
Abstract
This study reports the evolutionary history and in-silico functional characterization of a novel water-deficit and ABA-responsive gene in wheat. This gene has remote sequence similarity to known abiotic stress-related genes in different plants, including CAP160 in Spinacia oleracea, RD29B in Arabidopsis thaliana, and CDeT11-24 in Craterostigma plantagineum. The study investigated if these genes form a close homologous relationship or if they are a result of convergent evolutionary processes. The results indicated a closely shared homologous relationship between these genes. Bayesian phylogenetic analysis of the protein sequences of the remotely related CAP160 proteins from various plant species indicated the presence of three distinct clades. Further analyses indicated that CAP160 homologous genes have predominantly evolved through neutral processes, with multiple regions experiencing signatures of purifying selection, while others were indicated to be the result of episodic diversifying selection events. Functional predictions revealed that these genes might share at least two functions related to abiotic stress conditions: one similar to the cryoprotective function of LEA protein, and the other a signalling molecule with phosphatidic acid binding specificity. Studies focused on the identification of cold-responsive genes are essential for the development of cold-tolerant crop plants, if we are to increase agricultural productivity throughout temperate regions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Global transcriptome analysis is an essential step to identify genes controlling specific traits. RNA sequencing (RNA-seq) on the crowns of the field-grown wheat (Triticum aestivum) revealed many genes of interest that are differentially expressed among wheats of different genetic backgrounds (Li et al. 2018). One gene (XLOC_002283) has been annotated as Traes_1AL_5184A1376 through the International Wheat Genome Sequencing Consortium as specified in the supplementary data of Li et al. (2018). This wheat gene (Traes_1AL_5184A1376) shows distant homology with the CAP160 (So-CAP160) gene from spinach, the CDeT11-24 (Cp-CAP160) gene from Craterostigma plantagineum, and the At-RD29B gene from Arabidopsis. Although the BLAST search shows that these genes are distantly related, their expressions are similarly affected by different abiotic stressors that elicit water-deficit conditions. Arabidopsis RD29 (AtRD29) genes are abiotic stress marker genes that are upregulated by ABA and water-deficit stress conditions (Hoth et al. 2002; Yamaguchi-Shinozaki et al. 1992) and have a remote homology with the cold acclimation protein, CAP160, from spinach that accumulates as a phosphoprotein in the plant leaves during cold acclimation (Guy and Haskell 1989, 1987; Kaye et al. 1998).
The protein profile of cold-acclimated spinach leaves versus non-acclimated plants showed the accumulation of many new polypeptides. Four of these proteins had been named CAP proteins and were denoted numbers for their molecular weights. For example, CAP160 is a protein that has been identified as one of the four major accumulated proteins in the cold-acclimated spinach leaves, where 32P-radiolabelling on cold-acclimated spinach seedlings indicates that CAP160 with its characteristic isoelectric point (pI) 4.5 is a phosphoprotein (Guy and Haskell 1989, 1987). The So-CAP160 has seven exons that tend to be shorter than the intervening introns, and its product is noticed in the soluble fraction with a characteristic acidic N-terminal region, which has similarity with some distantly related proteins. The low mobility of So-CAP160 on the gel may be owing to its acidic nature where the predicted molecular mass is 83.9 kDa and its isoelectric point is 4.5 (Guy and Haskell 1989). Eventually, the function of the accumulated So-CAP160, as enhanced by a low-temperature stressor, is still unpredictable.
Although of the low similarity between So-CAP160 protein and RD29 gene products from Arabidopsis, Kaye et al. (1998) suggested a homologous relationship due to the presence of a common acidic region among these proteins. Moreover, the CDeT11-24 gene from Craterostigma plantagineum has a distant homology with both So-CAP160 and AtRD29 genes (Velasco et al. 1998). The CDeT11-24 gene is upregulated in response to dehydration, salinity, and abscisic acid, and its product has a highly conserved lysine-rich domain, which resembles K-segments in dehydrins. Liquid chromatography-tandem mass spectrometry techniques confirmed that its protein has many phosphorylation sites, which are subject to phosphorylation only in dehydrated tissues (Röhrig et al. 2006). This low-water activity inducing protein phosphorylation is similar to what had been preliminarily demonstrated to its distant homologous So-CAP160 (Guy and Haskell 1989).
Though diverse abiotic stressors regulate RD29, CAP160, and CDeT11-24 genes, the common characteristics of these genes have not yet been identified. A newly identified water-deficit and ABA-regulated gene from wheat is distantly related to this group of genes which all have remote homology to one another, and because of this distant homology, only a limited number of genes have been documented. Thus, this study aimed to test whether the water-deficit and ABA-responsive gene from wheat is an authentic homologous gene of So-CAP160, Cp-CDeT11-24, and At-RD29B genes, where these genes were selected as examples due to their available experimental data from different studies. If these genes were found to be homologs, this study further aimed to identify additional homologous genes from various monocot and dicot plants to scrutinize the common and distinctive characteristics of this gene family.
While identity, e-value, and bit-score are the common ways to infer homology, e-value and bit-score are more useful, since a 30% identity cut-off may ignore many homologs that can be found with an e-value < 10–10 for nucleotide to nucleotide sequence comparison or e-value < 10–3, and bit-score > 50 for protein to protein sequence comparison (Pearson 2013). Moreover, the presence of the same domain among proteins may infer function relatedness but not authentic homologous relationships. Thus, the presence of a protein domain confirms homology but does not validate it as many non-homologous proteins may share domains (Joseph and Durand 2009). One limitation of inferring authentic homology among protein sequences using e-value, bit-score, and identity cut-offs is the availability of information in the query database during a blast search. Thus, a reciprocal blast should be used to confirm identities and avoid misleading results (Agostino 2013). Also, synteny can be used to confirm homology and verify the occurrence of homologous genes within the flanking regions surrounding the gene of interest in different species.
Furthermore, looking into the selective pressure on the molecular sequence evolution of many CAP160 homologous genes from dicots and monocots, fixed effects likelihood approaches (FEL) and Mixed-effects models of evolution (MEME) are two algorithms that are used to assess pervasively or episodically selected regions of proteins, respectively (Kosakovsky Pond and Frost 2005a; Murrell et al. 2012). Since selection pressure is a group of factors that drive nucleotide sequence mutations to enhance protein adaptability, the variable regions across protein sequences are weakly selected (positively selected) to improve the adaptability of the protein function, while stable regions are strongly selected (negatively selected) to maintain the structural stability and functions of these regions (Marsh 2009). The FEL algorithm uses the maximum-likelihood statistics to extrapolate a significant difference between the non-synonymous (β) and synonymous (α) nucleotide substitutions across the given nucleotide alignment for a specified phylogenetic tree. The FEL algorithm has a prior assumption of a constant selection pressure per site across the phylogenetic tree; thus, it only infers the pervasive selection. Giving this limitation, MEME algorithm uses two ω (β/α) rate classes to detect sites under positive selection where the rate of ω not only varies from site to site, but also from branch to branch. Therefore, the MEME algorithm is more sensitive in revealing sites under episodic diversifying selection through the same maximum-likelihood approach. One limitation of MEME is that it does not assign specific branches where individually selected sites exhibit episodic positive selection.
Consequently, to assess which branches and nodes demonstrated episodic positive selection during the evolution of this gene family, the adaptive branch site-random effects likelihood (aBSREL) method is used (Smith et al. 2015). The aBSREL test requires no prior knowledge about lineages-of-interest, and thus all the lineages across the entire phylogenetic tree will be tested. Using these methods should identify sites under positive or negative pervasive selection and sites under episodic positive selection, which is crucial for recognizing functionally essential regions in the CAP160 proteins.
Ultimately, identification of stress-inducible genes is essential for developing molecular markers, genetic improvements and understanding the molecular pathways of stress resistance in plants. This article answers the question on whether these remotely related genes share a common ancestry and attempt to predict their functions during water-deficit stress conditions. The results showed that So-CAP160 gene homologs constitute a family that is postulated to have evolved by vertical descent from a single common ancestor. The identification of this unique gene family not only improves the annotation of many remotely related genes but also supports future molecular and functional characterization studies.
Materials and Methods
Sequence Retrieval
A novel water-deficit and ABA-responsive gene (XLOC_002283; Traes_1AL_5184A1376) was retrieved from the RNA sequencing (RNA-seq) dataset of a cold experiment on wheat (Li et al. 2018). The wheat protein sequence was used as a query in a tblastn search with a cut-off e-value < 1e−04 to retrieve other homologous genes from wheat and other monocots (Pearson 2013), as well as against the Theobroma cacao genome database hosted by Ensembl Plants (https://plants.ensembl.org). The tblastn search against the T. cacao genome identified only one gene sequence that has been confirmed as an authentic homologous protein of the wheat and barley genes by the reciprocal blast. The retrieved protein sequence from T. cacao was used in a manual PSI-tBLASTn with the same cut-off e-value against many dicot plant genomes from different databases to retrieve all other dicot sequences (Table S1).
Multiple Sequence Alignment and Structure Predictions
Retrieved protein sequences were aligned by MAFFT version 7 (https://mafft.cbrc.jp/alignment/server/), T-COFFEE (Di Tommaso et al. 2011; Notredame et al. 2000), and Decipher (Wright 2015). The quality of the alignments was assessed using the VerAlign online tool (Bawono et al. 2015). Protein secondary structure was predicted using Jalview software version 2.10.3b1 (Waterhouse et al. 2009). The PONDR program (https://www.pondr.com) was used to predict the folding states of some selected proteins. The amphipathic helical wheels were predicted (Gautier et al. 2008) and their tertiary structures were presented (Reißer et al. 2014; Schrödinger LLC 2016).
Phylogenetic Analysis and Gene Structure
The best-fitted amino acid substitution model was selected with the lowest Bayesian information criterion (BIC) score, using ProtTest v3.4.2 (Darriba et al. 2011). A Bayesian phylogenetic tree was constructed via MrBayes v3.2.6. (Ronquist et al. 2012) with the Jones–Taylor–Thornton (JTT) amino acid substitution model, and (+ I + G) for the rate of heterogeneity with (+ F) for the empirical equilibrium of amino acid frequencies from the alignment. The Markov Chain Monte Carlo parameters were: Ngen = 106, nchains = 12, burninfrac = 0.25. The reconstructed phylogenetic tree of clade III was done using the same protein substitution model for 6 × 105 generations, while the model JTT + G + F suits clade I and clade II were done with 5 × 105 and 105 generations, subsequently. The phylogenetic tree was imported to iTOL (Interactive Tree of Life) webserver for visualization (Letunic and Bork 2016) and the intron/exon structures of different genes were graphed using the GSDS online tool (Hu et al. 2015).
Comparative Genomics
Conserved gene order in microsyntenic chromosomal regions (100-Kb) containing CAP160 gene homologs was compared to Oryza sativa, Gossypium raimondii, Solanum tuberosum, Solanum lycopersicum, Sorghum bicolor, Capsella rubella, Fragaria vesca, Eucalyptus grandis, Carica papaya, Arabidopsis thaliana, and Amborella trichopoda against the matching genetic region in T. cacao using the Plant Genome Duplication Database (PGDD), freely available at https://chibba.agtec.uga.edu/duplication/ (Lee et al. 2013).
Analysis of Enriched Cis-Regulatory Elements in Promoter Regions
Upstream sequences (2-Kb) relative to the translation start site for each gene were retrieved from the blast service hosted by the NCBI database (NCBI Resource Coordinators 2016). Distribution and occurrence of enriched PLACE cis-elements were identified and graphed (Higo et al. 1999; Austin et al. 2016). Only motifs with Z-scores more than 5 were presented, where the functional depth of the position-specific scoring matrix (PSSM) was more than 0.35, and with the condition that a minimum of 50% of the tested promoters in each group must have the selected motif.
Protein Characters and Subcellular Localization Predictions
Protein molecular weights, isoelectric points, and the grand average of hydropathy (GRAVY) were estimated (Kozlowski 2017; Kyte and Doolittle 1982). Subcellular localization of each protein was predicted by five different tools, which are LOCALIZER (Sperschneider et al. 2017), PredSL (Petsalaki et al. 2006), CELLO (Yu et al. 2006), Predotar (Small et al. 2004), and LocTree3 (Goldberg et al. 2014).
Protein Conserved Motif Evaluation
Thirty conserved protein motifs were identified using Multiple EM for Motif Elicitation (MEME) at the website https://meme.sdsc.edu/meme/intro.html (Bailey et al. 2009), with default parameters. The predicted functional sites in these motifs were retrieved from the Eukaryotic Linear Motif (ELM) web-interface https://elm.eu.org/ (Dinkel et al. 2016).
In-Silico Differential Gene Expression and Co-expression Analyses
Differential gene expression analyses of Arabidopsis, rice, barley, maize, and wheat CAP160 homologous genes within various microarray-based experiments and across different treatments were retrieved from Genevestigator software (Zimmermann et al. 2014), using the appropriate gene identifiers. In Arabidopsis, AT5G52300.1 and AT4G25580.1 were used to represent the genes Ath1.1 and Ath1.2, respectively. For the rice, Loc-Os10g36180.1 was used to represent the Os gene. In barley, Mloc_74793.3 was used to represent the Hv gene. While in maize, GRMZM2G376743 was used to represent both Zm1.1 and Zm1.2 genes. For wheat, the Ta.8085.1.S1_at probe was used to represent Ta1.1, Ta1.2, and Ta1.3 genes, while the TaAffx.51089.1.S1_at probe was used to represent Ta1.3, Ta1.4, and Ta1.5 genes. Positively co-expressed genes of CAP160 homologs in Arabidopsis and rice were only presented by using the same software, highlighting the annotations of the highly connected genes and their differential expression profiles.
Estimation of the Selection Pressure
The alignment of protein-coding sequences in different species was used to evaluate the effect of natural selection on protein evolution across this gene family. Two methods, MEME (Murrell et al. 2012) and FEL (Kosakovsky Pond and Frost 2005a), were used to assess selection on individual sites that may be under episodic diversifying selection, as determined by MEME, or pervasive selection, as measured by FEL. To specify that the branches and nodes demonstrated episodic positive selection during the evolution of this gene family, the aBSREL method was used (Smith et al. 2015). These tools for estimating selection pressures are implemented in the Datamonkey web server (Kosakovsky Pond and Frost 2005b).
Results
Authentic Homology Assessment
A reciprocal blast search was performed against different monocot plant species, such as barley and wheat, to validate the homologous relationships. Using the cacao protein as a query in a tBLASTn search with an e-value parameter < 1e−04, returns all known plant CAP160 genes from various dicot species. These include So-CAP160, At-RD29, and Cp-CDeT11-24. The inability of wheat or barley proteins to retrieve these sequences by a simple tBLASTn may be due to the difficulty of establishing a reliable alignment score among distantly related proteins by using its algorithm. Though, comparing cacao with barley proteins yields a low identity value (26%), the cacao protein query covers 83%, with a bit-score of 62.4, which indicates the limitation of using 30% identity cut-off in overlooking many homologs (Pearson 2013). Finally, using cacao or barley protein as queries in a tblastn search with cut-off e-values < 1e−04 against different dicot and monocot plant genome databases, respectively, retrieved CAP160 homologous gene sequences from the different dicot or monocot species (Table S1).
To further confirm the orthologous relationship between the retrieved cacao sequence gene and other genes from monocot or dicot clades, comparative genomics was used for the chromosomal segment-containing TCM_015165 from cacao with the corresponding chromosomal segments from eleven different plant species. Results showed that many syntenic anchors in cacao match those in Amborella, rice, sorghum, and many other genes from dicot plants (Fig. S1). Moreover, microsyntenic analysis confirms the presence of duplication events of the whole protein-coding genes; as examples, this includes the two genes from Capsella and for genes Ath-RD29B and Ath-CAP160. Although two adjacent markers are enough to infer homologous synteny blocks, the analysis revealed at least the presence of four syntenic markers and confirmed the divergence of matching microsyntenic anchors among distantly related species (Fig. S1).
Molecular Phylogenetics
Many homologous genes available in the public databases were retrieved (Table S1) and aligned to estimate the evolutionary history of the newly identified water-deficit and ABA-responsive gene in wheat. Testing the quality of different alignments by the VerAlign tool (Bawono et al. 2015) indicated that Decipher as a test alignment presents a higher sum of pairs (SP) score, column score (CS), and SPdist score (Table S2). Therefore, the output alignment from Decipher was used for further analyses.
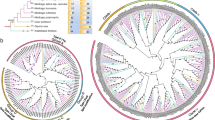
By taking the protein from the basal angiosperm Amborella trichopoda, as an outgroup, the Bayesian analysis showed that all the retrieved protein sequences could be clustered within three major clades (Fig. 1). All monocot species from clade II share two common ancestors, Phoenix dactylifera (Pd) and Musa acuminate (Ma1.1), which is in line with the phylogeny inference of Poales based on their plastid genome (Givnish et al. 2010) and phylogenetic analysis of angiosperms families based on fossil records (Magallón et al. 2015), where Arecales and Zingiberales are presented with older ages, respectively. Additionally, cold- and drought-sensitive monocot species form a separate clade originated from node 42 (N 42) and can be distinguished from cold- and drought-tolerant monocot species from node 21 (N 21). Clade III represents only species from dicots, while clade I forms a unique monophyletic group near the common outgroup. The intron/exon organization showed pervasive symmetric exons, where all symmetric exons are surrounded with phase 1 introns, while asymmetric exons are bordered by phase 0 and phase 1, and numbers of introns ranged from 0 to 7 (Fig. 1).

Bayesian phylogenetic analysis and gene structures with intron phase distributions of CAP160 homologous genes from different plant species. The phylogenetic tree is rooted by using the homologous sequence from Amborella trichopoda (Atr). The analysis reveals three distinctive clades which are coloured as green, violet, blue for clade I, clade II, and clade III, respectively. The phylogenetic tree nodes were named according to Datamonkey web server, and branches with red colour and nodes with stars show episodic diversifying selection with a p value threshold of 0.05 as estimated by aBSREL method (Color figure online)
Signatures of Natural Selection
The FEL analysis showed that 373 sites are significantly under pervasive negative selection (Table S4), while only five sites are positively selected with a p value threshold of 0.05 (Table S3). This result indicated that the evolution of the plant CAP160 genes could be best described by neutral theory where only 12.6% are under constrained selection, and a small portion of around 0.17% are positively selected. MEME detected twenty sites under episodic diversifying selection (Table 1); four of them were also significantly detected by FEL under pervasive positive selection (Table S3). Furthermore, aBSREL revealed that 34% of branches could be modelled with a single ω rate class, while 66% of branches can be only modelled with multiple ω rate classes, and 24 branches have a proportion of sites under diversifying selection (Table S5). A total of twelve lineages and twelve nodes showed episodic positive selection. In most lineages, the portion of sites under positive selection was less than 30%, while the remaining sites exhibit neutral or negative selection where ω1 ranges from 0.022 to 1 (Table S6).
Functional Predictions
The predicted isoelectric points of CAP160 homologous proteins reveal acidic tendency, except for Br1.4 (Table S7). Sizes of the proteins revealed that Br1.4 is the smallest predicted protein, with only 220 amino acids. Thus, the presence of the K-like segment within its small size may accord its predicted pI at a higher pH value. The biggest protein was Pv1.2, with 141.4 kDa, while other protein predicted sizes ranged from 34.1 to 100.7 kDa. The narrow range of sizes and isoelectric points suggest common biochemical characters among diverse CAP160 proteins.
GRAVY values showed that all the tested proteins are hydrophilic. Localization predictions show that all clade I proteins are nuclear proteins; however, PredSl predicts Kl1.4, Kf1.2, and Kl1.3 as mitochondrial proteins and Localizer predicts this set of proteins to be targeted to the nucleus or the chloroplast (Table S7). Md1.3 protein is predicted by LocTree3 to localize in the plasma membrane based on a weak homology to the mucin HKR1 in yeast. Clade I proteins are frequently predicted as nuclear proteins. All tools predict Atr, clade II, and III proteins as either nuclear or cytoplasmic proteins. Only Gr protein in clade III was predicted with a signal peptide by PredSl and Predotar, while Fv was predicted as a secretory protein only by PredSl; this might be because both use a neural network-based approach that is trained only on known proteins.
The promoter analysis of species from clade I with the promoter of Atr did not show significant enrichment (Z score > 5) of cis-elements that may regulate the expression of abiotic stress-induced genes (Fig. S3). The promoters of genes Mg1.2, Cs1.2, Md1.3, and Ac1.2 from clade I did not show motifs related to either the CBF-regulatory pathway or the ABA-dependent pathway to be significantly enriched. The gene promoters of clade II confirmed the enrichment of ABA-responsive elements, as shown in groups IIA and IIB (Fig. S3). Groups from III to VII represent plant gene promoters of clade III, where their promoter analysis displayed the prevalence of ABA-responsive cis-acting elements. DRE/CRT elements were also detected in Pv1.2, So1.1, and So1.2 of group VII promoters. Most groups showed the enrichment of one or more elements related to light-dependent development. The circadian regulation was enriched only in group IVA promoters, where the CIACADIANLELHC motif was found in all of its members. These results show that this gene family embraces many ABA-regulated genes, although of the diminishment of ABA-responsive cis-elements in the analysed clade I and Atr gene promoters.
Microarray-based differential expression analysis was explored across four different species: two genes from Arabidopsis that represent clade III, and four genes from barley, wheat, and rice that represent clade II. The results revealed that almost all genes are upregulated by similar conditions such as abscisic acid (ABA), cold, drought, and dehydration (Fig. S4). Positive co-expression analysis indicated that many LEA and dehydrins are positively co-expressed with CAP160 genes from Arabidopsis or rice (Dataset S1). This result signifies the functional similarity of the target genes to dehydrins. The analysis of mutually correlated genes from the explored co-expression networks showed that hub genes from Arabidopsis and rice share significant and co-ordinated upregulation of their expression under ABA treatment or with conditions that instigate a decline in water activity. Moreover, the target genes with these hub genes were downregulated during germination, hypoxia, and anoxia (Dataset S1). These results are consistent with the promoter analysis of the genes, where it revealed the presence of many ABRE and CRT/DRE cis-regulatory elements in their promoter region.
The protein motif analysis showed the presence of motif MEME-1 in all the tested sequences (Table S8); this motif is found in the N-termini of all proteins (Figs. S5–S7) where it localizes in or borders to the lysine-rich segment. The motif consensus sequence is SVLK (Fig. S2) and has a corresponding homology with the [GS]IL[RK] sequence that is known as the SILK motif (Table S8). The SILK motif is a feature of Protein Phosphatase 1-interacting proteins (PIPs), and is essential for inhibiting Protein Phosphatase 1 (PP1) activity, as the phosphorylated form of some PIPs acts as pseudosubstrates to PP1 (Dinkel et al. 2016; Hendrickx et al. 2009). Motif MEME-4 has a PPXY motif that interacts with a WW-domain when the S/T motif in the P(S/T)P sequence is phosphorylated (Dinkel et al. 2016). Additionally, motif (MEME-30) that is found only in some proteins of clade III and II recognizes the SH2 domain when the tyrosine of this motif is phosphorylated. Other motifs are related to the clathrin-mediated pathway, where MEME-3 and MEME-13 recruit adaptor proteins for clathrin-coated vesicle formation, and MEME-6 binds the VHS domain of clathrin-related adaptor proteins through its acidic dileucine motif.
Many degron motifs were found across all the tested proteins; these are MEME-5, MEME-9, MEME-18, MEME-24, MEME-10, MEME-26, and MEME-28. Two motifs were found as a recognition site for the Inhibitor of Apoptosis Proteins (IAPs): MEME-12 and MEME-27. Four short linear motifs were identified by their predicted ability to recognize protease enzymes such as separase: MEME-14, MEME-16, MEME-21, and MEME-22, which may guide the cleavage of the target protein. More motifs were found to mediate protein–protein interaction, such as MEME-8, MEME-15, and MEME-17 that recognize the UEV domain, and MEME-23 that binds to WD40 repeat domain. Only MEME-7 and MEME-20 were detected as DNA binding motifs. Motif MEME-29 was predicted to bind with DEAD-box helicase to mediate the degradation of aberrant RNAs, while MEME-11 and MEME-25 were predicted as possible sites for C-terminal amidation. Motif MEME-19 is found in the N-terminal half of the cytosolic soluble Pex5 protein to mediate protein import into peroxisomes. Finally, MEME-2 was predicted as a binding site for TRAF2, a cytosolic tumour necrosis factor receptor (TNFR).
The overall functions of various motifs within different proteins were found to mediate and regulate miscellaneous classes of protein–protein interactions where phosphorylation regulates many of them. The presence of the sequence motif MEME-1 near the K-like segment and its conservation in all the tested proteins implies that all the tested proteins may at least share one common biochemical function although of their divergence. The hypothesis of sharing functions among these proteins was also confirmed by the presence of many signature sequences with the same conserved functions, such as the four short linear motifs MEME-14, MEME-16, MEME-21, and MEME-22 among different proteins from various clades. Although these results may infer the convergent evolution among these proteins, the phylogenetic analysis results confirmed their divergent evolution.
Many large unstructured regions (> 30 amino acids) were detected in all the tested proteins (Fig. 2). The CDeT11-24 gene product maintains its solubility with heat treatment and adopts many α-helical contents upon dehydration, as experimented by trifluoroethanol (TFE) (Petersen et al. 2012). Thus, these natively disordered regions might hold α-helical structures upon dehydration; the thick black lines in Fig. 2 designate these regions.

Predictions of intrinsically disordered regions in some plant CAP160 homologous proteins from different clades as predicted by PONDR server
The identified K-like segment in the CDeT11-24 gene product was suggested to harbouring an amphipathic helix (AH) (Petersen et al. 2012). As the lysine-rich segments maintain their conservation through purifying selection, similar amphipathic helices may be found in other homologous proteins. Giving the essential function of this K-like segment (Petersen et al. 2012) and its highly conservation, the results confirmed a high similarity between the helices in the K-like segments from CAP160 homologous proteins in wheat and Craterostigma plantagineum, with six hydrophobic amino acids constitute the hydrophobic surface, and the hydrophilic surface establishes approximately 60% of the wheel (Fig. 3).

A visual comparison of the helical wheel representations from Cp-CAP160 (CDeT11-24) and Ta-CAP160 (Ta1.1) proteins
Discussion
The results revealed that So-CAP160, CDeT11-24, At-RD29B, and a novel cold-responsive gene from wheat are homologous genes, which belong to one gene family. The Bayesian phylogenetic inference grouped a sample of plant CAP160 proteins in three distinct clades (Fig. 1). All genes from clade I have intraspecies duplicates that have diverged in either clade II or III. This early origin of “clade I” indicates that clade II and III alleles may have arisen by divergent evolution from clade I. Additionally, authentic homology assessment and synteny analysis reject the hypotheses of convergent or parallel evolution. Many syntenic blocks surrounding CAP160 genes were inherited across different species (Fig. S1), which supports their evolution from a common ancestor. Given the long divergence time between clades II and III, a posterior probability value of 91% should be reasonable to support divergent evolution (Fig. 1).
Based on the Bayesian phylogenetic analysis, FEL indicated a lack of both background and differentiating selections, as supported by the similarity of non-synonymous (β) and synonymous (α) substitution rates per site-basis (Table S4). The presence of only 12.6% sites under pervasive negative selection indicates the presence of selective pressures that change the features of the encoded proteins and reveals the higher evolutionary rate inside this gene family. Table S4 shows that the codons which encode the K-like segments are under negative selection, signifying the existence of selective constraints that preserve the features of this alpha-helix fragment. Although, of the typical appearance of neutral evolution, some signatures of pervasive negative selection were apparent, with only five sites experiencing positive selection as detected by FEL and 20 sites by MEME. Thus, these sites could be essential for protein function, as their evolutionary changes can be explained by natural selection (Kimura 1991). The results denote the presence of adaptive protein evolution on specific sites in some plant species, which may be related to a specific function. The recency of this adaptive evolution can be seen by the absence of episodic diversifying signatures over the ancestral alleles from clade I (Fig. 1).
The high numbers of substitutions did not affect most protein characters such as solubility and acidic nature among proteins from different clades. All subcellular predictions were pointed towards predicting this protein family as either nuclear or cytoplasmic proteins. The nuclear-targeting predictions were based only on the most conserved alpha-helical region (K-like segment), as seen by Localizer (Table S7). If the function of the K-like segments is not to target these proteins to the nucleus, these proteins can be favourably predicted as cytoplasmic proteins. To compare these results with the experimentally verified proteins, the So-CAP160 protein was detected in the soluble fraction and mitochondria, rather than other subcellular fractions of the spinach leaf and the hypocotyl tissue (Kaye et al. 1998). Furthermore, the GFP-tagged At-RD29A or At-RD29B cDNAs trace these proteins in the cytoplasm only (Msanne et al. 2011). Immunolocalization shows that CDeT11-24 gene product is localized in the cytoplasm (Velasco et al. 1998). Accordingly, the experimental validations and in-silico predictions agree on the cytoplasmic subcellular localization of CAP160 proteins.
The promoter cis-elements analysis shows that the CAP160 gene family encompasses mainly ABA-regulated genes (Fig. S3), with a prominent enrichment of ABRE elements in the derived alleles (Uno et al. 2000); this may indicate an adaptation to abiotic stress conditions. These results are in line with the meta-analysis gene expressions where orthologous genes from Arabidopsis, rice, barley, and wheat were upregulated by ABA (Fig. S4). Furthermore, results in Fig. S3 show that CAP160 genes from Arabidopsis and rice are co-expressed with many LEA proteins, mostly in the ABA-dependent pathway (van Dam et al. 2017). Additionally, the enrichment of CRT/DRE elements in Group IIB that represents monocot crops with a winter background, rather than Group IIA of monocot summer crops, highlights the importance of the CAP160 genes in increasing tolerance to cold conditions (Gusta et al. 2005). This result matches the marked log2 expression values of CAP160 genes from barley and wheat, in comparison with rice in response to cold (Fig. S4). Motifs related to development, embryogenesis, and light regulation are enriched along promoters from different clades, which follows the in-silico expression analysis of many CAP160 homologous genes from different plants (Fig. S4). Moreover, the enrichment of CArG cis-elements in the promoters of ancestral alleles, rather than the derived alleles, supports the upstream regulation of the plant MADS-box transcription factors in the latter (de Folter et al. 2005).
The folding state of CAP160 proteins emphasizes their role as molecular shields and their flexibility to engage in different biological interactions (Wright and Dyson 2015). Also, many of these regions, such as the K-like segments, can be structured upon dehydration or biochemical interactions (Fig. 2). Remarkably, intrinsically disordered proteins (IDPs) can tolerate mutations and maintain similar functions (Zarin et al. 2017); this feature was noticed from the analysis of protein motifs where many motifs with similar functions were well conserved among separated clades. This is in line with the variation of selective pressure on different regions of CAP160 proteins where some ordered regions such as the K-like segment are negatively selected, whereas many disordered regions are under neutral or positive selection. This variance in evolutionary rates between ordered and disordered regions for the same protein is a feature for IDPs (Chi and Liberles 2016).
The protein motif predictions confirm the fact that this family encompasses a class of phosphoproteins. These results are supported by the mass spectrometric analysis results of CDeT11-24 (Röhrig et al. 2006). Moreover, three verified phosphorylated sites correspond with three different MEMEs from the in-silico motif predictions. Phosphorylation site 473A is predicted within an alpha-helix (Fig. S3) and is harboured by MEME-2 which binds to TRAF proteins; this site is significantly under negative selection (Table S4). Many plant TRAF proteins were identified with roles in either plant development or during abiotic stress, such as SINA2, a TRAF-like family protein that is involved in ABA-mediated drought stress signalling in Arabidopsis (Bao et al. 2014). Phosphorylation site 318P within MEME-18, a degron motif that has redundant functions with other MEMEs (MEME-5, MEME-9, MEME-24, MEME-10, MEME-26, and MEME-28), leads to the same function outputs among the diverged proteins. Phosphorylation controls the protein interaction-guided degradation; for example, stress-induced phosphorylation constrains KEAP1 inhibitor protein binding to the phosphorylated degron motif of the target protein (Atia and Abdullah 2014). Finally, MEME-6 embodies the phosphorylation site 552S, which is located within a well-conserved alpha-helix that may bind to the VHS domain when the serine residue is phosphorylated. This serine site is maintained by negative selection, as detected by FEL under codon number 2807 (Table S4). Although the function of the VHS domain is not fully elucidated yet, it is mainly characterized by its interaction with phosphoinositides and their ability to recruit adaptor proteins into clathrin-coated vesicle formation (Demmel et al. 2008; Zouhar and Sauer 2014).
Interestingly, the presence of MEME-1 in all the tested proteins speculates the function of plant CAP160 phosphoproteins as negative regulators of protein phosphatases. Also, the K-like segment, which adopts an amphipathic helical structure where the hydrophilic face can be clearly distinguished from the hydrophobic one (Fig. 3), is in vitro verified to bind specifically with phosphatidic acid rather than other lipids (Petersen et al. 2012). These results may indicate a conserved signalling function in the ABA-dependent pathway and may provide a clue to the mechanism by which low-temperature or ABA treatment inactivates protein phosphatases, such as ABSCISIC ACID-INSENSITIVE 1 (ABI1) in Arabidopsis, to promote ABA signalling. The PA binding to ABI1 is most likely inhibiting the negative effect of ABI1 on ABA production (Zhang et al. 2004). However, this binding was not efficient in vitro, which indicates the need for other molecules that may stabilize this binding. These predictions adopt that plant CAP160 proteins may mediate protein phosphatase binding with phosphatidic acid in their activated/phosphorylated form that is initiated only under abiotic stressors such as low-temperature conditions.
In conclusion, plant CAP160 proteins have evolved in most land plant species, showing neutral evolution patterns with many signatures of negative selection to maintain their essential functions and few sites showing episodic positive selection. The expression and co-expression analyses show that this family of proteins are upregulated mostly through the ABA signalling pathway and under water-deficit stress conditions. This regulation was confirmed by the enrichment of many ABRE motifs and CRT/DRE motifs in their promoters. The plant CAP160 proteins adopt many phosphorylation sites (Röhrig et al. 2006), have intrinsic disorder structures (Fig. 2), act as local shields by protecting the target enzyme from desiccation damage, and tend to bind specifically with PA through their highly conserved K-like segment (Petersen et al. 2012). Interestingly, the functional predictions adopt that these proteins can be involved in essential pathways that are mainly binding with PA, clathrin coat formation, and in facilitating the formation of a stable complex assembly, which may include phosphatidic acid, phosphorylated CAP160, and the target protein phosphatase. The presence of many intrinsically disordered regions is a characteristic of protein phosphatase1-interacting proteins, by which they can associate with the large surface of protein phosphatases to inhibit their activity (Heroes et al. 2013). Thus, the phosphorylated forms of the plant CAP160 proteins may activate the inhibition of protein phosphatases, and therefore retains the generation of the ABA signalling pathway. Additionally, they may act as local shields to protect phosphatases from degradation during desiccation conditions. Although it is experimentally verified that ABA releases the inhibitory effect of the protein phosphatase type 2C (HAB1) on the Arabidopsis RD29B promoter (Saez et al. 2008), the function of plant CAP160 proteins during abiotic stress is still questionable. Eventually, plant CAP160 protein–lipid and protein–protein interactors may still to be identified in vivo to facilitate our understanding of their functions during water-deficit conditions.
References
Agostino M (2013) Reciprocal BLAST: confirming identities. In: Practical bioinformatics. Garland Science, Taylor & Francis Group, LLC, New York, pp 127–130
Atia A, Abdullah A (2014) The Nrf2-Keap1 signalling pathway: mechanisms of ARE transcription regulation in antioxidant cellular defence. Int. J. PharmTech Res. 6:154–167
Austin RS, Hiu S, Waese J, Ierullo M, Pasha A, Wang TT, Fan J, Foong C, Breit R, Desveaux D, Moses A, Provart NJ (2016) New BAR tools for mining expression data and exploring Cis-elements in Arabidopsis thaliana. Plant J. 88:490–504. https://doi.org/10.1111/tpj.13261
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 37:W202–W208. https://doi.org/10.1093/nar/gkp335
Bao Y, Wang C, Jiang C, Pan J, Zhang G, Liu H, Zhang H (2014) The tumor necrosis factor receptor-associated factor (TRAF)-like family protein SEVEN IN ABSENTIA 2 (SINA2) promotes drought tolerance in an ABA-dependent manner in Arabidopsis. New Phytol. 202:174–187. https://doi.org/10.1111/nph.12644
Bawono P, Van Der Velde A, Abeln S, Heringa J (2015) Quantifying the displacement of mismatches in multiple sequence alignment benchmarks. PLoS ONE 10(5):e0127431. https://doi.org/10.1371/journal.pone.0127431
Chi PB, Liberles DA (2016) Selection on protein structure, interaction, and sequence. Protein Sci. 25(7):1168–1178. https://doi.org/10.1002/pro.2886
Darriba D, Taboada G, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. https://doi.org/10.1093/bioinformatics/btr088Epub 2011
de Folter S, Immink RGH, Kieffer M, Pařenicová L, Henz SR, Weigel D, Busscher M, Kooiker M, Colombo L, Kater MM, Davies B, Angenent GC (2005) Comprehensive interaction map of the Arabidopsis MADS Box transcription factors. Plant Cell 17:1424–1433. https://doi.org/10.1105/tpc.105.031831
Demmel L, Gravert M, Ercan E, Habermann B, Müller-Reichert T, Kukhtina V, Haucke V, Baust T, Sohrmann M, Kalaidzidis Y, Klose C, Beck M, Peter M, Walch-Solimena C (2008) The clathrin adaptor Gga2p is a phosphatidylinositol 4-phosphate effector at the Golgi exit. Mol. Biol. Cell. 19(5):1991–2002. https://doi.org/10.1091/mbc.E06-10-0937
Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang JM, Taly JF, Notredame C (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res 39:W13–W17. https://doi.org/10.1093/nar/gkr245
Dinkel H, Roey K, Michael S, Kumar M, Uyar B, Altenberg B, Milchevskaya V, Schneider M, Kühn H, Behrendt A, Dahl SL, Damerell V, Diebel S, Kalman S, Klein S, Knudsen AC, Mäder C, Merrill S, Staudt A, Thiel V, Welti L, Davey NE, Diella F, Gibson TJ (2016) ELM 2016—data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res 44:D294–D300. https://doi.org/10.1093/nar/gkv1291
Gautier R, Douguet D, Antonny B, Drin G (2008) HELIQUEST: A web server to screen sequences with specific α-helical properties. Bioinformatics 24:2101–2102. https://doi.org/10.1093/bioinformatics/btn392
Givnish TJ, Ames M, McNeal JR, McKain MR, Steele PR, dePamphilis CW, Graham SW, Pires JC, Stevenson DW, Zomlefer WB, Briggs BG, Duvall MR, Moore MJ, Heaney JM, Soltis DE, Soltis PS, Thiele K, Leebens-Mack JH (2010) Assembling the tree of the monocotyledons: plastome sequence phylogeny and evolution of Poales. Ann Missouri Bot Gard 97:584–616. https://doi.org/10.3417/2010023
Goldberg T, Hecht M, Hamp T, Karl T, Yachdav G, Ahmed N, Altermann U, Angerer P, Ansorge S, Balasz K, Bernhofer M, Betz A, Cizmadija L, Do KT, Gerke J, Greil R, Joerdens V, Hastreiter M, Hembach K, Herzog M, Kalemanov M, Kluge M, Meier A, Nasir H, Neumaier U, Prade V, Reeb J, Sorokoumov A, Troshani I, Vorberg S, Waldraff S, Zierer J, Nielsen H, Rost B (2014) LocTree3 prediction of localization. Nucleic Acids Res. https://doi.org/10.1093/nar/gku396
Gusta LV, Trischuk R, Weiser CJ (2005) Plant cold acclimation: the role of abscisic acid. J. Plant Growth Regul. https://doi.org/10.1007/s00344-005-0079-x
Guy CL, Haskell D (1987) Induction of freezing tolerance in spinach is associated with the synthesis of cold acclimation induced proteins. Plant Physiol 84:872–878. https://doi.org/10.1104/pp.84.3.872
Guy CL, Haskell D (1989) Preliminary characterization of high molecular mass proteins associated with cold acclimation in spinach. Plant Physiol Biochem 27:777–784
Hendrickx A, Beullens M, Ceulemans H, Den Abt T, Van Eynde A, Nicolaescu E, Lesage B, Bollen M (2009) Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem Biol 16:365–371. https://doi.org/10.1016/j.chembiol.2009.02.012
Heroes E, Lesage B, Görnemann J, Beullens M, Van Meervelt L, Bollen M (2013) The PP1 binding code: a molecular-lego strategy that governs specificity. FEBS J 280:584–595. https://doi.org/10.1111/j.1742-4658.2012.08547.x
Higo K, Ugawa Y, Iwamoto M, Korenaga T (1999) Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. https://doi.org/10.1093/nar/27.1.297
Hoth S, Morgante M, Sanchez J-P, Hanafey MK, Tingey SV, Chua N-H (2002) Genome-wide gene expression profiling in Arabidopsis thaliana reveals new targets of abscisic acid and largely impaired gene regulation in the abi1-1 mutant. J Cell Sci 115(Pt 24):4891–4900. https://doi.org/10.1242/jcs.00175
Hu B, Jin J, Guo AY, Zhang H, Luo J, Gao G (2015) GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31:1296–1297. https://doi.org/10.1093/bioinformatics/btu817
Joseph JM, Durand D (2009) Family classification without domain chaining. Bioinformatics. https://doi.org/10.1093/bioinformatics/btp207
Kaye C, Neven L, Hofig A, Li QB, Haskell D, Guy C (1998) Characterization of a gene for spinach CAP160 and expression of two spinach cold-acclimation proteins in tobacco. Plant Physiol 116:1367–1377. https://doi.org/10.1104/pp.116.4.1367
Kimura M (1991) The neutral theory of molecular evolution: a review of recent evidence. Jpn J Genet. https://doi.org/10.1266/jjg.66.367
Kosakovsky Pond SL, Frost SDW (2005a) Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222. https://doi.org/10.1093/molbev/msi105
Kosakovsky Pond SL, Frost SDW (2005b) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533. https://doi.org/10.1093/bioinformatics/bti320
Kozlowski LP (2017) Proteome-pI: proteome isoelectric point database. Nucleic Acids Res 45:D1112–D1116. https://doi.org/10.1093/nar/gkw978
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132. https://doi.org/10.1016/0022-2836(82)90515-0
Lee TH, Tang H, Wang X, Paterson AH (2013) PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res. https://doi.org/10.1093/nar/gks1104
Letunic I, Bork P (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. https://doi.org/10.1093/nar/gkw290
Li Q, Byrns B, Badawi MY, Diallo AB, Danyluk J, Sarhan F, Laudencia-Chingcuanco D, Zou J, Fowler DB (2018) Transcriptomic insights into phenological development and cold tolerance of wheat grown in the field. Plant Physiol 176:2376–2394. https://doi.org/10.1104/pp.17.01311
Magallón S, Gómez-Acevedo S, Sánchez-Reyes LL, Hernández-Hernández T (2015) A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytol 207:437–453. https://doi.org/10.1111/nph.13264
Marsh L (2009) A model for protein sequence evolution based on selective pressure for protein stability: application to hemoglobins. Evol. Bioinform 5:107–118
Msanne J, Lin J, Stone JM, Awada T (2011) Characterization of abiotic stress-responsive Arabidopsis thaliana RD29A and RD29B genes and evaluation of transgenes. Planta 234:97–107. https://doi.org/10.1007/s00425-011-1387-y
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL (2012) Detecting individual sites subject to episodic diversifying selection. PLoS Genet. https://doi.org/10.1371/journal.pgen.1002764
NCBI Resource Coordinators (2016) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 44:D7–D19. https://doi.org/10.1093/nar/gks1189
Notredame C, Higgins DG, Heringa J (2000) T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217. https://doi.org/10.1006/jmbi.2000.4042
Pearson WR (2013) An introduction to sequence similarity (“homology”) searching. Curr Protoc Bioinform. https://doi.org/10.1002/0471250953.bi0301s42
Petersen J, Eriksson SK, Harryson P, Pierog S, Colby T, Bartels D, Röhrig H (2012) The lysine-rich motif of intrinsically disordered stress protein CDeT11-24 from Craterostigma plantagineum is responsible for phosphatidic acid binding and protection of enzymes from damaging effects caused by desiccation. J Exp Bot 63:4919–4929. https://doi.org/10.1093/jxb/ers173
Petsalaki EI, Bagos PG, Litou ZI, Hamodrakas SJ (2006) PredSL: a tool for the N-terminal sequence-based prediction of protein subcellular localization. Genomics Proteomics Bioinform 4:48–55. https://doi.org/10.1016/S1672-0229(06)60016-8
Reißer S, Strandberg E, Steinbrecher T, Ulrich AS (2014) 3D hydrophobic moment vectors as a tool to characterize the surface polarity of amphiphilic peptides. Biophys J 106:2385–2394. https://doi.org/10.1016/j.bpj.2014.04.020
Röhrig H, Schmidt J, Colby T, Bräutigam A, Hufnagel P, Bartels D (2006) Desiccation of the resurrection plant Craterostigma plantagineum induces dynamic changes in protein phosphorylation. Plant Cell Environ 29:1606–1617. https://doi.org/10.1111/j.1365-3040.2006.01537.x
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. https://doi.org/10.1093/sysbio/sys029
Saez A, Rodrigues A, Santiago J, Rubio S, Rodriguez PL (2008) HAB1-SWI3B interaction reveals a link between abscisic acid signaling and putative SWI/SNF chromatin-remodeling complexes in Arabidopsis. Plant Cell Online. https://doi.org/10.1105/tpc.107.056705
Schrödinger LLC (2016) The PyMOL molecular graphics system. Schrödinger LLC, New York
Small I, Peeters N, Legeai F, Lurin C (2004) Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4:1581–1590. https://doi.org/10.1002/pmic.200300776
Smith MD, Wertheim JO, Weaver S, Murrell B, Scheffler K, Kosakovsky Pond SL (2015) Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol 32:1342–1353. https://doi.org/10.1093/molbev/msv022
Sperschneider J, Catanzariti AM, Deboer K, Petre B, Gardiner DM, Singh KB, Dodds PN, Taylor JM (2017) LOCALIZER: subcellular localization prediction of both plant and effector proteins in the plant cell. Sci Rep. https://doi.org/10.1038/srep44598
Uno Y, Furihata T, Abe H, Yoshida R, Shinozaki K, Yamaguchi-Shinozaki K (2000) Arabidopsis basic leucine zipper transcription factors involved in an abscisic acid-dependent signal transduction pathway under drought and high-salinity conditions. Proc Natl Acad Sci USA 97:11632–11637. https://doi.org/10.1073/pnas.190309197
van Dam S, Võsa U, van der Graaf A, Franke L, de Magalhães JP (2017) Gene co-expression analysis for functional classification and gene–disease predictions. Brief Bioinform 19(4):575–592. https://doi.org/10.1093/bib/bbw139
Velasco R, Salamini F, Bartels D (1998) Gene structure and expression analysis of the drought- and abscisic acid-responsive CDeT11-24 gene family from the resurrection plant Craterostigma plantagineum Hochst. Planta 204:459–471. https://doi.org/10.1007/s004250050280
Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ (2009) Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. https://doi.org/10.1093/bioinformatics/btp033
Wright ES (2015) DECIPHER: harnessing local sequence context to improve protein multiple sequence alignment. BMC Bioinformatics. https://doi.org/10.1186/s12859-015-0749-z
Wright PE, Dyson HJ (2015) Intrinsically disordered proteins in cellular signaling and regulation. Nat Rev Mol Cell Biol 16:18–29. https://doi.org/10.1038/nrm3920
Yamaguchi-Shinozaki K, Koizumi M, Urao S, Shinozaki K (1992) Molecular cloning and characterization of 9 cDNAs for genes that are responsive to desiccation in Arabidopsis thaliana: sequence analysis of one cDNA clone that encodes a putative transmembrane channel protein. Plant Cell Physiol 33:217–224. https://doi.org/10.1093/oxfordjournals.pcp.a078243
Yu C-S, Chen Y-C, Lu C-H, Hwang J-K (2006) Prediction of protein subcellular localization. Proteins 64:643–651. https://doi.org/10.1002/prot.21018
Zarin T, Tsai CN, Nguyen Ba AN, Moses AM (2017) Selection maintains signaling function of a highly diverged intrinsically disordered region. Proc Natl Acad Sci USA 114:E1450–E1459. https://doi.org/10.1073/pnas.1614787114
Zhang W, Qin C, Zhao J, Wang X (2004) Phospholipase D 1-derived phosphatidic acid interacts with ABI1 phosphatase 2C and regulates abscisic acid signaling. Proc Natl Acad Sci USA 101:9508–9513. https://doi.org/10.1073/pnas.0402112101
Zimmermann P, Bleuler S, Laule O, Martin F, Ivanov NV, Campanoni P, Oishi K, Lugon-Moulin N, Wyss M, Hruz T, Gruissem W (2014) ExpressionData—a public resource of high quality curated datasets representing gene expression across anatomy, development and experimental conditions. BioData Min. https://doi.org/10.1186/1756-0381-7-18
Zouhar J, Sauer M (2014) Helping hands for budding prospects: ENTH/ANTH/VHS accessory proteins in endocytosis, vacuolar transport, and secretion. Plant Cell Online. https://doi.org/10.1105/tpc.114.131680
Acknowledgements
We want to express our gratefulness to Professor Selvadurai Dayanandan and Kyle Grant from Concordia University for their valuable comments and suggestions.
Author information
Authors and Affiliations
Contributions
MZ—Conceptualization, methodology, formal analysis, and writing. MAB—Conceptualization, Validation and review.
Corresponding author
Additional information
Handling editor: David Alvarez-Ponce.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zayed, M., Badawi, M.A. In-Silico Evaluation of a New Gene From Wheat Reveals the Divergent Evolution of the CAP160 Homologous Genes Into Monocots. J Mol Evol 88, 151–163 (2020). https://doi.org/10.1007/s00239-019-09920-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-019-09920-5