al (2009) Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics. https://doi.org/10.1093/bioinformatics/btp033
Webb B, Sali A (2016) Comparative protein structure modeling using MODELLER. Curr Protoc Bioinform. https://doi.org/10.1002/cpbi.3
Wilkins A, Erdin S, Lua R, Lichtarge O (2012) Evolutionary trace for prediction and redesign of protein functional sites. Methods Mol Biol. https://doi.org/10.1007/978-1-61779-465-0_3
Wilson AC, Carlson SS, White TJ (1977) Biochemical evolution. Annu Rev Biochem 46:573–639. https://doi.org/10.1146/annurev.bi.46.070177.003041
Wolf Y, Novichkov P, Karev G et al (2009) The universal distribution of evolutionary rates of genes and distinct characteristics of eukaryotic genes of different apparent ages. Proc Natl Acad Sci USA 106:7273–7280. https://doi.org/10.1073/pnas.0901808106
Zhang Z, Nadeau P, Song W et al (2000) Presenilins are required for γ-secretase cleavage of β-APP and transmembrane cleavage of Notch-1. Nat Cell Biol. https://doi.org/10.1038/35017108
Zhang Y, Thompson R, Zhang H, Xu H (2011) APP processing in Alzheimer’s disease. Mol Brain 4:3. https://doi.org/10.1186/1756-6606-4-3
Acknowledgements
We are grateful to Professor Dr. Khalid J. Siddiqui for spending time in improving the grammar and other language-related aspects of our manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Joana Projecto-Garcia.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Khan, A.A., Ali, R.H. & Mirza, B. Evolutionary History of Alzheimer Disease-Causing Protein Family Presenilins with Pathological Implications. J Mol Evol 88, 674–688 (2020). https://doi.org/10.1007/s00239-020-09966-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-020-09966-w