
Abstract
Fossil evidence suggests that cetaceans evolved from artiodactylans. Thus, there was a major dietary change from herbivorous to carnivorous during their transition from a terrestrial to an aquatic environment. However, the molecular evolutionary mechanisms underlying this dietary switch have not been well investigated. Evidence of positive selection of digestive proteinases and lipases of cetaceans was detected: (1) For the four pancreatic proteinase families (carboxypeptidase, trypsin, chymotrypsin, and elastase) examined in this study, each family included only a single intact gene (e.g., CPA1, PRSS1, CTRC, and CELA3B) that had no ORF-disrupted or premature stop codons, whereas other members of each family had become pseudogenized. Further selective pressure analysis showed that three genes (PRSS1, CTRC, and CELA3B) were subjected to significant positive selection in cetaceans. (2) For digestive proteinases from the stomach, PGA was identified to be under positive selection. (3) Intense positive selection was also detected for the lipase gene PLRP2 in cetaceans. In addition, parallel /convergent amino acid substitutions between cetaceans and carnivores, two groups of mammals that have evolved similar feeding habits, were identified in 10 of the 12 functional genes. Although pseudogenization resulted in each family of pancreatic proteinases only retaining one intact gene copy in cetacean genomes, positive selection might have driven pancreatic proteinases, stomach proteinases, and lipases to adaptively evolve a stronger ability to digest a relatively higher proportion of proteins and lipids from animal foods. This study can provide some novel insights into the molecular mechanism of cetacean dietary changes during their transition from land to sea.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cetaceans re-entered water from land approximately 50 million years ago and became a dominant group of marine mammals (Uhen 2007). Cetaceans evolved from artiodactyl ancestors, thus, they underwent a major dietary change from herbivorous to carnivorous (e.g., feeding on fishes, squids, zooplanktons, and euphausiids) (Thewissen et al. 2007, 2009). Dietary changes have occurred not only in cetaceans but also in other mammals (e.g., dog, giant panda, and columbine monkeys) (Axelsson et al. 2013; Jin et al. 2011; Zhang et al. 2002). Some evolutionary biologists have been attracted to this intriguing phenomenon and have sought evidence behind the adaptation of dietary changes from paleontological, morphological, anatomical, and molecular traits. For example, the pandas are a representative of a dietary switch in carnivores, and its feeding habit transformed into bamboo consumption. Dental remains of the earliest giant pandas have a similar structure to herbivores, revealing that the time of their dietary change was approximately 2 million years ago (Jin et al. 2007). The sixth finger, called the manus, made the giant panda better adapted to acquiring bamboo. However, the digestive system of the giant panda, which is still a carnivore-like digestive system, is more suitable for a carnivorous diet than a vegetarian one (Dierenfeld et al. 1982). Further studies have found an association of the panda’s dietary switch with the pseudogenization of the umami taste receptor gene and some defects of catecholamine metabolic pathways (Jin et al. 2011; Zhao et al. 2010).
Similar to the specialized manus of the giant panda, some morphological changes in cetaceans have also occurred during the adaption to their dietary change. For example, baleen whales engulf schools of small fish or krill along with a large amount of water and then expel the water through their baleen plates, which are unique to baleen whales. Thus, the baleen serves as a filter and is crucial for these whales to obtain food. Although some changes have occurred, cetaceans still retain a multiple chambered stomach that is similar to that of other artiodactyls (Mead 2007). It is still unclear whether genetic changes have occurred in their digestive enzymes in response to their dietary change from herbivorous to carnivorous. Thus, to explore the molecular mechanisms of their adaptations of digestion, Wang et al. (2016) discussed this problem using 10 digestive enzyme genes from representative mammals. He noted that some proteinases and lipases were found to have undergone positive selection, indicating that cetaceans have developed an enhanced ability to digest dietary protein and fat. However, there are other proteinases, lipases, and proteins associated with digestion that should be analyzed in addition to the above-mentioned 10 digestive enzymes.
The gastrointestinal tract (GIT) is an important location for digestion of dietary protein and fat (Schneeman 2002). Food must undergo mechanical and chemical digestion processes in the GIT to be transformed into small molecules that can be absorbed by the intestines. Chemical digestion is mainly performed by digestive enzymes in the digestive tract, bile from the liver and hydrochloric acid from the stomach (Duke 1986). As for digestive enzymes, they consist of proteinases, lipases, and amylases, which digest dietary protein, fat, and starch, respectively. In addition, each digestive enzyme can act on a specific substrate (Whitcomb and Lowe 2007). As for bile, its role in the digestion process is mainly accomplished by bile salts. Bile salts play a crucial role in promoting fat digestion and absorption (Maldonado-Valderrama et al. 2011). As for gastric acid, the gastric H+, K+-ATPase, which contains α and β subunits, is the only enzyme that generates gastric acid by catalyzing H+ into the stomach (Shin et al. 2005). Mechanical digestion refers to grinding food, mixing food and enzymes, and pushing food into the digestive tract by the motor function of the gastrointestinal muscle. It is worth noting that gastrointestinal hormones play an important role during the mechanical digestion process by regulating gastrointestinal motility. In addition, gastrointestinal hormones can also stimulate the secretion of gastric acid and a variety of digestive enzymes (Sato et al. 2010; Zhao et al. 2007).
In brief, the genes encoding proteinases, lipases, gastrointestinal hormones and gastric H+, K+-ATPase can be screened to further explore the molecular mechanism of the cetacean dietary switch. Interestingly, both proteinase types exist as multigene families, whereas lipase, gastrointestinal hormone, and the gastric H+, K+-ATPase are encoded by only one gene each (Carginale et al. 2004; Steinert et al. 2013; Whitcomb and Lowe 2007). Furthermore, we found that some proteinase genes are inactivated in certain cetacean lineages, which is puzzling. However, a previous study clarified that specific gene losses were likely beneficial for cetaceans to adapt to diving habits, such as vasoconstriction-, DNA damage repair- and unihemispheric sleep-related genes (Matthias 2019). It was not clear whether the proteinase genes exist in inactive forms owing to consumption of higher protein diets. To solve this problem and explore the molecular adaptive mechanism of other genes associated with digestion for the cetacean dietary switch, the present study investigated ten proteinase genes, four lipase genes, one gastrointestinal hormone gene, and two gastric H+, K+-ATPase genes in seven representative cetaceans and compared them with orthologous sequences in representative terrestrial mammals.
Methods
Source of Data and Validation of Pseudogenes
In our study, the protein-coding sequences of 16 genes (Table 1) associated with digestion were acquired from 27 representative mammalians (including 7 cetaceans, 7 artiodactyls, 5 carnivores, 1 chiroptera, 2 insectivores, and 5 euarchontoglires, Supplementary Material, Table S2). These 27 mammalians were grouped into 3 data sets, including the cetacean, mammalian, and cetartiodactyl data sets for subsequent analysis (Supplementary Material, Table S8). Cetaceans and some mammals used sequences of kinship as a query to run a BLAST to obtain single exons in the whole genome obtained from NCBI (https://www.ncbi.nlm.nih.gov/; Supplementary Material, Table S1). The single exons were integrated by concatenation into the complete gene, and sequences of other mammalians were derived from NCBI or Ensemble (https://www.ensembl.org/index.html?redirect=no), with details please refer to Supplementary Material, Table S2. Genes that were missing more than one exon were not used for further analysis. By comparing the spliced and downloaded sequences using MEGA software, we found that the pancreatic proteinase genes had frameshift mutations or premature stop codons in cetartiodactylans (Supplementary Material, Fig. S1). To exclude the impact of the genome assembly on the sequences, further experiments for PCR verification were indispensable. Owing to the scarcity of cetacean samples, our laboratory only has a subset of the above-mentioned cetartiodactylans with pseudogenes, and thus, further experiments could only validate these species that our laboratory had collected, including Tursiops truncatus, Orcinus orca, Neophocaena phocaenoides, Lipotes vexillifer, Physeter catadon, and Balaenoptera acutorostrata. In addition, for genes with multiple sites of frameshift mutations or premature stop codons, we only validated one site, which was enough to prove the gene was a pseudogene. All animal samples were gifts or from dead individuals collected in the field, and the preservation of these samples complied with ethical standards and Chinese law requirements.
The cetacean muscle tissue used to extract genomic DNA was stored frozen at − 40 °C. The experimental methods were based on the standard phenol/chloroform extraction method, followed by ethanol precipitation. The DNA concentration and quality were measured by a UV spectrophotometer (Thermo scientific, NANODROP 2000). Primers were designed in the conserved regions at both ends of the mutational locus for the 7 representative cetaceans, and the optimal primers were further selected by primer 3 (https://primer3.ut.ee/) (Supplementary Material, Table S3). The PCR was run on an AB1 9700 with a 25-μl reaction system, including 1 μl of DNA (100 ng/μl), 1 μl of each primer (10 μM) and 15 μl of 2 × EasyTaq PCR SuperMix (Takara). PCR amplification conditions were 95 °C for 5 min, 35 cycles of 94 °C for 30 s, 50–57 °C for 40 s, and 72 °C for 1–2 min, followed by 72 °C for 10 min. The PCR products were detected by agarose gel electrophoresis and the products with the appropriately sized band were sent to Sangon Biotech for sequencing. The results of sequencing were consistent with the blast.
Data Consolidation and Analysis of Selective Pressure
The sequences of each gene were aligned with MEGA5, and then, negligible insertions and deletions were manually adjusted according to the results of the comparison to facilitate subsequent analysis. Maximum likelihood (ML) phylogenies of 12 intact genes were inferred using IQ-TREE (Nguyen et al. 2015) under the model automatically selected by IQ-TREE ('Auto' option in IQ-TREE) for 1000 ultrafast (Minh et al. 2013) bootstraps, as well as the Shimodaira–Hasegawa-like approximate likelihood ratio test (Guindon et al., 2010). Homologous sequences of Dipodomys ordii for each gene served as the outgroup. In contrast to the processing of the intact genes described above, for the pseudogenes, we chose a sequence of orthologous genes that were complete as a reference to determine the location of inDels and premature stop codons. Then, to assess the selective pressure on pseudogenes for later analysis, we removed each inDel and premature stop codon.
The ω value calculated by the CODEML program in the PAML package, which is the ratio of the nonsynonymous substitution rate and the synonymous substitution rate (ω = dN/dS), can be used as a criterion for evaluating selective pressure, where ω = 1, ω > 1, and ω < 1 represent neutral selection, positive selection and negative selection, respectively (Yang 2007). The detection of selective pressure was based on the mutual comparison of nested models and the significant P values calculated by the likelihood ratio test (LRT) with a chi-square distribution between 2ΔL and a degree of freedom of less than 0.05, and at the same time, the posterior probability of the positive selective sites by the BEB approach was greater than 0.8 (Yang et al. 2005). The species tree used for analysis was a well-accepted phylogenetic relationship for Primates (Perelman et al. 2011) and Laurasiatheria (Zhou et al. 2012). Notable, pseudogenes and intact genes have different models for analyzing selective pressure.
For functional genes, the site model assumed that each of the clades was subjected to the same selective pressure but had different evolutionary rates and different ω values for each site. The site model was used to detect positively selected sites in the cetacean and all mammal data sets. In particular, one of the site models, M8a vs M8, was selected to test positive selection by comparing these two models. Further analysis of the branch-site model was used to detect the existence of positive selective sites and positive selection on all lineages, and this model has obvious advantages for evaluating the episodic evolution that is common in nature (Zhang 2000). In the specific analysis, the mammalian and cetartiodactyl data sets were used to analyze the selective pressure and positively selected sites encountered in the different lineages. Moreover, in the branch-site model analyses, we corrected the p value by FDR, and the genes whose p value was still less than 0.05 after the correction were used for subsequent analysis. In addition to PAML, the online server Datamonkey, which calculates synonymous and nonsynonymous substitutions for each site, can also be used to test the positively selected sites in the cetacean data set. Fixed-effect likelihood (FEL), single likelihood ancestor counting (SLAC) and random-effect likelihood (REL) were selected to validate the results of PAML (Pond and Frost 2005). The significance level of SLAC and FEL was set at 0.2, and the Bayesian factor for REL was set at 50. The next analysis at the protein-level was TreeSAAP, which was performed using the sites detected by the branch-site model and the sites detected simultaneously by the site model and at least one likelihood method by Datamonkey. This program presumes the magnitude of physicochemical property changes of nonsynonymous amino acids by comparing sequences with their closest common ancestor. The magnitudes are classified into eight categories according to the changes from conservative (1–3) to very radical substitutions (6–8) (Woolley et al. 2003).
For pseudogenes, its analytical method was based on an analysis of the selective pressure of the giant panda Taslr1 pseudogene to construct a series of models for the mammalian data set (Zhao et al. 2010a). We first constructed model A, which assumes that all the branches have a common ω value in the phylogenetic tree, and model B, which assumes that all the branches have a fixed ω = 1. By comparing model A with the null hypothesis model B, we can evaluate the selective pressure on the pseudogenes in the entire phylogenetic tree. To further understand the selective pressure on the branch of pseudogenes, we constructed model C and compared it to model A. Model C assumes that the branch where the pseudogenes occurred has a common ω2, whereas the branches that do not have pseudogenes have a common ω1. Finally, the construction of model D and the comparison with model C assessed whether the functional constraint was completely relaxed, and model D assumes that the pseudogenized clade has a fixed ω2 = 1 and the other branches have a common ω1.
Locate Positively Selected Sites of Cetaceans to Protein Structure
To visualize the importance of positively selected sites of cetaceans, we mapped these sites into 3D structure of the protein. Firstly, we used protein sequence of PGA, CELA3B, and PLRP2 of bottlenose dolphin (T. truncatus) to predict 3D structure via the online serve I-TASSER (https://zhanglab.ccmb.med.umich.edu/services/). Then, these sites were mapped to obtained 3D structure using Pymol (https://pymol.org/2/) and Adobe illustrator. Finally, the Uniprot website (https://www.uniprot.org/) was used to review the specific functional domains of each gene and organize the positively selected sites that were located in important domains.
Determination of Parallel/Convergent Amino Acid Sites in Carnivorous Lineages
In addition to cetaceans, the vast majority of carnivores are carnivorous. In particular, walruses and seals, like whales, live in marine environment and rely on predation of fish and other marine organisms to survive. Thus, we used the previous method to find out whether there are parallel/convergence amino acid substitution in two Carnivorous Lineages (Foote et al. 2015). First, the ancestral sequences in the mammalian data set were reconstructed by Bayesian method in the PAML (Yang et al. 1995; Zhang and Kumar 1997). Then, specific convergent/parallel sites were found in internal ancestry, ancestors, and terminal nodes between cetaceans and carnivores. Finally, to determine if these sites were preserved due to selective pressure rather than random substitution, we used the CONVERG2 to calculate p values, and random substitution can be ruled out when p < 0.05 (Zhang and Kumar 1997).
Results
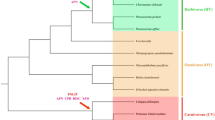
In our study, a total of 16 genes related to digestion were chosen as candidate loci to explore the molecular mechanism of the dietary switch in cetaceans. To detect mutations that disrupt the protein open reading frames (premature stop codons and frameshifting insertions or deletions), we used a comparative method by corresponding orthologous sequences in representative terrestrial mammals. Note that no BLAST hits of entire exons can be caused by genome assembly issues and were not considered evidence confirming pseudogenes in this study. After the comparison and PCR validation, the CPA2, PRSS2, CTRL, and CELA2A genes were identified to be pseudogenized in most cetartiodactylans, and the CPB1 and CELA1 genes were pseudogenized in some cetaceans, and for other genes, were partial or intact sequences that had no ORF-disrupted or premature stop codons (Fig. 1 and Supplementary Material, Fig. S1). In detail, among these 6 proteinase genes, only the bottlenose dolphin’s CTRL did not have BLAST hits, and for the remaining genes we were able to identify specific ORF-disrupted mutations (Supplementary Material, Table S7). The CPA2 and CTRL genes exhibit inactivating mutations shared between cetaceans and artiodactylans, such as a 4-bp deletion at site 314 and a 1-bp insertion at site 33 for the CPA2 gene and a premature stop codon (at the 154th site) shared by Physeter macrocephalus, Bos taurus and Ovis aries in the CTRL gene. Additionally, sequences from hippopotamus, the closest living relative to cetaceans, were used to further confirm which gene was inactivated during the transition from land to water in the cetacean stem lineage. Although these results found that many entire exons failed to BLAST, a mutation site (4-bp deletion at site 314) shared by cetartiodactylans was also present in the hippopotamus CPA2 gene. Alignment sequences are provided in the supplementary file, and the mutations sites that disrupt the protein open reading frames of some genes are retained in the sequences.

Existence of pseudogenes and positive selection of functional genes at pancreatic protease genes The sequences of blast and experimental validation were displayed to the right of phylogenetic tree, each gene family is represented by particular colors, e.g., carboxypeptidase (brown), trypsin (green), chymotrypsin (red), and elastase (yellow). And open circles indicate pseudogenes, solid circles indicate functional gene, half circles indicate partial sequences, and N indicate no consequence of BALST. Please note that the analyses of PRSS1 and CTRC genes had done in our laboratory, the relevant results could refer to Wang et al. (2016). PRSS1, CTRC, and CELA3B genes on orange background were detected positively selected signal in cetaceans
Using the ML method incorporated in PhyloSuite software to construct phylogenetic trees of the mammalian data set of functional genes, it was found that the achieved mammalian phylogenetic relationship was basically consistent with the generally accepted tree, both of which supported the close affinity between artiodactylans and cetaceans (Perelman et al. 2011; Zhou et al. 2012; Supplementary Material, Fig. S2). For this reason, this study used the generally accepted phylogenetic mammalian tree for subsequent evolutionary analyses.
Molecular Evolution of Digestion-related Genes in Cetaceans
Site Model (M8 vs M8a)
To examine whether specific codons of digestion-related genes in mammals were subjected to positive selection, a pair of site models (M8 vs. M8a) in the PAML package were used. The results showed that model M8 of 6 genes (PGA, CELA1, CPA1, CPB1, PLA2G1B, and PLRP2) was significantly better than the neutral M8a. The ω values of these 6 genes ranged from 1.926 to 9.208, and a total of 30 codons identified to be positively selected had a posterior probability ≥ 0.80 by the BEB approach (Supplementary Material, Table S4).
Further, to test whether a similarly selective pattern existed in cetaceans, we used the same approach to detect positive selection in a data set that only included cetaceans. The results showed that, similar to the results for the all mammalian data set, the PGA gene was also found to be under positive selection in the cetacean data set (ω = 11.942), and its LRTs of the site model were statistically significant (p = 0). In addition, the ATP4A, CELA3B, and PLA2G1B genes were also detected to be under positive selection in the cetacean data set (ω = 79.468, 309.201, and 5.826, respectively), and the M8 model was significantly better than the neutral M8a model (p = 0, 0, 0.005, respectively). The M8 model detected 1, 9, 6, and 3 positively selected sites with a posterior probability ≥ 0.80 by the BEB approach for ATP4A, PGA, CELA3B and PLA2G1B, respectively (Table2 and Supplementary Material, Table S4). Another important piece of evidence to support positive selection was the implementation of three ML methods, including SLAC, FEL, and REL in Datamonkey, which showed a total of 12 positively selective sites (1 in ATP4A, 6 in PGA, 4 in CELA3B, and 1 in PLA2G1B) not only in the M8 model but also in one or two ML methods in Datamonkey (Table 3). Moreover, to explore whether the amino acid mutation was radical, TreeSAAP was used to analyze the 12 positively selected sites that were simultaneously identified by two different methods, the M8 model and Datamonkey. Notably, 4 positively selected sites (ATP4A: 15, PGA: 266, CELA3B: 246, 259) were identified with mutations of more than 10 amino acid properties, which provided evidence of positive selection at the protein level (Table 3).
Branch-Site Model
We further selected the branch-site model to identify positive selection of specific lineages in the mammalian data set for each terminal mammalian branch, the ancestral and each internal node of cetaceans and the ancestral branch of other groups. Consistent with the results of the site model of the cetacean data set, positive selection of the PGA gene was identified in cetacean-specific lineages, and particularly, positive selection was identified in the lineage of common ancestors of cetaceans (Table 2). In addition, PGA and PLRP2 in carnivores, CELA1and CELA3B in primates, and CELA3B in rodentia were also found to be under positive selection (Supplementary Material, Table S5).To exclude the influence of the background lineage and validate the above results, a similar branch-site model was used in the cetartiodactylan data set to explore positive selection for the terminal, ancestral, and each internal node of cetaceans. Positive selection was identified in the PGA, ATP4A, and PLRP2 genes for the cetacean lineage. Especially, evidence for positive selection of PGA and PLRP2 was identified along the lineages leading to the ancestral node of cetaceans (Table2). Taken together, based on the results of the above branch-site model, a total of 12 potential positively selected sites with a posterior probability ≥ 0.80 according to the BEB approach were detected in the cetacean lineages (PGA:6, PLRP2:6). Moreover, 58% (7/12) of the positively selected sites were detected as radical AA changes by TreeSAAP, which further provided evidence of positive selection at the protein level (Table 3).
Distribution of Positively Selected Sites in the 3D Structure of Proteins
To visually indicate whether the positively selected sites were located in important functional domains of proteins, we mapped these positively selected sites to the three-dimensional structure of the proteins using Pymol. The results showed that CELA3B (site: 259), PGA (site: 106, 330) and PLRP2 (site: 59, 88, and 226) were located in the alpha-helical area; PGA (site: 154, 225) was located in the beta-sheet area; and the other sites were located in areas with random coils (Fig. 2). Further, according to the results of the Uniport website, we found that some of the sites were located in important domains. For example, one positively selected site of PGA (site: 23) was located in the activation peptide, and another site of CELA3B (site: 13) was located in the signal peptide. In addition, the remaining sites were intensively located on the disulfide bridge (Table 3).

3D structures of proteins The sites in red are positively selected sites
Convergent Analysis in Carnivorous Lineages
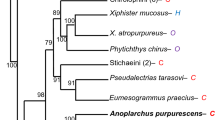
Although a similar pattern of evolution was detected in the PGA and PLRP2 genes of Carnivorous lineages, e.g., ancestral node of cetaceans and carnivores, there was no evidence that the remaining genes undergo convergent evolution. Thus, we reconstructed the ancestral sequences via the PAML package to find specific convergent/parallel sites of amino acid substitution between cetaceans and carnivores. Taken together, 10 of 12 functional genes were found to have 36 parallel and 13 convergent amino acid changes, which were supported by significant statistical support (p < 0.001) confirmed by Converg2 (Supplementary Material, Table S6). To clearly display the position of the positively selected sites, we mapped all parallel/convergent sites to the phylogenetic tree, which mainly consisted of carnivorous lineages (Fig. 3).

As shown in right of figure, 10 genes were detected parallel/convergent amino acid substitutions with the gene from left to right are PGA (puce), CELA3B (yellow), CELA1 (bottle green), CPA1 (wathet), CPB1 (mazarine), ATP4A (gray), ATP4B (purple), PLA2G1B (pink), PLRP2 (white), and CEL (reseda)
The Relaxation of Selective Pressure in Pseudogenes
To assess whether the selective pressure was relaxed in pseudogenes, such as CPA2, PRSS2, CTRL, and CELA2A, a series of ω values were calculated to evaluate the degree of selective constraint in the mammalian data set. The ω values of the A model, which assume that all the branches in the phylogenetic tree have a common ω, were 0.33439, 0.19987, 0.24710, and 0.22592 for the genes CPA2, PRSS2, CTRL, and CELA2A, respectively. The A model was significantly better than the B model, which assumed a single ω of 1 for all the clades for the four genes, indicated that these genes were subjected to purifying selection in the phylogenetic tree. Moreover, the C model was constructed to investigate whether the functional constraint was relaxed, which assumes that branches with pseudogenes have an independent ω2 and the other branches have a common ω1. By comparison, the C model fits the data significantly better than the A model, which indicates that the functional constraint on the pseudogenes was indeed nearly relaxed for these four genes. Finally, D model which assumes that the pseudogenized clade has a fixed ω2 = 1 and the other branches have a common ω1, was constructed and compared with the C model to assess whether the functional constraint was completely relaxed. It is worth noting that the significant p values indicated a slight relaxation but otherwise was completely relaxed. The results of the comparison showed that the selection on the branches of pseudogenes, such as CPA2, PRSS2, and CELA2A, were completely relaxed (see Table 4 for the specific ω values).
Discussion
Unique Evolutionary Pattern of Cetacean Pancreatic Proteinase Genes
Based on the morphological evidence of the cranial and dental findings of Eocene south Asian raoellid artiodactyls, which are the transition branches between cetaceans and artiodactyls, Thewissen et al. (2007) showed that there was a major dietary switch during the origin of cetaceans. Compared with the vegetarian diet of the ancestors of cetaceans, extant cetaceans mainly eat fishes and aquatic invertebrates for food, which means the food composition of extant cetaceans is richer in protein and fat than that of ancestral cetaceans, and there must have been a physiological adaptation during the process of the dietary switch. However, the stomach of cetaceans, which serves as the main location for food digestion, still retains the artiodactyls-like stomach. Subsequent studies have found that the real driving force of the cetacean dietary switch is adaptive evolution of certain proteinases and lipases that can have a positive impact on absorption of dietary protein and fat (Wang et al. 2016). However, only 10 digestive enzymes were examined by Wang et al. (2016), and it is still uncertain whether other digestion-related genes, such as other proteinases, lipases, gastrointestinal hormones and gastric H+, K+-ATPase, have a similar pattern of evolution as the ten enzymes that have been studied. In particular, the pseudogenization of certain proteinase genes in cetaceans has not been explained.
Notably, the pancreas proteinase gene family, including carboxypeptidase (CPA1, CPA2, and CPB1), trypsin (PRSS1 and PRSS2), chymotrypsin (CTRC and CTRL) and elastase (CELA2A,CELA1 and CELA3B), was produced by gene duplication, but the function of these proteinases have differentiated. In details, these proteinases have different specificity pockets that allow only suitable peptide bonds to be hydrolyzed (Szabo et al. 2016; Whitcomb and Lowe 2007; Table 1). Carboxypeptidase hydrolyzes the C-terminal amino acid residues of the peptide chain. Unlike carboxypeptidase, the other proteinase families are endopeptidases and have a similar structure and mechanism for hydrolyzing the peptide chains of dietary proteins, but with different hydrolysis substrates. For example, the specific hydrolysis substrate of the trypsin-like proteinase is a basic amino acid, the chymotrypsin-like proteinase hydrolyzes aromatic amino acids and elastase-like proteinases hydrolyze uncharged amino acids (Whitcomb and Lowe 2007).
Previous studies have found that duplication is the main approach to generate new genes (Qian et al. 2010), because the duplicated new gene copy might acquire new or subfunctions (He and Zhang 2005). In other cases, duplicated genes might degrade into pseudogenes due to functional redundancy (Petrov and Hartl 2000). Interestingly, duplicated pancreas proteinase genes had both evolutionary fates in mammals (Fig. 1). The phenomenon of pseudogenization events of pancreatic proteinase genes was concentrated in cetartiodactylans, and the CPA2 and CTRL genes probably lost their function before cetartiodactyl radiation. Two lines of evidence support the above conclusion. First, the selective pressure on four pseudogenized genes (CPA2 in representative cetaceans and artiodactylans, PRSS2 in four cetaceans, CTRL in representative cetaceans and three artiodactylans, CELA2A in five cetaceans and Ovis aries) was close to relaxed. Second, the CPA2 and CTRL genes shared ORF-disrupted or premature stop codons, respectively, in cetartiodactylans, e.g., CPA2 had a 4-bp deletion (at the 321st place) and a 2-bp deletion (at the 630th place) shared by the representative cetaceans, B. taurus and O. aries, and the mutation site (4-bp deletion at site 314) is also present in hippopotamus. CTRL had a premature stop codon (at 154th place) shared by P. macrocephalus, B. taurus and O. aries. Thus, the most parsimonious hypothesis for inactivating mutations of CPA2 and CTRL genes shared by cetaceans and artiodactylans is that they occurred before the split of these two clades. The inactivating mutations of other genes only shared by cetaceans, including PRSS2, CELA2A, CPB1, and CELA1, may have occurred after the formation of the cetacean branch. In addition to the phenomena of pseudogenization events, concerted evolution of pancreatic proteinase genes might exist in mammals apart from cetartiodactylans, that is, the duplicated genes product new or sub-function (Zhang 2003). For example, human elastase II has the specific function of hydrolyzing elastin, but the hydrolysis substrate is large hydrophobic amino acids, similar to chymotrypsin (Del Mar et al. 1980). Compared to the other elastase zymogens, the elastase 3B zymogen binds more closely to the carboxypeptidase zymogen, which is beneficial for enhancing carboxypeptidase stability (Szabo et al. 2016).
In fact, the phenomenon of functional redundancy, wherein proteinase genes degrade into pseudogenes, is clearly puzzling owing to the higher protein diets in cetaceans. However, for cetartiodactylans, their common ancestors between cetaceans and artiodactylans were herbivorous and their diet contained less protein. Thus, it was not difficult to infer that the inactivating mutations in the CPA2 and CTRL genes might have occurred before the split between cetaceans and artiodactylans owing to the less protein diets. And, based on the available data, we found that the ratio of pseudogenization of cetaceans (62%) was higher than that of artiodactylans (33%), that is, pseudogenization of some genes exists only in cetaceans, such as CPB1, PRSS2, CELA2A, and CELA1. We hypothesized that with the formation of the cetacean species, the functional genes of the ancestral pancreatic proteinases might not have been sufficient to digest more protein in foods and it was not possible for the pseudogenized genes to return to being functional. Thus, cetaceans have evolved a unique evolutionary pattern for pancreatic proteinase genes, that is, only one gene copy was maintained for trypsin, chymotrypsin, and elastase, which evolved to enhance their ability to digest protein more efficiently, whereas other genes in the family became pseudogenes to save energy and resources (Drummond and Wilke 2008; Wagner 2005). In support of this hypothesis, the PRSS1 and CTRC genes were subjected to a strong positive selection in cetaceans (Wang et al. 2016), and the CELA3B gene was identified to have a positive selection signal in cetaceans in our study. For example, a series of positively selected sites were identified in the CELA3B gene in cetaceans by the site model and Datamonkey. Further, among the 4 positively selected sites, the 246th and 259th sites were detected to have more than 10 physicochemical property changes. In the course of evolution, more amino acid changes in properties means a greater impact on function (Yampolsky and Stoltzfus 2005). In addition, the location of the positively selected sites on the important protein domain of proteinases further supported the adaptive evolution of the CELA3B gene. For example, the 13th positively selected site was located on the signal peptide, which plays an import role in the transportation, modification and maturation of the proteinase (Kiraly et al. 2007). The remaining sites were located in the protein domain, which is significant for the binding and hydrolysis of the substrate. Of course, further experiments are necessary to examine the enhancement of hydrolytic ability of the maintained functional genes in cetaceans (PRSS1, CTRC and CELA3B) to test this hypothesis.
Adaptive Evolution of the Cetacean Gastric PGA Gene
For genes related to the digestion of proteins in the stomach, PGA, PGC, ATP4A, and ATP4B, were selected as candidates because the PGA and PGC genes are members of the pepsin family, which has an important function in digestion (Kageyama 2002), and the ATP4A and ATP4B are genes of the α submit and β submit of gastric H+, K+-ATPase, respectively. This ATPase generates gastric acid, which can not only activate pepsinogen but also provide an acidic environment for effective digestion in stomach (Fellenius et al. 1981; Korbova and Kohout 1981; Sachs et al. 1995; Samloff et al. 1975). Interestingly, unlike the pancreatic proteinase family, no pseudogene was detected in the gastric proteinase family, which might be explained by pepsin being a unique enzyme that digests protein in the stomach (Foltmann 1981). In addition, pepsin not only has an important physiological function to digest protein but can also digest amino acids in food (Liu et al. 2015). Thus, it is possible that the phyletic uniqueness and functional diversity of the gastric proteinase made all the pepsin genes retain their function. Further selection analysis found that PGA was identified to be under strong positive selection for all four genes. Previous studies have reported adaptive evolution of PGA in apes to effectively digest a wide range of foods (Narita et al. 2000), whereas the adaptive evolution in cetaceans to adapt to a dietary switch was positive selection of PGA, which was different from apes. Three lines of evidence support the molecular evolution of PGA to adapt to the dietary switch in cetaceans. First, it was not hard to find a single copy of PGA in representative cetaceans by a local BLAST of the genome. Second, a branch-site model identified positive selection in lineages of the ancestors of cetaceans and carnivores, which are highly differentiated species, yet have similar feeding habits. Finally, 5 of the 7 positively selective sites had changes of their physicochemical properties. Particularly, site 225 was located in the backbone of the active-site region, which is an important part involved in binding to the substrate (Sielecki et al. 1990). Thus, cetacean pepsin A may have evolved a stronger ability to digest food proteins by enhancing affinity with the substrate. In summary, although cetaceans still retain the multiple chambered stomach that is common in artiodactylans, the gastric PGA gene has adaptively evolved in response to the dietary switch.
Adaptive Evolution of Cetacean Lipid Digestion
Dietary triglycerides and phospholipids have a variety of complex forms; for example, they are composed of different chemical and stereochemical structures, and for different triglycerides and phospholipids, the fatty acids that compose them differ in many ways, such as their length and degree of esterification and saturation (Breckenridge et al. 1969; Carey et al. 1983; Freeman et al. 1965). To completely hydrolyze complex dietary lipids, a variety of lipases are required to work together, such as gastric lipase (LIPF), pancreatic triglyceride lipase (PNLIP), colipase (CLPS), pancreatic lipase-related protein (PLRP2), carboxyl ester lipase(CEL) and pancreatic phospholipase A2 (PLA2G1B). Thus, we selected these genes as candidates to explore the molecular mechanism of cetacean adaptation to a higher fat diet. In this study, none of the lipase genes had a disrupted ORF or premature stop codon, which suggested that lipases might express functional enzymes in cetaceans. Further selection detection showed that the PLRP2 gene underwent strong positive selection in cetaceans. PLPR2 has a lipolysis function, as it is responsible not only for the specific hydrolysis of long-chain monoglycerides but also lipolysis of galactolipids, retinol phospholipids, phospholipids and cholesterol (Sias et al. 2004). It is noteworthy that previous studies have found that expression of the PLRP2 gene in newborns is high (Yang et al. 2000), suggesting that the PLRP2 gene in the newborn plays a very important role in fat digestion. In addition, the fat content of cetacean milk was significantly higher than that of terrestrial mammals (White 1953), so it is reasonable to suggest that the positive selection on this gene in cetaceans might be related to their adaptation to a higher fat milk. Notably, codon 88 of PLRP2 is located in the PFAM domain and had 14 radical changes in properties, which suggests that this site might be important to enhance the esterification reactions of the enzyme. In addition to the PLRP2 gene, the LIPF, PNLIP and CYP7A1 genes were also detected to be under strong positive selection in cetaceans (Wang et al. 2016). In summary, cetaceans might have evolved a complex and effective mechanism in response to high-fat foods, and the above-mentioned four lipase genes have played an important role in this adaptive process to a higher fat diet after the dietary switch in cetaceans.
Convergent Evolution in Carnivorous Lineages
There are two lines of evidence showing that convergent evolution might exist in carnivorous lineages (cetaceans and carnivores). First, 10 of 12 functional genes were found to have specific parallel/convergent amino acid substitution between cetaceans and carnivores, and further statistic detection showed 36 parallel and 13 convergent nonsynonymous mutations. Because the existence of parallel/convergent evolution at the amino acid sequence level is regarded as a consequence of adaptive evolution (Zhang and Kumar 1997), the identification of parallel/convergent nonsynonymous changes in cetaceans and carnivores suggested convergent evolution related to a dietary switch for consuming higher protein and fat. Second, PGA and PLRP2 genes showed similar selection pressures in cetaceans and carnivores. Taken together, different carnivorous lineages, although having different evolutionary history, have evolved a similar mechanism at the molecular level in response to the carnivorous feeding habits.
References
Axelsson E et al (2013) The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 495:360–364
Breckenridge WC, Marai L, Kuksis A (1969) Triglyceride structure of human milk fat. Can J Biochem 47:761–769
Carey MC, Small DM, Bliss CM (1983) Lipid digestion and absorption. Pediatrics 45:651–677
Carginale V, Trinchella F, Capasso C, Scudiero R, Riggio M, Parisi E (2004) Adaptive evolution and functional divergence of pepsin gene family. Gene 333:81–90
Del Mar EG, Largman C, Brodrick JW, Fassett M, Geokas MC (1980) Substrate specificity of human pancreatic elastase 2. Biochemistry 19:468–472
Dierenfeld ES, Hintz HF, Robertson JB, Van Soest PJ, Oftedal OT (1982) Utilization of bamboo by the giant panda. The Journal of nutrition 112:636–641
Drummond DA, Wilke CO (2008) Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134:341–352
Duke GE (1986) Alimentary Canal: Secretion and Digestion, Special Digestive Functions, and Absorption. Avian Physiol 16:289–302
Fellenius E, Berglindh T, Sachs G, Olbe L, Elander B, Sjostrand SE, Wallmark B (1981) Substituted benzimidazoles inhibit gastric acid secretion by blocking (H+ + K+)ATPase. Nature 290:159–161
Foltmann B (1981) Gastric proteinases: structure, function evolution and mechanism of action. Essays Biochem 17:52–84
Foote AD et al (2015) Convergent evolution of the genomes of marine mammals. Nat Genet 47:272–275
Freeman CP, Jack EL, Smith LM (1965) Intramolecular fatty acid distribution in the milk fat triglycerides of several species. J Dairy Sci 48:853–858
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
He X, Zhang J (2005) Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics 169:1157–1164
Jin C, Ciochon RL, Dong W, Hunt RM Jr, Liu J, Jaeger M, Zhu Q (2007) The first skull of the earliest giant panda. Proc Natl Acad Sci USA 104:10932–10937
Jin K et al (2011) Why does the giant panda eat bamboo? A comparative analysis of appetite-reward-related genes among mammals. PLoS ONE 6:e22602
Kageyama T (2002) Pepsinogens, progastricsins, and prochymosins: structure, function, evolution, and development. Cell Mol Life Sci CMLS 59:288–306
Kiraly O et al (2007) Signal peptide variants that impair secretion of pancreatic secretory trypsin inhibitor (SPINK1) cause autosomal dominant hereditary pancreatitis. Hum Mutat 28:469–476
Korbova L, Kohout J (1981) Gastric acid proteinases and their zymogens. Acta Univ Carol Med Monogr 101:1–144
Liu Y et al (2015) Digestion of nucleic acids starts in the stomach. Sci Rep 5:11936
Maldonado-Valderrama J, Wilde P, Macierzanka A, Mackie A (2011) The role of bile salts in digestion. Adv Coll Interface Sci 165:36–46
Mead JG (2007) Stomach anatomy and use in defining systemic relationships of the Cetacean family Ziphiidae (beaked whales). Anat Rec (Hoboken) 290:581–595
Matthias H et al (2019) Genes lost during the transition from land to water in cetaceans highlight genomic changes associated with aquatic adaptations. Sci Adv 5(9):eaaw6671
Minh BQ, Nguyen MA, von Haeseler A (2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 30:1188–1195
Narita Y, Oda S, Takenaka O, Kageyama T (2000) Multiplicities and some enzymatic characteristics of ape pepsinogens and pepsins. J Med Primatol 29:402–410
Perelman P et al (2011) A molecular phylogeny of living primates. PLoS Genet 7:e1001342
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274
Petrov DA, Hartl DL (2000) Pseudogene evolution and natural selection for a compact genome. J Hered 91:221–227
Pond SL, Frost SD (2005) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533
Qian W, Liao BY, Chang AY, Zhang J (2010) Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet 26:425–430
Sachs G, Shin JM, Briving C, Wallmark B, Hersey S (1995) The pharmacology of the gastric acid pump: the H+, K+ ATPase. Annu Rev Pharmacol Toxicol 35:277–305
Samloff IM, Secrist DM, Passaro E Jr (1975) A study of the relationship between serum group I pepsinogen levels and gastric acid secretion. Gastroenterology 69:1196–1200
Sato M, Shibata C, Kikuchi D, Ikezawa F, Imoto H, Sasaki I (2010) Effects of biliary and pancreatic juice diversion into the ileum on gastrointestinal motility and gut hormone secretion in conscious dogs. Surgery 148:1012–1019
Schneeman BO (2002) Gastrointestinal physiology and functions. Br J Nutr 88(Suppl 2):S159–163
Shin JM, Grundler G, Senn-Bilfinger J, Simon WA, Sachs G (2005) Functional consequences of the oligomeric form of the membrane-bound gastric H, K-ATPase. Biochemistry 44:16321–16332
Sias B et al (2004) Human pancreatic lipase-related protein 2 is a galactolipase. Biochemistry 43:10138–10148
Sielecki AR, Fedorov AA, Boodhoo A, Andreeva NS, James MN (1990) Molecular and crystal structures of monoclinic porcine pepsin refined at 1.8 A resolution. J Mol Biol 214:143–170
Steinert RE, Feinle-Bisset C, Geary N, Beglinger C (2013) Digestive physiology of the pig symposium: secretion of gastrointestinal hormones and eating control. J Anim Sci 91:1963–1973
Szabo A, Pilsak C, Bence M, Witt H, Sahin-Toth M (2016) Complex formation of human proelastases with procarboxypeptidases A1 and A2. J Biol Chem 291:17706–17716
Thewissen JG, Cooper LN, Clementz MT, Bajpai S, Tiwari BN (2007) Whales originated from aquatic artiodactyls in the Eocene epoch of India. Nature 450:1190–1194
Thewissen JGM, Cooper LN, George JC, Bajpai S (2009) From land to water: the origin of whales, dolphins, and porpoises. Evol Educ Outreach 2:272–288
Uhen MD (2007) Evolution of marine mammals: back to the sea after 300 million years. Anat Rec 290:514–522
Wagner A (2005) Energy constraints on the evolution of gene expression. Mol Biol Evol 22:1365–1374
Wang Z et al (2016) Evolution of digestive enzymes and RNASE1 provides insights into dietary switch of cetaceans. Mol Biol Evol 33:3144–3157
Whitcomb DC, Lowe ME (2007) Human pancreatic digestive enzymes. Dig Dis Sci 52:1–17
White JC (1953) Composition of whales' milk. Nature 171:612
Woolley S, Johnson J, Smith MJ, Crandall KA, McClellan DA (2003) TreeSAAP: selection on amino acid properties using phylogenetic trees. Bioinformatics 19:671–672
Yampolsky LY, Stoltzfus A (2005) Untangling the effects of codon mutation and amino acid exchangeability. Pacif Symp Biocomput 10:433–444
Yang Y, Sanchez D, Figarella C, Lowe ME (2000) Discoordinate expression of pancreatic lipase and two related proteins in the human fetal pancreas. Pediatr Res 47:184–188
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang Z, Kumar S, Nei M (1995) A new method of inference of ancestral nucleotide and amino acid sequences. Genetics 141:1641–1650
Yang Z, Wong WS, Nielsen R (2005) Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Zhang J (2000) Rates of conservative and radical nonsynonymous nucleotide substitutions in mammalian nuclear genes. J Mol Evol 50:56–68
Zhang J (2003) Evolution by gene duplication: an update. Trends Ecol Evol 18:292–298
Zhang J, Kumar S (1997) Detection of convergent and parallel evolution at the amino acid sequence level. Mol Biol Evol 14:527–536
Zhang J, Zhang YP, Rosenberg HF (2002) Adaptive evolution of a duplicated pancreatic ribonuclease gene in a leaf-eating monkey. Nat Genet 30:411–415
Zhao H, Yang JR, Xu H, Zhang J (2010) Pseudogenization of the umami taste receptor gene Tas1r1 in the giant panda coincided with its dietary switch to bamboo. Mol Biol Evol 27:2669–2673
Zhao JH, Dong L, Hao XQ (2007) Small intestine motility and gastrointestinal hormone levels in irritable bowel syndrome. J South Med Univ 27:1492–1495
Zhou X, Xu S, Xu J, Chen B, Zhou K, Yang G (2012) Phylogenomic analysis resolves the interordinal relationships and rapid diversification of the laurasiatherian mammals. Syst Biol 61:150–164
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (NSFC) Grant No. 31872219 and 31370401, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Contributions
WR, GY, SX, GL and LL contributed to the study conception and design. Material preparation, data collection, analysis and experiment were performed by GL, HW, JB and XD. The first draft of the manuscript was written by GL. GY and WR revised previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling editor: Peter Chi.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, G., Wei, H., Bi, J. et al. Insights into Dietary Switch in Cetaceans: Evidence from Molecular Evolution of Proteinases and Lipases. J Mol Evol 88, 521–535 (2020). https://doi.org/10.1007/s00239-020-09952-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-020-09952-2