
Abstract
Copy number variants (CNVs), deletions and duplications of segments of DNA, account for at least five times more variable base pairs in humans than single-nucleotide variants. Several common CNVs were shown to change coding and regulatory sequences and thus dramatically affect adaptive phenotypes involving immunity, perception, metabolism, skin structure, among others. Some of these CNVs were also associated with susceptibility to cancer, infection, and metabolic disorders. These observations raise the possibility that CNVs are a primary contributor to human phenotypic variation and consequently evolve under selective pressures. Indeed, locus-specific haplotype-level analyses revealed signatures of natural selection on several CNVs. However, more traditional tests of selection which are often applied to single-nucleotide variation often have diminished statistical power when applied to CNVs because they often do not show strong linkage disequilibrium with nearby variants. Recombination-based formation mechanisms of CNVs lead to frequent recurrence and gene conversion events, breaking the linkage disequilibrium involving CNVs. Similar methodological challenges also prevent routine genome-wide association studies to adequately investigate the impact of CNVs on heritable human disease. Thus, we argue that the full relevance of CNVs to human health and evolution is yet to be elucidated. We further argue that a holistic investigation of formation mechanisms within an evolutionary framework would provide a powerful framework to understand the functional and biomedical impact of CNVs. In this paper, we review several cases where studies reveal diverse evolutionary histories and unexpected functional consequences of CNVs. We hope that this review will encourage further work on CNVs by both evolutionary and medical geneticists.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Copy Number Variants Explain the Majority of Human Genetic Variation
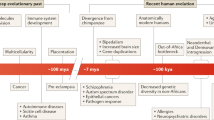
The majority of human genetic variation is imperfectly studied (Eichler 2019). One of the least understood types of genetic variation is copy number variants (CNVs), which refer to deletions and duplications of relatively large genomic segments (Fig. 1, often larger than 1 kb in relevant studies). Previous approaches to detect common genetic variation have focused on single-nucleotide polymorphisms. It is only recently with the advent of array comparative genomic hybridization and whole-genome sequencing that we can visualize and appreciate the extent of CNVs in the human genome. CNVs account for at least five times more variable base pairs compared to single-nucleotide variants when two human genomes are compared to each other (Conrad et al. 2010; Redon et al. 2006; Sudmant et al. 2015a; Pang et al. 2010). In addition to the extent of their impact on the landscape of genetic variation, follow-up studies have linked copy number variation to several important complex diseases (Girirajan et al. 2011), as well as adaptively relevant phenotypes (Redon et al. 2006; Iskow et al. 2012). These initial glimpses hail a promising new avenue to collectively understand the genetic basis of human health and evolution; recent studies have linked putatively adaptive CNVs to biomedically relevant traits (Iskow et al. 2012; Stankiewicz and Lupski 2010; Girirajan et al. 2011). This observation is concordant with the notion called antagonistic pleiotropy, which was first proposed by Williams (1957) that one gene can control multiple traits with both beneficial and detrimental effects. Recent genome-wide studies have found suggestive evidence that genetic variants which affect disease susceptibility are also associated with adaptation to environmental conditions and longevity (Voskarides 2018; Byars and Voskarides 2019).

Characteristics of gene copy number variants compared to single-nucleotide variants
One challenge in studying copy number variation is that CNVs evolve through different formation mechanisms and thus vary significantly in their size, genomic location, and potential functional impact. In other words, CNVs as a category should be considered a collection of multiple different types of variants with different properties. For example, a large deletion that evolved through nonallelic homologous recombination has substantially different functional consequences than a smaller gene duplication that occurred through retrotranscription (i.e., a retrogene) (Abyzov et al. 2013; Hastings et al. 2009). In addition, recurrence, gene conversion, and methodological identification biases (false positives/negatives) affect different types of CNVs in varied ways and add to the challenges of studying the evolutionary and biomedical impacts of CNVs (Fig. 2). In the first part of this review, we will provide examples of these challenges within the context of anthropologically relevant CNVs. Then, to highlight the contributions of CNVs to human phenotypic variation, we will discuss examples of evolutionarily relevant CNVs and their potential health consequences. Last, we will briefly touch on the broad functional categories that CNVs have a particular impact. Overall, we hope that this review will provide a general picture of the current state of the field.

Mechanistic characteristics of copy number variants. a Copy number variations are often formed in repeat-rich regions, which makes the haplotype-based analysis challenging. b Gene conversion. c Recurrent formation of copy number variations can break the flanking haplotypes and consequently the linkage disequilibrium, which reduces the statistical power of GWAS and neutrality tests based on haplotypes
The Different Mechanisms of Copy Number Variant Formation Lead to Diversity in Their Evolutionary and Biomedical Impact
Segmental Duplication-Associated Nonallelic Homologous Recombination
Copy number variations can be formed by multiple mechanisms. One well-described mechanism through which large CNVs form is nonallelic homologous recombination. When two relatively large (generally > 1 kb) repeats exist nearby on a chromosome, they increase the chances of chromosomal misalignment during meiosis, leading to unequal crossing over (Hurles 2004) (Fig. 1a). The result is that one of the chromosomes may end up losing while the other gaining a DNA segment. Empirical data show that copy number variants are significantly enriched in segmental duplication-rich regions of the genome (Redon et al. 2006). Segmental duplications are large segments of DNA sequence (1–400 kb) with high (more than 90%) sequence similarity that occur more than one site within the human genome. Segmental duplications account for 5% of the human genome (Sharp et al. 2005; Bailey et al. 2001, 2002) and are particularly prone to the nonallelic homologous recombination, especially because they are larger than short tandem repeats and most retrotransposons. It is estimated that about 28% of copy number variants likely evolved through segmental duplication-mediated nonallelic homologous recombination (Kim et al. 2008).
These nonallelic homologous recombination-based CNVs have certain commonalities with each other. They cluster in segmental duplication-rich portions of the genome, which happen to be gene-rich (Bailey et al. 2002) and can harbor well-described gene families (Sudmant et al. 2010). Thus, the majority of CNVs that lead to polymorphic gene deletions or duplications form in such areas (Bailey et al. 2002). This genic nature of segmental duplications is relevant to human evolution. Earlier studies reported a “burst” of segmental duplications in the great ape lineage (Marques-Bonet et al. 2009), and moreover, these great ape-specific segmental duplications harbor genes related to neuronal development, potentially explaining the unusual large brains of great apes and humans in particular (Levchenko et al. 2018). From a biomedical perspective, the same mutational mechanisms that lead to gene duplications related to neurological functions may have led a predisposition to higher CNV mutation rate in these regions. Thus, it is not surprising that nonallelic homologous recombination underlies a considerable number of rare de novo CNVs linked with neurological disorders (Mefford et al. 2010). A last but important point is the formation of new CNVs can further increase the segmental duplication content in a genome, subsequently leading to even more CNVs. In fact, some segmental duplication-rich regions of the primate genomes are hot spots of common CNVs. Such hot spots were implicated in the maintenance of functional variation in some immunity-related gene families (Gokcumen et al. 2011; Lin and Gokcumen 2019). In sum, CNVs that evolve through nonallelic homologous recombination include variants that are more likely to be larger, genic, and recurrent, and their genomic context often includes other repetitive sequences. They have been the major focus of research within the field and remain as the best-understood type of CNVs.
Retrotransposons and Retrotransposon-Driven CNVs
Retrotransposition is arguably the most important factor shaping eukaryotic genomes (Kazazian 2004; Deininger et al. 2003; Cordaux and Batzer 2009). In humans, retrotransposon activity can affect CNV formation at three levels. First, some retrotransposons in the human genome are still active and thus remain polymorphic (Cordaux and Batzer 2009). These can be considered as CNVs themselves. These retroposons often carry their own regulatory machinery and as such are discussed as major players in the regulation of expression (Cordaux and Batzer 2009; Feschotte 2008). MicroRNAs (miRNAs), a distinct class of ~ 22 nt single-stranded noncoding endogenous RNAs, are also related to retrotransposons. For example, the large, primate specific miRNA family of mir-548 were derived from Made1 miniature inverted-repeat transposable elements (Piriyapongsa and Jordan 2007). The mir-548 family members have high sequence similarity with each other and are widely spread across the human genome as slightly different copies from the same template sequence (Liang et al. 2012).
Second, retrotransposition mechanism can occasionally lead to the integration of processed mRNAs back into the genome through retrotranscriptase activity (Trizzino et al. 2017). These “retrogenes” can be polymorphic and often express an RNA molecule (Abyzov et al. 2013; Chuong et al. 2017). The function of these expressed RNA molecules is unknown, but anecdotal findings indicated that these retrogenes can replace the original genes (Ciomborowska et al. 2013) and once retrogenes acquire introns, they can lead to increase in gene dosage (Fablet et al. 2009). It is also possible that retrotransposons can facilitate the formation of large CNVs either by facilitating nonallelic homologous recombination or by generating structural plasticity in the genomic regions where they are inserted (Hastings et al. 2009). However, this last mechanism has not been definitively shown at a genome-wide level in humans and only reported in specific cases with major biomedical consequences (Gu et al. 2015). If we consider polymorphic Alu and LINE elements as CNVs, then the majority of CNVs by sheer number can be categorized as such, but their size is generally smaller than segmental duplication-associated CNVs. The polymorphic retrogenes and retrotransposon events are much rarer than the fixed ones; however, anecdotal findings suggest that CNVs that have evolved through retrotransposition machinery can contribute significantly to human biological variation.
Tandem Repeats Smaller than Segmental Duplications
CNVs are often observed with other types of repeats besides segmental duplications (Repnikova et al. 2013). For example, smaller tandem repeats also contribute significantly to the CNV landscape in various ways. However, the relationship between CNVs and such tandem repeats is multifaceted as in the relationship between retrotransposition and CNVs. It is a matter of definition if one considers the variation in the number of short tandem repeats can be considered CNVs (e.g., triplets in Huntington region which can be repeated dozens (healthy) to hundreds (pathogenic) times) (Möncke-Buchner et al. 2002). In addition to the copy number variation of these shorter tandem repeats themselves, these arrays of repeated sequence can lead to increase in genomic instability, which in turn can facilitate the formation of larger CNVs (Nguyen et al. 2006).
A more clear-cut example of how such repeats can contribute to copy number variation landscape of the genome involves sub-exonic repeats. These repeats can involve multiple copies of dozens of nucleotides to hundreds of nucleotides, contributing to the coding sequence. The formation of sub-exonic repeats polymorphism is often thought to occur through strand-slippage replication (Fan and Chu 2007). Sub-exonic repeats in mucin genes provide a case example, where they are often polymorphic and code for highly glycosylated PTS-rich peptides (Dekker et al. 2002). As such, they contribute to the variation in the glycosylation of these proteins. Kirby et al. identified an insertion of a C in the coding repeat region in the MUC1 gene leads the frameshift which introduces a stop codon shortly beyond the variable number tandem repeat domain. This variation is thought to be responsible for medullary cystic kidney disease type 1 (Kirby et al. 2013). In addition to mucins, thousands of other mammalian genes harbor such subexonic repeats (Schaper et al. 2014). Thus, the biological relevance of the polymorphisms in the copy number of these repeats remains a fascinating area of future study.
Other Mechanisms
Non-homologous end-joining pathway is the key mechanism to repair DNA double-strand breakage in mammalian cells (Chang et al. 2017). When double strands are broken, the related proteins are recruited and ligate the DNA strands together. If two fragments from different chromosomes ligate together, it can result in gene deletions and duplications (Currall et al. 2013; Bickhart and Liu 2014; Weckselblatt and Rudd 2015). Korbel et al. (2007) suggest that 56% of the copy number variations are caused by nonhomologous end joining (NHEJ). However, the specifics of this broad mechanistic category remain relatively understudied among healthy individuals. Generally, CNVs are considered to evolve through NHEJ if the formation mechanisms cannot be categorized as nonallelic homologous recombination or retrotransposition. This broad mechanistic category of CNVs, which vary widely for their size, functional impact, chromosomal location, and sequence content, remains the least understood. With the advent of longer reads and a better understanding of these mechanisms, recent studies begin to tackle these mechanisms more systematically (Zhao et al. 2016; Chaisson et al. 2019).
A Note on the Evolution of CNVs Through the Different Formation Mechanisms in the Human Lineage
The genome evolution is led by different mechanisms between lineages. For example, there are many more gene loss and gene gain events in the primate lineage as compared to other mammalian lineages (Hahn et al. 2007). Moreover, a “burst” of segmental duplications documented in the great ape lineage distinguishes their genomes from other primates (Marques-Bonet et al. 2009). These major shifts in genome evolution trends are likely due to the change in the rate of nonallelic homologous recombination-based events (Gokcumen et al. 2013). Indeed, there is a significantly higher proportion of large CNVs, which likely evolved through recombination-based mechanisms, within great ape species as compared to those found within rhesus macaques (Gokcumen et al. 2013). Moreover, the recombination trends (e.g., the localization of recombination hot spots) evolve rapidly between primate species, further contributing to the notion that shifts in recombination-based formation mechanisms may explain the differences noted in the CNV landscape within different primate species (Stevison et al. 2016).
Another major contributor to the differences in CNV landscapes across primate species is the rate of retrotransposition-based mechanisms (Hedges and Batzer 2005). Comparative genomics revealed that Alu-mediated formation of copy number variation was the predominant mechanism 40 million years ago. Indeed, polymorphic Alu elements are the predominant type of CNVs within rhesus macaque individuals, but their rate of new Alu insertions is significantly lower within great ape species (Gokcumen et al. 2013). To study the underlying factors that determine the extent of polymorphic and active retrotransposition in a given species is not a trivial task. The rate and maintenance of retrotransposition depend on multiple factors: the demographic history of the species (e.g., effective population size (Gurdasani et al. 2019)), the selection acting on the genome as a whole (Enard et al. 2010), the multiple defense mechanisms that work to dampen retrotransposition activity [e.g., APOBEC3 gene activity, piwi RNAs, etc. (Stenglein and Harris 2006; Yang and Kazazian 2006)]. Thus, the mechanistic bases and the potentially widespread (Chuong et al. 2017) functional impact of polymorphic retrotransposons in different species remain mostly unknown. However, it is clear that the primate genomes evolve differently than those of other mammals and from each other when it comes to CNVs (Derti et al. 2006).
Examples
Several CNVs have been thoroughly studied within the context of human evolution and disease. Deletions or duplications of exonic sequences have been a particular interest. However, because there are multiple mechanisms to form CNVs as we discussed above, studying the evolutionary and functional impact of these exonic CNVs can be challenging. Specifically, recurrence and gene conversion lead to sometimes unexpected methodological complications. Here, we provide a relatively comprehensive list of common exonic CNVs (Table S1). Further, we summarize recent work on some of these variants to highlight the diverse evolutionary histories of CNVs and also to show how researchers tackle unique methodological challenges in each case.
Lack of Linkage Disequilibrium with Nearby Variants: APOBEC3B and UGT2B17 Deletions
Linkage disequilibrium (LD) is one of the most relevant properties of genetic variation for evolutionary and phenotypic analyses (Slatkin 2008). It refers to the non-random co-occurrence of two or more variants in a given population. This is primarily because physically close alleles are inherited together and only rarely separated by the action of recombination. Most of the genome-wide association studies, which are critical for investigating the genetic basis of human traits, including diseases, depend on the LD architecture of the genome in their experimental design (Visscher et al. 2017). Similarly, the majority of the modern population genetics analysis uses different ramifications of LD across the genome to reconstruct evolutionary history and identify potentially adaptive sections of the genome (Slatkin 2008). However, only 73% of CNVs with > 1% frequency are in LD with nearby single-nucleotide polymorphisms even when relatively modest LD threshold is used (r2 > 0.6) (Sudmant et al. 2015b). Thus, the contribution of the majority of the CNVs to the genetic basis of human traits remains unexplored in genome-wide association studies (Wellcome Trust Case Control Consortium et al. 2010). Two of the best examples for this phenomenon are the deletion polymorphisms of APOBEC3B and UGT2B17 genes, both of which have been shown to have important evolutionary and biomedical consequences, but their impact was missed in genome-wide single-nucleotide variant-based interrogations.
The APOBEC3B deletion: The APOBEC3 (Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) gene family plays a key role in innate cellular immunity against retroviral infection (Cullen 2006). Moreover, it was argued to be a major suppressor of active retrotransposition (Kinomoto et al. 2007). One of the best-studied members of this family is APOBEC3B, being linked to HIV resistance, regulation of retrotransposons in soma, and other innate immunity functions (Cullen 2006). Despite its important functions, APOBEC3B is deleted in millions of human genomes, with allele frequencies of 0.9% in African, 6% in European, 36.9% in East Asian, and 57.7% in American populations (Kidd et al. 2007; Redon et al. 2006; Sharp et al. 2005). This common deletion of APOBEC3B gene spans 29.5 kb from the fifth exon of APOBEC3A to the eighth exon of APOBEC3B (Kidd et al. 2007; Redon et al. 2006; Sharp et al. 2005). This deletion essentially eliminates APOBEC3B, while replacing the 3′ UTR of APOBEC3A with the 5′ UTR of APOBEC3B. It was shown that APOBEC3B deletion is associated with susceptibility to HBV infection (An et al. 2009; Zhang et al. 2013) (but see Itaya et al. 2010; Imahashi et al. 2014), malaria (Jha et al. 2012), and hepatocellular carcinoma (Zhang et al. 2013). The exact mechanisms underlying these associations are not known.
The high allele frequency with population structure and important immune functions presents APOBEC3 deletion as a potential target for natural selection to act on. Indeed, haplotype-level analyses of the single-nucleotide variants flanking the deletion polymorphism suggested weak signals of selection, particularly in the Yoruba population (Kidd et al. 2007). However, this observation is not consistent with allele frequency distribution and the expectation would be that the deletion is selected in Eurasian populations where the allele frequency is much higher. One plausible reason as to why the haplotype-based analyses failed to detect any selection in Eurasian populations is that the single-nucleotide variants around the deletion did not have strong LD (r2 > 0.8) with the deletion variant. In other words, the lack of signal of selection on this exonic deletion was not necessarily because there was no selection, but the tests of selection had considerably diminished power because of the lack of LD. The lack of LD between the deletion and nearby variants cannot be explained by recurrence given the exact breakpoints of this deletion shared by many individuals point that the deletion variants in humans are identical by descent (Kidd et al. 2007). Instead, a more likely scenario to explain the lack of LD in this locus is gene conversion events (Fig. 2b), which are common in gene families with similar sequence content where homologous sequences can be swapped between chromosomes without changing the copy number. To this day, the adaptive role of APOBEC3B deletion, if any, and the details of its evolutionary history remain mostly unknown.
UGT2B17 Deletion
UDP-glucuronosyltransferase (UGT) is a group of enzymes that contribute significantly to the metabolism of several xenobiotic molecules and are as such studied within the context of pharmacogenomics (Oda et al. 2015; Burchell et al. 1997). The entire UGT2B17 gene, which codes one of these UGT enzymes, is commonly deleted in human populations. This deletion has been associated with prostate cancer and endometrial cancer (Karypidis et al. 2008; Hirata et al. 2010). Thus, similar to the APOBEC3B deletion described above, an immediate question is why this common deletion polymorphism was not eliminated from the population by the action of negative selection. In other words, would there be a fitness benefit to this deletion to maintain its presence in the human population? Suggesting non-neutral evolution, UGT2B17 deletion shows unusual population differentiation: Non-deleted allele is extremely common in Africa, Europe, and the Middle East (~75% allele frequency), but relatively rare in Asia and Oceania (~20% allele frequency) (Xue et al. 2008). Moreover, this gene showed an expression difference between different human populations (Spielman et al. 2007).
Similar to APOBEC3B deletion, the LD architecture of the UGT2B17 is complicated. Specifically, the UGT2B17 gene is flanked by two segmental duplications and it is assumed that nonallelic homologous recombination formed this gene deletion (Xue et al. 2008). Moreover, none of the downstream single-nucleotide variants are found to be in LD with the deletion polymorphism, which indicates past gene conversion events similar to the observations in APOBEC3 locus (Xue et al. 2008). However, Xue et al. (2008) found strong LD between the deletion and some upstream single-nucleotide variants only after the close, locus-specific inspection. By studying the upstream haplotypic variation linked to the deletion variant, Xue et al. (2008) documented signatures of balancing and positive selection in Europe and East Asia, respectively. Again, the exact evolutionary history and the underlying adaptive reasons for these signatures of non-neutral evolution observed for UGT2B17 deletion remain unknown. However, we argue that UGT2B17 deletion provides a strong case for locus-specific analysis of CNVs where it is possible to resolve the haplotype architecture of the locus and, by doing so, to better understand the evolutionary history of the variant. Intriguingly, later studies have found that UGT2B17 is also polymorphically deleted in chimpanzees—raising the possibility that complex adaptive pressures have been shaping this locus in different species (Saitou et al. 2018a). This brings us to the next challenge in studying CNVs recurrence.
Recurrent Formations of Structural Variations: HP and GSTM1
Unlike single-nucleotide variants, CNVs tend to recur in the same loci because of mechanistic predispositions. This is primarily due to the fact that certain repeat content, such as segmental duplications, predispose certain loci for rapid CNV formation (van Ommen 2005). Such loci were implicated in the formation of rare, congenital CNVs with dire medical consequences. However, common CNVs can also show recurrence either within or between species, both breaking the expected LD patterns in the locus and also posing interesting questions of evolutionary impact of recurrence.
Glutathione Transferases
GSTM1 belongs to a superfamily of metabolic enzymes, called glutathione transferases (Hayes et al. 2005). This particular gene metabolizes cancer chemotherapeutic agents, carcinogens, and by-products of oxidative stress (Hayes et al. 2005). It has been known that the GSTM1 deletion allele is commonly observed in human populations (~50%) (Xu et al. 1998) and the deletion allele is associated with bladder cancer (Rothman et al. 2010). As one of the most common polymorphic deletions in the human lineage and also because of its reported functional relevance, this gene deletion is also a potential target for natural selection. However, careful dissection of the haplotypic variation in this locus revealed an extremely complicated evolutionary picture, riddled with gene conversions and recurrent mutations (Saitou et al. 2018b; Khrunin et al. 2016). As a result, no strong LD could be observed between the deletion and the nearby single-nucleotide variants and traditional population genetics approaches based on haplotypic variation are underpowered to detect any selective pressure in this locus with any level of definitiveness.
To complicate the picture further, Saitou et al. (2018a) found that chimpanzees have the same gene deleted polymorphically with very similar breakpoints. Genome-wide analysis revealed that GSTM1 is not alone, and another metabolism gene UGT2B17, which we described above, is also polymorphically deleted in chimpanzees. This is a rather remarkable observation. To put it in context, in a breakthrough paper, Leffler et al. showed that a number of shared single-nucleotide polymorphisms between chimpanzee and human populations in the HLA locus are identical by descent, and thus they invoked inter-species balancing selection to explain this observation. This is one of the very few best-established cases for such selection in the human–chimpanzee lineage (Leffler et al. 2013). In a similar manner, the crucial question is whether UGT2B17 and GSTM1 deletions are polymorphic in these two species because they are identical by descent—a very strong case for inter-specific balancing selection, or because they were recurrently evolved in these two species. After several experimental and bioinformatic analyses, Saitou et al. (2018b) provided evidence that the GSTM1 deletion evolved recurrently in chimpanzee and human lineages. However, the evolutionary history of UGT2B17 with regard to allele sharing between humans and chimpanzees remains unexplored.
Globins
Free hemoglobins in human plasma can be toxic. Haptoglobins bind to these free hemoglobins and perturb their toxicity (Schaer et al. 2014). It has been reported that deletion polymorphisms overlap with parts of the haptoglobin gene and alter the structure of the coded proteins (Langlois and Delanghe 1996; Smithies and Walker 1955; Wejman et al. 1984). Moreover, locus-specific analysis of these deletions linked them with diabetes (MacKellar and Vigerust 2016). However, haptoglobin locus is repeat-rich and similar to the APOBEC3, GSTM1, and UGT2B17 loci described above in that it presents complex LD patterns. Not only that the deletion variants in this locus do not show strong LD with the nearby single-nucleotide variants, but it was not clear until recently whether these variants are recurrent or the lack of LD is due to gene conversion events.
Unlike SNVs, CNVs are often formed recurrently due to genomic architecture. Recurrently formed CNVs can be verified when different breakpoints overlapping CNVs are identified (Fig. 2). However, to identify the breakpoint is not straightforward in most cases. For example, in the GSTM1 case above, the GSTM1 deletion is formed by the recombination of two highly similar flanking sequences (Xu et al. 1998); thus, the two highly similar sequences in the original allele and the fused sequence of the two sequences in the deletion allele are similar to each other. As such, it was not possible to identify where the exact breakpoints were located at the nucleotide level (Saitou et al. 2018a). Haptoglobin locus presents a similar repeat richness. As such, Boettger et al. (2016) devised a more indirect strategy. To resolve the complex haplotype architecture in this locus in humans, they analyzed the 20 kb flanking sequences of this locus from 264 human samples. They deduced that gene conversion or similar recombination-based mechanism would break the LD, not only between the deletion and the nearby single-nucleotide variants but also between the single-nucleotide variants on both sides of the deletion. In contrast, in the case of recurrence, the LD patterns between the single-nucleotide variants on each side of the deletion remain intact. Indeed, they observed highly preserved linkage disequilibrium between single-nucleotide variants across the deletion, even though the deletion(s) itself is not in strong LD with these variants. They concluded the recurrence is the major force in breaking the LD in this locus. Not only that this strategy helped resolve an evolutionary puzzle, but it allowed the authors to identify the haplotypes that harbor specific recurrent deletion variants. They then conducted a meta-analysis of genome-wide association studies with the newly resolved haplotypes and found that haplotypes harboring the deletions are associated with lower blood cholesterol levels as compared to controls (Boettger et al. 2016). This analysis is representative of both the difficulties in studying CNVs but also highlights that many CNVs may be underappreciated when it comes to their evolutionary and biomedical impact.
Multiple Structural Variants at the Same Locus: DMBT1
There are hot spots and deserts of CNVs across the genome shaped by mechanistic forces along with adaptive constraints that shape their distribution (Perry et al. 2006; Fu et al. 2010; Lin and Gokcumen 2019) Interestingly, some genic hot spots of CNV formation were linked to human phenotypes. For example, immune-related loci such as HLA, globin, defensin gene families are CNV hot spots and balancing and diversifying selection were implied in the maintenance of the unusually high number of common CNVs in these loci (Lin and Gokcumen 2019). The functional and evolutionary impact of such recurrent CNV loci (Fig. 2c) can be difficult to resolve because the recurrence, combined with overall excess single-nucleotide variation in these loci, can hinder both direct genotyping and indirect resolution of haplotypes harboring the individual CNVs. However, as in the case of Boettger et al (2016), recent studies have scrutinized some of these loci and identify complex and fascinating evolutionary stories.
DMBT1
The variation affecting the DMBT1 gene exemplifies the complexity of studying loci where multiple multiallelic CNVs frequently and recurrently form. DMBT1 codes for a salivary agglutinin gene, which belongs to a superfamily of glycoproteins. This family is defined by ~ 100 amino acid long scavenger receptor cysteine-rich (SRCR) domains (Mollenhauer et al. 1997). These SRCR domains are repetitive, and dozens of CNVs were reported affecting these domains in the DMBT1. These CNVs were scrutinized further using a combination of bioinformatics, quantitative PCR, long-range traditional PCR, and fiber-FISH (Polley et al. 2015). This study was able to characterize two of the most common CNVs, one of which involves diploid copy number variation ranging from 0 to 5 of a segment encompassing four SRCR repeats spanning SRCR3 to SRCR6. The second involves diploid copy number variation ranging from 0 to 11 copies of a segment encompassing three SRCR repeats spanning SRCR9 to SRCR11 (Polley et al. 2015). This study further showed that the primary source of the frequent structural variation within this locus is unusually high mutation rate (~5% of the gametes were estimated to have de novo copy number gain or loss involving the DMBT1 SRCR domains).
DMBT1 binds bacteria and viruses and acts as a pattern recognition receptor in innate immunity, and these functions are thought to be facilitated by the SRCR domain (Ligtenberg et al. 2010; Mollenhauer et al. 2000). One question is then whether the CNVs that change the number of the SRCR domains are adaptively maintained in human populations. One clue comes from the same study where Polley et al. (2015) were able to show that human populations that are historically agricultural have more copies of SRCR domains as compared to those that are not. Another clue comes from the observation that particular SRCR domains bind to oral bacterium Streptococcus mutans and hydroxyapatite (Ambatipudi et al. 2010). Thus, it is plausible that the higher copy number of SRCR domains in the DMBT1 gene may confer some advantage in agricultural diets protecting against particular oral pathogens. It is also possible that the actual number of these domains are not that crucial for fitness and the observed variation is just the effect of genetic drift. These questions are hard to answer, especially given the pleiotropic functions of DMBT1 (e.g., it is also involved in epithelial differentiation (Mollenhauer et al. 2000). Regardless, CNVs of the SRCR domains in DMBT1 likely contribute to human phenotypic variation and exemplify the diverse ways in which multi-allelic CNVs evolve, highlighting the multiple challenges in fully resolving the evolutionary history of such loci.
Discovery Bias: AMY and MUC7
The capabilities of different CNV detection methods vary depending on the type of CNV (Pirooznia et al. 2015). There is a fine methodological balance between sensitivity (i.e., low false-negative rates) and accuracy (low false-positive rates). Thus, it is not uncommon for even well-designed comprehensive studies to miss otherwise well-described and common copy number variations. Here, we give two examples of evolutionarily important CNVs affecting amylase (AMY) and mucin 7 (MUC7) genes.
Mucins
Mucins are a functional category of proteins that are abundantly observed in epithelial tissues and confer the mucusy properties of several bodily fluids, interact with microbes, and are even involved in signaling (Fábián et al. 2012). Mucins are defined by their O-glycosylation potential determined by tandem-repeated proline-, threonine-, and serine-rich domains (PTS domains). These domains can often be polymorphic (Table S1), and this variation is associated with differences in susceptibility to certain pathogens and other diseases (Kumar et al. 2017; Behera et al. 2015). One of the best-studied polymorphic mucin repeats is the salivary MUC7 with regard to its evolutionary history (Kirkbride et al. 2001). It has been shown that MUC7 has evolved in the ancestor of placental mammals and since then retains its 69-base-pair-long PTS domain repeats in different numbers across lineages (Xu et al. 2016). Indeed, the amino acid content of these PTS domains has evolved under negative selection, while their copy number showed signatures of lineage-specific adaptive pressures. One explanation is that the number of these repeats fine-tunes the glycosylation potential of the protein, perhaps to adjust to specific pathogenic pressures.
In humans, MUC7 PTS domains are repeated five and six (Kirkbride et al. 2001). It was shown that the minor five-repeat allele has evolved at least twice in the human lineage (Xu et al. 2017). Moreover, the same study showed that one of the haplotypes carrying the five-repeat allele splits more than 2 million years before presenting from the other MUC7 haplotypes. Based on the empirical and simulation-based investigation, the authors argued that this haplotype was introgressed into ancestors of modern humans ago from an archaic “ghost” hominin population in sub-Saharan Africa. In sum, MUC7, in particular, and mucins, in general, provide a fascinating glimpse into a rapid evolution of subexonic repeats and their functional consequences.
Despite the potential relevance to biological processes, the PTS repeat copy number variation of mucins is notoriously difficult to discover and genotype within conventional methods, such as array comparative genomic hybridization or short-read sequencing-based approaches. Specifically, it is difficult to design unique probes to target individual repeat sequences for array-based approaches and the mapping of the short sequence reads to the tandem repeats is often not specific enough to discover variations with enough statistical power. As such, it is not surprising that 1000 Genomes did not report the common variation involving MUC7 PTS-repeat copy number, even though this project documented common CNVs involving three other mucin genes (Table S1).
As well as MUC7, as we noted above, MUC1 has a frameshift variation in its exonic repeat structure due to one-nucleotide insertion (Kirby et al. 2013). The MUC1 variation they found by molecular cloning and capillary sequencing is likely to be causal to kidney disease, and the variation has been difficult to resolve by massively parallel sequencing (next-generation sequencing) (Kirby et al. 2013). To resolve the repeat architecture of this loci, several approaches have been presented, such as Pac-bio sequencing and illumina sequencing paired with dedicated bioinformatic analyses (Živná et al. 2018; Wenzel et al. 2018). To highlight the underappreciated importance of mucins at a more genome-wide manner, it is also important to touch on the results from a recent study of Pan-African genomes (Sherman et al. 2018). This study identified close to 50 genic insertions (i.e., genic sequences that are not present in the current version of the human reference genome). These sequences are likely variably present in extant human genomes, and three of these involve mucin genes with unknown functional implications. Thus, mucin genes and their PTS domains exemplify yet another subtype of copy number variations that are likely evolutionarily and biomedically relevant but underappreciated due to technical challenges. In addition to mucins, thousands of other mammalian genes harbor such subexonic repeats (Schaper et al. 2014). Thus, the biological relevance of the polymorphisms in the copy number of these repeats remains a fascinating area of future study.
Amylase
Dissecting the evolutionary history of the amylase copy number variation has been a major focus area for biology and anthropology. Amylase digests dietary starch and glycogen. Amylase is present in multiple copies in humans, and some of these copies are expressed in the pancreas and others are expressed in salivary glands (Duane et al. 1972; Robyt and French 1967; Hagenbüchle et al. 1980; Merritt et al. 1973). The copy number of the amylase gene varies extensively between different human populations (Perry et al. 2007; Yang et al. 2015; Bank et al. 1992; Santos et al. 2012; Mandel et al. 2010) and between different mammalian species (Boehlke et al. 2015; Paudel et al. 2013; Pezer et al. 2015; Pajic et al. 2019). This copy number variation contributes to the dosage of its expression (Atkinson et al. 2018) in both pancreas and in salivary gland (Pajic et al. 2019).
It has been shown that duplication from the ancestral pancreatic amylase (β-amylase) in the ancestor of great apes leads to an almost identical copy of this gene that is specifically expressed in the salivary glands, independent from mouse (Meisler and Ting 1993). Furthermore, this salivary amylase (α-amylase) further increased in copy number in the human lineage, increasing the dosage of salivary expression of this enzyme (Perry et al. 2007). This recent increase was considered a likely adaptation to higher-starch consumption among modern human ancestors. In fact, even very recent change in the human diet with the advent of agriculture was linked to a further increase in copy number of α-amylase. Given that the majority of starch digestion happens in the gastrointestinal tract, many studies investigate the functional impact of α-amylase on human phenotypes, linking the copy number of this gene to starch perception, microbiome composition from the oral cavity all the way to anus, immunity, and metabolic disorders (Santos et al. 2012; Poole et al. 2019; Pruimboom et al. 2014; Falchi et al. 2014). There are also reports that AMY copy number variations are associated with body mass index (Viljakainen et al. 2015) and obesity (Marcovecchio et al. 2016; Falchi et al. 2014). In sum, AMY locus is one of the most fascinating loci in the human genome when it comes to recent human evolution and its consequences for human health (Varki et al. 2008). However, the variation in this locus is invisible to most genome-wide association studies and even to comprehensive catalogs of human genetic variation such as 1000 Genomes variation database.
The amylase locus is shaped by segmental duplications and near-identical lineage-specific retrotransposons. Thus, the sequence homology complicates mapping of short sequencing reads to this locus to the extent that even single-nucleotide variant discovery in this locus proves difficult. On top of this complication, copy number variation in this locus is extensive. It has been reported that β-amylase can range in copy number from 2 to 8 and the α-amylase can range in copy number from 2 to 17 (Usher et al. 2015). Further complicating the investigation of this locus is the likely recurrence of gene copy number mutations and gene conversion events, both of which disrupt the expected LD patterns (Popadić and Anderson 1995; Gumucio et al. 1988; Pajic et al. 2019). Indeed, careful dissection of the haplotype architecture of this locus using digital PCR estimation of the copy number and imputation of the CNVs with the nearby smaller variants reveals a complex structure in this locus (Usher et al. 2015). In sum, although amylase locus is a poster child that highlights the biomedical and evolutionary relevance of CNVs, it also presents some of the most difficult methodological challenges involving CNVs. Now, with the advent of long-read sequence technologies, it is likely that a better and more direct picture of the variation will emerge. This will allow investigating the extent and timing of the selection on the copy number variation in this locus as well as statistically robust associations with different traits.
A Note on Function, Evolutionary Relevance, and Disease Impact
There are several reviews on the functional and biomedical impact of CNVs (Girirajan et al. 2011; Schrider and Hahn 2010; Stankiewicz and Lupski 2010; Iskow et al. 2012). They often focus on large, rare, and common gene deletions and duplications. Common CNVs among healthy individuals are ontologically enriched for perception, skin barrier function, immunity, and metabolism (Table S1). In this section, we briefly discuss the roles of CNVs in shaping variation in these functional categories.
Perception
Chemosensory receptor genes and their pseudogenes have an enormous variation in their copy numbers among animal species. This variation is explained by a combination of evolutionary arguments, including genetic drift, adaptation to different environments, and variation in diets (Nei et al. 2008; Hayden et al. 2014). For example, primates have a significantly higher copy number variation in affecting their olfactory receptor genes and harbor significantly more pseudogenes as compared to other mammals (Niimura et al. 2018; Liman 2006; Go and Niimura 2008; Kawamura and Melin 2017). It is also reported that primates have undergone acceleration of gene loss of olfactory receptors during their evolution (Niimura et al. 2018; Liman 2006; Go and Niimura 2008; Kawamura and Melin 2017) including specific gene losses in the human lineage (Go and Niimura 2008). This general reduction and variability in functional olfactory repertoire due to copy number variation within and between species may indicate a general lack of selection acting on these genes. In fact, one evolutionary explanation was that the acquisition of full trichromatic vision in primates led to a reduction in the strength of selection on olfactory receptor genes (Gilad et al. 2004). Other researchers emphasized shifts in diets in primates as a more important driver of the olfactory receptor variation (Gilad et al. 2004; Matsui et al. 2010).
Given the copy number variation in olfactory receptor genes within and across primate species, it is not surprising that there is also an enormous variation in copy number of olfactory receptors between human individuals (Young et al. 2008). Even though this variation likely has very little fitness effect in present-day human populations (but see Hoover et al. 2015), CNVs overlapping olfactory receptor genes undoubtedly contribute to phenotypic variation. It is known that some of the CNVs that affect olfactory receptors are associated with odor perception sensitivity (Keller et al. 2007; Turner 2014; Reed and Knaapila 2010). For example, there are 16 common CNVs in the 1KGP phase 3 dataset that overlap with olfactory receptors (Table S1). Moreover, the recently published pan-genome from 910 African individuals mentioned above also highlights three new, non-reference insertions affecting olfactory receptor genes (Sherman et al. 2018). Putting these together and given that most of the ligands of olfactory receptors are yet to be determined, it is perceivable a considerable portion of inherited variation in smell perception is due to CNVs.
Immune System
Dozens of immune genes including major histocompatibility complex genes, defensins, KIRs, and immunoglobulins are copy number variable in humans (Table S1). These variations have been discussed within the context of response to pathogens and autoimmune disorders (Hollox and Armour 2008; Jiang et al. 2012; Bournazos et al. 2009; Wellcome Trust Case Control Consortium et al. 2010). One well-resolved evolutionary example of copy number variation affecting the immune system in an adaptive manner is the case of the chemokine receptor CCR-5. This gene acts as a co-receptor for HIV-1 (Samson et al. 1996a). A 32-base-pair deletion within the coding region (CCR5Δ32), which is commonly observed in Northern European populations, generates a non-functional receptor and thus prevents infection by some HIV-1 strains (Samson et al. 1996b). Since the geographic distribution of this variant is unusual and it affects the disease susceptibility, this deletion polymorphism has been extensively studied by evolutionary anthropologists as well as medical scientists (de Silva and Stumpf 2004; Novembre et al. 2005; Galvani and Novembre 2005).
Although its frequency and distribution suggest natural selection on the CCR5Δ32 allele in Northern European populations, there was no clear evidence from linkage-disequilibrium-based neutrality tests (Sabeti et al. 2005). Oleksyk et al. (2010) argued that multiple variants in this locus that was reported to be evolving under balancing selection (Bamshad et al. 2002) may have disrupted the linkage disequilibrium in this region. Recently, using the 409,693 British cohort dataset, Wei and Nielsen (2019) estimated a 21% increase in the all-cause mortality rate in individuals with the homozygous ∆32 alleles, suggesting a strong fitness cost. Thus, it follows that there has to be a fitness advantage, likely protection against certain retroviruses, that balances this fitness cost. To generalize this observation, it is our opinion that CNVs that affect immune system gene families likely evolve rapidly as a response to fast-evolving pathogenic pressures as was documented for single-nucleotide variants (Daugherty and Malik 2012). It is not surprising that immune system-related CNVs were shown in a case-by-case basis to confer to biomedically relevant phenotypes involving resistance to infectious diseases (Hollox and Armour 2008) and autoimmune disorders (Schaschl et al. 2009).
Skin-Related Genes
Hair and skin protect organisms from physical stimuli, temperature change, and ultraviolet radiation and thus have adaptively evolved to different environments (Jablonski 2008). For example, human skin color varies around the world and is associated with the adaptation to ultraviolet (Jablonski and Chaplin 2000; Relethford 2002; Quillen et al. 2019). In addition to skin color, hair keratin structure shows different patterns between human populations and between primate species (Hrdy and Baden 1973). There are several gene families involved in skin structure and copy number variable, including filaggrins (Eaaswarkhanth et al. 2016), keratins (Table S1), and late cornified envelope genes (Pajic et al. 2016).
Keratins are filament proteins of epithelia, skin, hair follicles, and nails with a remarkable diversity of their chemical nature (Bragulla and Homberger 2009). Heteropolymeric filaments are formed by pairing of type 1 keratin proteins (the gene cluster is located on human chromosome 17) and type 2 keratin proteins (the gene cluster is located on human chromosome 12) (Moll et al. 2008). In addition to four keratin-associated proteins, KRT34 has a duplication polymorphism (Table S1). The KRT34 duplication polymorphism has a population differentiation (3% in Eurasian populations and 13% in African populations) and is linked with flanking variants (R2 = 0.96), which makes KRT34 duplication a promising candidate for further evolutionary studies (Saitou and Gokcumen 2019).
The late cornified envelope (LCE) gene cluster is located in the epidermal differentiation complex on human chromosome 1. LCE genes respond to environmental stimuli to skin such as calcium levels and ultraviolet irradiation (Jackson et al. 2005). The deletion polymorphism of LCE3B and LCE3C is commonly observed (56%) in human populations and reported to affect a psoriasis susceptibility (de Cid et al. 2009). These deletions are also observed in archaic hominins (Lin et al. 2015) and showed the signature of balancing selection and a strong LD with the neighboring variants (Pajic et al. 2016). LCE1D and 1E are also commonly (20%) deleted (Table S1), and it is an open question why the LCE1D and 1E show the polymorphism and whether they have functional relevance. Considering the diversity of skin phenotypes between different populations (Jablonski 2008), at least some CNVs relevant to skin development and function may have been maintained as a result of adaptation to local environments and pathogens. This argument is not dissimilar to the case of CNVs affecting immune system genes. It is also important to note that skin is one of the most divergent organs in humans as compared to nonhuman primates (Arakawa et al. 2019). As such, it is plausible that some of the CNVs related to skin function may have been maintained in the human population as they were under reduced purifying selection, similar to the case of olfactory receptor CNVs, as a legacy of the rapid evolutionary transition of human skin from the ape common ancestor.
Metabolic Genes
Biotransformation of xenobiotics (foreign substances to the body) to molecules that are safe and readily usable within an organismal system is a crucial metabolic process relevant to both fitness and health. As such, the variation in the function of metabolic enzymes is an interesting area of evolutionary study. For example, it was recently shown that a particular haplotype of the arsenic [+3 oxidation state] methyltransferase (AS3MT) gene shows unusually high allele frequency among the Argentinean Andes population as compared to otherwise closely related Peruvian population (Schlebusch et al. 2015). Most of such enzymes that are involved in metabolizing often toxic xenobiotics are clustered into four gene families, namely cytochromes P450s, UDP-glucuronosyltransferases (UGTs), sulfotransferases (SULTs), and glutathione S-transferases (GSTs) (Jancova et al. 2010). As you may have noticed, we have already discussed CNVs affecting two genes belonging to these gene families, UGT2B17 and GSTM1. Indeed, several other CNVs are reported affecting other genes in these families as well (Table S1). Thus, one plausible scenario is that some of these CNVs increase in allele frequency in certain populations due to local adaptation. Of course, it should be noted that these gene families, which harbor several segmental duplications, have high rates of CNV formation rates, which may also explain the high number of copy number variable genes. Regardless, these CNVs contribute to phenotypic variation in human populations and are highly relevant to the field of pharmacogenomics (Li and Bluth 2011; Mazaleuskaya et al. 2015).
Conclusion
We argue in this paper that CNVs have a considerable but largely unexplored impact on human disease and adaptive evolution. We then laid out the methodological challenges in studying the function of CNVs. We specifically underlined the multiple formation mechanisms of CNVs, which can lead to CNVs with different functional properties. The various formation mechanisms of CNVs can also lead to differences in LD structure and complicate discovery and genotyping. Then, we summarized several examples, where locus-specific, careful dissection of the haplotypic variation of CNVs led to resolution of evolutionary history and functional relevance of these variants, explaining different phenotypic variations among humans. We then summarized some functional categories, including immunity, perception, skin, and metabolism, where CNVs are particularly relevant to understand phenotypic variation. We hope that our review serves as a primer for future studies, which will be increasingly more powerful with the advent of long-read sequencing technologies.
Methods
We used Ensembl human exon information (https://useast.ensembl.org/index.html), bedtools v2.27.1 (Quinlan and Hall 2010) and 1KGP phase 3 dataset (Sudmant et al. 2015b) to find exonic structural variants (Table S1).
References
Abyzov A et al (2013) Analysis of variable retroduplications in human populations suggests coupling of retrotransposition to cell division. Genome Res 23:2042–2052
Alkan C, Coe BP, Eichler EE (2011) Genome structural variation discovery and genotyping. Nat Rev Genet 12:363–376
Ambatipudi KS et al (2010) Human common salivary protein 1 (CSP-1) promotes binding of Streptococcus mutans to experimental salivary pellicle and glucans formed on hydroxyapatite surface. J Proteome Res 9:6605–6614
An P et al (2009) APOBEC3B deletion and risk of HIV-1 acquisition. J Infect Dis 200:1054–1058
Arakawa N et al (2019) Expression changes of structural protein genes may be related to adaptive skin characteristics specific to humans. Genome Biol Evol 11:613–628. https://doi.org/10.1093/gbe/evz007
Atkinson FS, Hancock D, Petocz P, Brand-Miller JC (2018) The physiologic and phenotypic significance of variation in human amylase gene copy number. Am J Clin Nutr 108:737–748. https://doi.org/10.1093/ajcn/nqy164
Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE (2001) Segmental duplications: organization and impact within the current human genome project assembly. Genome Res 11:1005–1017
Bailey JA et al (2002) Recent segmental duplications in the human genome. Science 297:1003–1007
Bamshad MJ et al (2002) A strong signature of balancing selection in the 5′ cis-regulatory region of CCR9. Proc Natl Acad Sci U S A 99:10539–10544
Bank RA et al (1992) Variation in gene copy number and polymorphism of the human salivary amylase isoenzyme system in Caucasians. Hum Genet 89:213–222
Behera SK, Praharaj AB, Dehury B, Negi S (2015) Exploring the role and diversity of mucins in health and disease with special insight into non-communicable diseases. Glycoconj J 32:575–613
Bickhart DM, Liu GE (2014) The challenges and importance of structural variation detection in livestock. Front Genet 5:37
Boehlke C, Zierau O, Hannig C (2015) Salivary amylase—the enzyme of unspecialized euryphagous animals. Arch Oral Biol 60:1162–1176. https://doi.org/10.1016/j.archoralbio.2015.05.008
Boettger LM et al (2016) Recurring exon deletions in the HP (haptoglobin) gene contribute to lower blood cholesterol levels. Nat Genet 48:359–366
Bournazos S, Woof JM, Hart SP, Dransfield I (2009) Functional and clinical consequences of Fc receptor polymorphic and copy number variants. Clin Exp Immunol 157:244–254
Bragulla HH, Homberger DG (2009) Structure and functions of keratin proteins in simple, stratified, keratinized and cornified epithelia. J Anat 214:516–559
Burchell B, Brierley CH, Monaghan G, Clarke DJ (1997) The structure and function of the UDP-glucuronosyltransferase gene family. In: Goldstein DS, Eisenhofer G, McCarty R (eds) Advances in pharmacology, vol 42. Academic Press, Cambridge, pp 335–338
Byars SG, Voskarides K (2019) Genes that improved fitness also cost modern humans: evidence for genes with antagonistic effects on longevity and disease. Evol Med Public Health 2019:4–6
Chaisson MJP et al (2019) Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun 10:1784
Chang HHY, Pannunzio NR, Adachi N, Lieber MR (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18:495–506
Chuong EB, Elde NC, Feschotte C (2017) Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet 18:71–86
Ciomborowska J, Rosikiewicz W, Szklarczyk D, Makałowski W, Makałowska I (2013) ‘Orphan’ retrogenes in the human genome. Mol Biol Evol 30:384–396
Conrad DF et al (2010) Origins and functional impact of copy number variation in the human genome. Nature 464:704–712
Cordaux R, Batzer MA (2009) The impact of retrotransposons on human genome evolution. Nat Rev Genet 10:691–703
Cullen BR (2006) Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J Virol 80:1067–1076
Currall BB, Chiang C, Talkowski ME, Morton CC (2013) Mechanisms for structural variation in the human genome. Curr Genet Med Rep 1:81–90
Daugherty MD, Malik HS (2012) Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet 46:677–700
de Silva E, Stumpf MPH (2004) HIV and the CCR29-Δ32 resistance allele. FEMS Microbiol Lett 241:1–12
de Cid R et al (2009) Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat Genet 41:211–215
Deininger PL, Moran JV, Batzer MA, Kazazian HH Jr (2003) Mobile elements and mammalian genome evolution. Curr Opin Genet Dev 13:651–658
Dekker J, Rossen JWA, Büller HA, Einerhand AWC (2002) The MUC family: an obituary. Trends Biochem Sci 27:126–131
Derti A, Roth FP, Church GM, Wu C-T (2006) Mammalian ultraconserved elements are strongly depleted among segmental duplications and copy number variants. Nat Genet 38:1216–1220
Duane WC, Frerichs R, Levitt MD (1972) Simultaneous study of the metabolic turnover and renal excretion of salivary amylase-125I and pancreatic amylase-131I in the baboon. J Clin Invest 51:1504–1513
Eaaswarkhanth M et al (2016) Atopic dermatitis susceptibility variants in filaggrin hitchhike hornerin selective sweep. Genome Biol Evol 8:3240–3255
Eichler EE (2019) Genetic variation, comparative genomics, and the diagnosis of disease. N Engl J Med 381:64–74
Enard D, Depaulis F, Roest Crollius H (2010) Human and non-human primate genomes share hotspots of positive selection. PLoS Genet 6:e1000840
Fábián TK, Hermann P, Beck A, Fejérdy P, Fábián G (2012) Salivary defense proteins: their network and role in innate and acquired oral immunity. Int J Mol Sci 13:4295–4320
Fablet M, Bueno M, Potrzebowski L, Kaessmann H (2009) Evolutionary origin and functions of retrogene introns. Mol Biol Evol 26:2147–2156
Falchi M et al (2014) Low copy number of the salivary amylase gene predisposes to obesity. Nat Genet 46:492–497
Fan H, Chu J-Y (2007) A brief review of short tandem repeat mutation. Genom Proteom Bioinform 5:7–14
Feschotte C (2008) Transposable elements and the evolution of regulatory networks. Nat Rev Genet 9:397–405
Fu W, Zhang F, Wang Y, Gu X, Jin L (2010) Identification of copy number variation hotspots in human populations. Am J Hum Genet 87:494–504
Galvani AP, Novembre J (2005) The evolutionary history of the CCR43-Δ32 HIV-resistance mutation. Microbes Infect 7:302–309
Gilad Y, Przeworski M, Lancet D (2004) Loss of olfactory receptor genes coincides with the acquisition of full trichromatic vision in primates. PLoS Biol 2:E5
Girirajan S, Campbell CD, Eichler EE (2011) Human copy number variation and complex genetic disease. Annu Rev Genet 45:203–226
Go Y, Niimura Y (2008) Similar numbers but different repertoires of olfactory receptor genes in humans and chimpanzees. Mol Biol Evol 25:1897–1907
Gokcumen O et al (2011) Refinement of primate copy number variation hotspots identifies candidate genomic regions evolving under positive selection. Genome Biol 12:R52
Gokcumen O et al (2013) Primate genome architecture influences structural variation mechanisms and functional consequences. Proc Natl Acad Sci U S A 110:15764–15769
Gu S et al (2015) Alu-mediated diverse and complex pathogenic copy-number variants within human chromosome 17 at p13.3. Hum Mol Genet 24:4061–4077
Gumucio DL, Wiebauer K, Caldwell RM, Samuelson LC, Meisler MH (1988) Concerted evolution of human amylase genes. Mol Cell Biol 8:1197–1205
Gurdasani D, Barroso I, Zeggini E, Sandhu MS (2019) Genomics of disease risk in globally diverse populations. Nat Rev Genet 20:520–535. https://doi.org/10.1038/s41576-019-0144-0
Hagenbüchle O, Bovey R, Young RA (1980) Tissue-specific expression of mouse α-amylase genes: nucleotide sequence of isoenzyme mRNAs from pancreas and salivary gland. Cell 21:179–187. https://doi.org/10.1016/0092-8674(80)90125-7
Hahn MW, Demuth JP, Han S-G (2007) Accelerated rate of gene gain and loss in primates. Genetics 177:1941–1949
Hastings PJ, Lupski JR, Rosenberg SM, Ira G (2009) Mechanisms of change in gene copy number. Nat Rev Genet 10:551–564
Hayden S et al (2014) A cluster of olfactory receptor genes linked to frugivory in bats. Mol Biol Evol 31:917–927
Hayes JD, Flanagan JU, Jowsey IR (2005) Glutathione transferases. Annu Rev Pharmacol Toxicol 45:51–88
Hedges DJ, Batzer MA (2005) From the margins of the genome: mobile elements shape primate evolution. BioEssays 27:785–794
Hirata H et al (2010) Function of UDP-glucuronosyltransferase 2B17 (UGT2B17) is involved in endometrial cancer. Carcinogenesis 31:1620–1626
Hollox EJ, Armour JAL (2008) Directional and balancing selection in human beta-defensins. BMC Evol Biol 8:113
Hoover KC et al (2015) Global survey of variation in a human olfactory receptor gene reveals signatures of non-neutral evolution. Chem Senses 40:481–488
Hrdy D, Baden HP (1973) Biochemical variation of hair keratins in man and non-human primates. Am J Phys Anthropol 39:19–24
Huddleston J et al (2017) Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res 27:677–685
Hurles M (2004) Gene duplication: the genomic trade in spare parts. PLoS Biol 2:E206
Imahashi M et al (2014) Lack of association between intact/deletion polymorphisms of the APOBEC3B gene and HIV-1 risk. PLoS ONE 9:e92861
Iskow RC, Gokcumen O, Lee C (2012) Exploring the role of copy number variants in human adaptation. Trends Genet 28:245–257
Itaya S et al (2010) No evidence of an association between the APOBEC3B deletion polymorphism and susceptibility to HIV infection and AIDS in Japanese and Indian populations. J Infect Dis 202:815–816 (author reply 816–817)
Jablonski NG (2008) Skin: a natural history. University of California Press, Berkeley
Jablonski NG, Chaplin G (2000) The evolution of human skin coloration. J Hum Evol 39:57–106
Jackson B et al (2005) Late cornified envelope family in differentiating epithelia—response to calcium and ultraviolet irradiation. J Invest Dermatol 124:1062–1070
Jancova P, Anzenbacher P, Anzenbacherova E (2010) Phase II drug metabolizing enzymes. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 154:103–116
Jha P et al (2012) Deletion of the APOBEC3B gene strongly impacts susceptibility to falciparum malaria. Infect Genet Evol 12:142–148
Jiang W et al (2012) Copy number variation leads to considerable diversity for B but not A haplotypes of the human KIR genes encoding NK cell receptors. Genome Res 22:1845–1854
Karypidis A-H, Olsson M, Andersson S-O, Rane A, Ekström L (2008) Deletion polymorphism of the UGT2B17 gene is associated with increased risk for prostate cancer and correlated to gene expression in the prostate. Pharmacogenom J 8:147–151
Kawamura S, Melin AD (2017) Evolution of genes for color vision and the chemical senses in primates. In: Saitou N (ed) Evolution of the human genome I: the genome and genes. Springer, Tokyo, pp 181–216
Kazazian HH Jr (2004) Mobile elements: drivers of genome evolution. Science 303:1626–1632
Keller A, Zhuang H, Chi Q, Vosshall LB, Matsunami H (2007) Genetic variation in a human odorant receptor alters odour perception. Nature 449:468–472
Khrunin AV et al (2016) GSTM1 copy number variation in the context of single nucleotide polymorphisms in the human GSTM cluster. Mol Cytogenet 9:30
Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE (2007) Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. https://doi.org/10.1371/journal.pgen.0030063
Kim PM et al (2008) Analysis of copy number variants and segmental duplications in the human genome: evidence for a change in the process of formation in recent evolutionary history. Genome Res 18:1865–1874
Kinomoto M et al (2007) All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res 35:2955–2964
Kirby A et al (2013) Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet 45:299–303
Kirkbride HJ et al (2001) Genetic polymorphism of MUC7: allele frequencies and association with asthma. Eur J Hum Genet 9:347–354
Korbel JO et al (2007) Paired-end mapping reveals extensive structural variation in the human genome. Science 318:420–426
Kumar S et al (2017) Genetic variants of mucins: unexplored conundrum. Carcinogenesis 38:671–679
Langlois MR, Delanghe JR (1996) Biological and clinical significance of haptoglobin polymorphism in humans. Clin Chem 42:1589–1600
Lee E et al (2012) Landscape of somatic retrotransposition in human cancers. Science 337:967–971
Leffler EM et al (2013) Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 339:1578–1582
Levchenko A, Kanapin A, Samsonova A, Gainetdinov RR (2018) Human accelerated regions and other human-specific sequence variations in the context of evolution and their relevance for brain development. Genome Biol Evol 10:166–188
Li J, Bluth MH (2011) Pharmacogenomics of drug metabolizing enzymes and transporters: implications for cancer therapy. Pharmgenom Pers Med 4:11–33
Liang T, Guo L, Liu C (2012) Genome-wide analysis of mir-548 gene family reveals evolutionary and functional implications. J Biomed Biotechnol 2012:679563
Ligtenberg AJM, Karlsson NG, Veerman ECI (2010) Deleted in malignant brain tumors-1 protein (DMBT1): a pattern recognition receptor with multiple binding sites. Int J Mol Sci 11:5212–5233
Liman ER (2006) Use it or lose it: molecular evolution of sensory signaling in primates. Pflugers Arch 453:125–131
Lin Y-L, Gokcumen O (2019) Fine-scale characterization of genomic structural variation in the human genome reveals adaptive and biomedically relevant hotspots. Genome Biol Evol 11:1136–1151
Lin Y-L, Pavlidis P, Karakoc E, Ajay J, Gokcumen O (2015) The evolution and functional impact of human deletion variants shared with archaic hominin genomes. Mol Biol Evol 32:1008–1019
MacKellar M, Vigerust DJ (2016) Role of haptoglobin in health and disease: a focus on diabetes. Clin Diabetes 34:148–157
Mandel AL, Peyrot des Gachons C, Plank KL, Alarcon S, Breslin PAS (2010) Individual differences in AMY1 gene copy number, salivary α-amylase levels, and the perception of oral starch. PLoS ONE 5:e13352
Marcovecchio ML et al (2016) Low AMY1 gene copy number is associated with increased body mass index in prepubertal boys. PLoS ONE 11(5):e0154961
Marques-Bonet T et al (2009) A burst of segmental duplications in the genome of the African great ape ancestor. Nature 457:877–881
Matsui A, Go Y, Niimura Y (2010) Degeneration of olfactory receptor gene repertories in primates: no direct link to full trichromatic vision. Mol Biol Evol 27:1192–1200
Mazaleuskaya LL et al (2015) PharmGKB summary: pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet Genom 25:416–426
Mefford HC et al (2010) Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet 6:e1000962
Meisler MH, Ting CN (1993) The remarkable evolutionary history of the human amylase genes. Crit Rev Oral Biol Med 4:503–509
Merritt AD, Rivas ML, Bixler D, Newell R (1973) Salivary and pancreatic amylase: electrophoretic characterizations and genetic studies. Am J Hum Genet 25:510–522
Mills RE et al (2011) Mapping copy number variation by population-scale genome sequencing. Nature 470:59–65
Moll R, Divo M, Langbein L (2008) The human keratins: biology and pathology. Histochem Cell Biol 129:705–733
Mollenhauer J et al (1997) DMBT1, a new member of the SRCR superfamily, on chromosome 10q25.3-26.1 is deleted in malignant brain tumours. Nat Genet 17:32–39
Mollenhauer J et al (2000) DMBT1 encodes a protein involved in the immune defense and in epithelial differentiation and is highly unstable in cancer. Cancer Res 60:1704–1710
Möncke-Buchner E et al (2002) Counting CAG repeats in the Huntington’s disease gene by restriction endonuclease EcoP15I cleavage. Nucleic Acids Res 30:e83
Narzisi G, Schatz MC (2015) The challenge of small-scale repeats for indel discovery. Front Bioeng Biotechnol 3:8
Nei M, Niimura Y, Nozawa M (2008) The evolution of animal chemosensory receptor gene repertoires: roles of chance and necessity. Nat Rev Genet 9:951–963
Nguyen D-Q, Webber C, Ponting CP (2006) Bias of selection on human copy-number variants. PLoS Genet 2:e20
Niimura Y, Matsui A, Touhara K (2018) Acceleration of olfactory receptor gene loss in primate evolution: possible link to anatomical change in sensory systems and dietary transition. Mol Biol Evol 35:1437–1450
Novembre J, Galvani AP, Slatkin M (2005) The geographic spread of the CCR110 Δ32 HIV-resistance allele. PLoS Biol 3:e339
Oda S, Fukami T, Yokoi T, Nakajima M (2015) A comprehensive review of UDP-glucuronosyltransferase and esterases for drug development. Drug Metab Pharmacokinet 30:30–51
Oleksyk TK, Smith MW, O’Brien SJ (2010) Genome-wide scans for footprints of natural selection. Philos Trans R Soc Lond B Biol Sci 365:185–205
Pajic P, Lin Y-L, Xu D, Gokcumen O (2016) The psoriasis-associated deletion of late cornified envelope genes LCE3B and LCE3C has been maintained under balancing selection since human Denisovan divergence. BMC Evol Biol 16:265
Pajic P et al (2019) Independent amylase gene copy number bursts correlate with dietary preferences in mammals. Elife 8:e44628. https://doi.org/10.7554/eLife.44628
Pang AW et al (2010) Towards a comprehensive structural variation map of an individual human genome. Genome Biol 11:R52
Paudel Y et al (2013) Evolutionary dynamics of copy number variation in pig genomes in the context of adaptation and domestication. BMC Genom 14:449
Perry GH et al (2006) Hotspots for copy number variation in chimpanzees and humans. Proc Natl Acad Sci U S A 103:8006–8011
Perry GH et al (2007) Diet and the evolution of human amylase gene copy number variation. Nat Genet 39:1256–1260
Pezer Ž, Harr B, Teschke M, Babiker H, Tautz D (2015) Divergence patterns of genic copy number variation in natural populations of the house mouse (Mus musculus domesticus) reveal three conserved genes with major population-specific expansions. Genome Res 25:1114–1124
Piriyapongsa J, Jordan IK (2007) A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS ONE 2007. https://doi.org/10.1371/journal.pone.0000203
Pirooznia M, Goes FS, Zandi pp. (2015) Whole-genome CNV analysis: advances in computational approaches. Front Genet 6:138
Polley S et al (2015) Evolution of the rapidly mutating human salivary agglutinin gene (DMBT1) and population subsistence strategy. Proc Natl Acad Sci U S A 112:5105–5110
Poole AC et al (2019) Human salivary amylase gene copy number impacts oral and gut microbiomes. Cell Host Microbe 25:553–564.e7
Popadić A, Anderson WW (1995) Evidence for gene conversion in the amylase multigene family of Drosophila pseudoobscura. Mol Biol Evol 12:564–572
Pruimboom L, Fox T, Muskiet FAJ (2014) Lactase persistence and augmented salivary alpha-amylase gene copy numbers might have been selected by the combined toxic effects of gluten and (food born) pathogens. Med Hypotheses 82:326–334
Quillen EE et al (2019) Shades of complexity: New perspectives on the evolution and genetic architecture of human skin. Am J Phys Anthropol 168:4–26
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842
Redon R et al (2006) Global variation in copy number in the human genome. Nature 444:444–454
Reed DR, Knaapila A (2010) Genetics of taste and smell. Prog Mol Biol Transl Sci. https://doi.org/10.1016/b978-0-12-375003-7.00008-x
Relethford JH (2002) Apportionment of global human genetic diversity based on craniometrics and skin color. Am J Phys Anthropol 118:393–398
Repnikova EA et al (2013) Characterization of copy number variation in genomic regions containing STR loci using array comparative genomic hybridization. Forensic Sci Int Genet 7:475–481
Robyt JF, French D (1967) Multiple attack hypothesis of α-amylase action: Action of porcine pancreatic, human salivary, and Aspergillus oryzae α-amylases. Arch Biochem Biophys 122:8–16
Rothman N et al (2010) A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nat Genet 42:978–984
Sabeti PC et al (2005) The case for selection at CCR133-Δ32. PLoS Biol 3:1963–1969
Saitou M, Gokcumen O (2019) Resolving the insertion sites of polymorphic duplications reveals a HERC2 haplotype under selection. Genome Biol Evol 11:1679–1690. https://doi.org/10.1093/gbe/evz107
Saitou M, Satta Y, Gokcumen O, Ishida T (2018a) Complex evolution of the GSTM gene family involves sharing of GSTM1 deletion polymorphism in humans and chimpanzees. BMC Genom 19:293
Saitou M, Satta Y, Gokcumen O (2018b) Complex haplotypes of GSTM1 gene deletions harbor signatures of a selective sweep in East Asian populations. G3 8:2953–2966. https://doi.org/10.1534/g3.118.200462
Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M (1996) Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry 35:3362–3367
Samson M, Libert F et al (1996) Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382:722–725
Santos JL et al (2012) Copy number polymorphism of the salivary amylase gene: implications in human nutrition research. J Nutrigenet Nutrigenom 5:117–131
Schaer DJ, Vinchi F, Ingoglia G, Tolosano E, Buehler PW (2014) Haptoglobin, hemopexin, and related defense pathways—basic science, clinical perspectives, and drug development. Front Physiol 5:415
Schaper E, Gascuel O, Anisimova M (2014) Deep conservation of human protein tandem repeats within the eukaryotes. Mol Biol Evol 31:1132–1148
Schaschl H, Aitman TJ, Vyse TJ (2009) Copy number variation in the human genome and its implication in autoimmunity. Clin Exp Immunol 156:12–16
Schlebusch CM et al (2015) Human adaptation to arsenic-rich environments. Mol Biol Evol 32:1544–1555
Schrider DR, Hahn MW (2010) Gene copy-number polymorphism in nature. Proc Biol Sci 277:3213–3221
Sharp AJ et al (2005) Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 77:78–88
Sherman RM et al (2018) Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat Genet 51:30–35. https://doi.org/10.1038/s41588-018-0273-y
Slatkin M (2008) Linkage disequilibrium–understanding the evolutionary past and mapping the medical future. Nat Rev Genet 9:477–485
Smithies O, Walker NF (1955) Genetic control of some serum proteins in normal humans. Nature 176:1265–1266
Spielman RS et al (2007) Common genetic variants account for differences in gene expression among ethnic groups. Nat Genet 39:226–231
Stankiewicz P, Lupski JR (2010) Structural variation in the human genome and its role in disease. Annu Rev Med 61:437–455
Stenglein MD, Harris RS (2006) APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem 281:16837–16841
Stevison LS et al (2016) The time scale of recombination rate evolution in Great Apes. Mol Biol Evol 33:928–945
Sudmant PH et al (2010) Diversity of human copy number variation and multicopy genes. Science 330:641–646
Sudmant PH, Mallick S et al (2015) Global diversity, population stratification, and selection of human copy number variation. Science. https://doi.org/10.1126/science.aab376
Sudmant PH, Rausch T et al (2015) An integrated map of structural variation in 2,504 human genomes. Nature 526:75–81
Trizzino M et al (2017) Transposable elements are the primary source of novelty in primate gene regulation. Genome Res 27:1623–1633
Turner T (2014) Faculty of 1000 evaluation for the missense of smell: functional variability in the human odorant receptor repertoire. Nat Neurosci 17(1):114–120
Usher CL et al (2015) Structural forms of the human amylase locus and their relationships to SNPs, haplotypes and obesity. Nat Genet 47:921–925
van Ommen G-JB (2005) Frequency of new copy number variation in humans. Nat Genet 37:333–334
Varki A, Geschwind DH, Eichler EE (2008) Explaining human uniqueness: genome interactions with environment, behaviour and culture. Nat Rev Genet 9:749–763
Viljakainen H et al (2015) Low copy number of the AMY1 locus is associated with early-onset female obesity in Finland. PLoS ONE 10(7):e0131883
Visscher PM et al (2017) 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet 101:5–22
Voskarides K (2018) Combination of 247 genome-wide association studies reveals high cancer risk as a result of evolutionary adaptation. Mol Biol Evol 35:473–485
Weckselblatt B, Rudd MK (2015) Human structural variation: mechanisms of chromosome rearrangements. Trends Genet 31:587–599
Wei X, Nielsen R (2019) CCR164-∆32 is deleterious in the homozygous state in humans. Nat Med 25:909–910
Wejman JC, Hovsepian D, Wall JS, Hainfeld JF, Greer J (1984) Structure and assembly of haptoglobin polymers by electron microscopy. J Mol Biol 174:343–368
Wellcome Trust Case Control Consortium et al. 2010. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 464:713–720.
Wenzel A et al (2018) Single molecule real time sequencing in ADTKD-MUC1 allows complete assembly of the VNTR and exact positioning of causative mutations. Sci Rep 8:4170
Williams GC (1957) Pleiotropy, natural selection, and the evolution of senescence. Evolution 11(4):398–411
Xu S, Wang Y, Roe B, Pearson WR (1998) Characterization of the human class Mu glutathione S-transferase gene cluster and the GSTM1 deletion. J Biol Chem 273:3517–3527
Xu D et al (2016) Recent evolution of the salivary mucin MUC7. Sci Rep 6:31791
Xu D et al (2017) Archaic hominin introgression in Africa contributes to functional salivary MUC7 genetic variation. Mol Biol Evol 34:2704–2715
Xue Y, Sun D, Daly A, Yang F, Zhou X (2008) Adaptive evolution of UGT2B17 copy-number variation. Am J Hum Genet 83:337–346
Yang N, Kazazian HH Jr (2006) L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat Struct Mol Biol 13:763–771
Yang Z-M et al (2015) The roles of AMY1 copies and protein expression in human salivary α-amylase activity. Physiol Behav 138:173–178
Young JM et al (2008) Extensive copy-number variation of the human olfactory receptor gene family. Am J Hum Genet 83:228–242
Zhang T et al (2013) Evidence of associations of APOBEC3B gene deletion with susceptibility to persistent HBV infection and hepatocellular carcinoma. Hum Mol Genet 22:1262–1269
Zhao M, Wang Q, Wang Q, Jia P, Zhao Z (2013) Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinform 14(Suppl 11):S1
Zhao X, Emery SB, Myers B, Kidd JM, Mills RE (2016) Resolving complex structural genomic rearrangements using a randomized approach. Genome Biol 17:126
Živná M et al (2018) Noninvasive immunohistochemical diagnosis and novel MUC1 mutations causing autosomal dominant tubulointerstitial kidney disease. J Am Soc Nephrol 29(9):2418–2431
Acknowledgements
We thank Izzy Starr and Skyler Resendez for careful reading of the manuscript. We are grateful for funding from the National Science Foundation (NSF) (Grant No. 1714867 (OG)).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Handling Editor: Konstantinos Voskarides.
Electronic supplementary material
Below is the link to the electronic supplementary material.
239_2019_9911_MOESM1_ESM.xlsx
Table S1. All the common (5% >) exonic genes in the 1000 Genomes project dataset (Sudmant et al. 2015b) with function annotation from OMIM (https://www.omim.org/). “Copy number” indicates the type of variation, CNV is multiallelic copy number variation, CN0 is deletion, and CN2 is duplication compared to the reference genome hg19. Supplementary file 1 (XLSX 13 kb)
Rights and permissions
About this article

Cite this article
Saitou, M., Gokcumen, O. An Evolutionary Perspective on the Impact of Genomic Copy Number Variation on Human Health. J Mol Evol 88, 104–119 (2020). https://doi.org/10.1007/s00239-019-09911-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-019-09911-6