
Abstract
It is generally considered that if an RNA World ever existed that it would be driven by an RNA capable of RNA replication. Whether such a catalytic RNA could emerge in an RNA World or not, there would need to be prior routes to increasing complexity in order to produce it. It is hypothesized here that increasing sequence variety, if not complexity, can in fact readily emerge in response to a dynamic equilibrium between synthesis and degradation. A model system in which T4 RNA ligase catalyzes synthesis and Benzonase catalyzes degradation was constructed. An initial 20-mer served as a seed and was subjected to 180 min of simultaneous ligation and degradation. The seed RNA rapidly disappeared and was replaced by an increasing number and variety of both larger and smaller variants. Variants of 40–80 residues were consistently seen, typically representing 2–4% of the unique sequences. In a second experiment with four individual 9-mers, numerous variants were again produced. These included variants of the individual 9-mers as well as sequences that contained sequence segments from two or more 9-mers. In both cases, the RNA products lack large numbers of point mutations but instead incorporate additions and subtractions of fragments of the original RNAs. The system demonstrates that if such equilibrium were established in a prebiotic world it would result in significant exploration of RNA sequence space and likely increased complexity. It remains to be seen if the variety of products produced is affected by the presence of small peptide oligomers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A key to understanding the origins of life is identifying the sources of the substantial increases in complexity required to transform a prebiotic environment to a living system. Nucleic acids in particular are relevant in this regard, because once available, they can produce increasing complexity through hybridization, ligation, and recombination. When RNAs were found capable of catalyzing reactions, the idea (Gilbert 1986; Lahav 1993; Orgel and Crick 1993; Rich 1962) of an RNA World quickly became popular (Cech 1993, 2009, 2015; Filipowicz 2017; Kawamura and Maurel 2017; Yeates et al. 2017). The key missing link, however, was thought to be the demonstration of the existence of a readily obtained self-copying RNA replicase (Higgs and Lehman 2015; Ma 2014; Robertson and Joyce 2012). Although the replicase problem remains unsolved, some progress has been made (Vaidya et al. 2012; Sczepanski and Joyce 2014; Horning and Joyce 2016).
The problem of origins is further compounded by the observation that evolutionary biases and constraints in the prebiotic world appear to be major factors resulting in an asphalt paradox (Benner et al. 2012). It would be necessary to maintain stability and information processing in a hostile water-based environment. To overcome the asphalt paradox, a primitive RNA or RNA-peptide world needs to rapidly produce structures with higher order complexity. Progress has been made. The origins of the RNA polymers have been linked to potential precursors including ammonium cyanide (Oró 1960) and nucleoside formation from 2-pyrimidinone (Bean et al. 2007). Formaldehyde has been shown to give rise to sugars including ribose (Decker et al. 1982; Eschenmoser 2011; Kim et al. 2011; Larralde et al. 1995; Sagi et al. 2012; Weber 1992), while formamide has been implicated as a precursor for prebiotic molecules (Saladino et al. 2007). Polymerization by itself, as a natural target process, has been demonstrated to occur in multiple ways. These include polymerization via a combination of condensation and dehydration reactions (Hud et al. 2013) and the ligation of random oligomers (Briones et al. 2009), Consistent with all of this, recent studies have made prebiotic synthesis of RNA more feasible (Cafferty et al. 2016; He et al. 2017).
Although progress has clearly been made, difficulty in finding a replicase along with the second problem of even creating RNAs under prebiotic conditions has led to increasing doubts about the existence of an RNA World. At this point, one might usefully consider that the first RNA replicase was never actually formed in an RNA World. Rather, it may have begun as a peptide produced by a primitive ribosome that emerged from the replicase-free RNA World (Fox 2016, 2010). At the least the absence of an easy path to an RNA replicase implies a long period of time in which information is not readily stored and mutations that are normally introduced by replication errors would seldom occur. The issue then is whether increasing complexity can occur in a pre-replicase RNA World.
The requisite increases in complexity might be facilitated by the wide variety of changing local environments on the prebiotic Earth. An example is wet/dry cycles (Forsythe et al. 2015). However, the potential effects of very long-term changes are not readily studied on laboratory time scales. To address this long-standing problem, a novel experimental approach based on dynamic combinatorial chemistry (DCC) has been discussed (Benner et al. 1996; Leal et al. 2006). It is hypothesized here that when subjected to a persistent equilibrium of ligation and cleavage, RNAs will naturally increase in variety and complexity despite the absence of frequent point mutations caused by replication errors. They also may gain resistance to degradation over time.
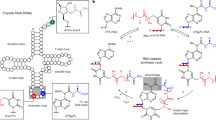
To test this hypothesis, a two enzyme system was employed to produce a dynamic equilibrium (Fig. 1). The cleavage enzyme is Benzonase, which is a commercial name for an extracellular endonuclease secreted by Serratia marcescens. This enzyme cleaves RNA, including double-stranded and circular forms, to produce products with a 3′ hydroxyl and 5′ phosphate. The cleavage was initially found to be non-specific resulting in products that are primarily small fragments of 2–4 residues but seldom monomers (Nestle and Roberts 1969). More recent studies have shown that within a single-stranded RNA there is a significant preference for duplex (secondary structure) regions (Meiss et al. 1999). This reflects the fact that the enzyme prefers substrates in their A-form structure. As a result, structures with significant secondary structure may not accumulate as well as others. Given the termini produced, Benzonase products are ideal for ligation by T4 RNA ligase (Tessier et al. 1986), which connects fragments with a 5′ phosphate to those with a 3′ OH non-specifically and utilizes ATP as a source of energy. The smallest acceptor is a trimer of the type NpNpNOH (Atencia et al. 2000). The smallest donor that will be ligated to an available 3′ OH is pNp. Hence, monomers including ATP will not be incorporated into the system. An increase in the chain length of either the acceptor or donor does not impact the rate or extent of the ligation reaction. However, it is reported that the presence of uridine in the acceptor greatly reduces the amount of product formed. In general, purines in the acceptor and pyrimidines in the donor have been shown to result in higher reactions yields. In order to monitor population changes, samples were extracted from the reaction mix and millions of individual sequences were determined using RNA-seq technology (http://support.illumina.com/downloads/nextseq-500-user-guide-15046563.html).

Overview of dynamic equilibrium system. (Figure adapted from https://www.sigmaaldrich.com)
Materials and Methods
Enzymes and Chemicals
Benzonase/Pierce Nuclease (~ 250U/µl), T4 RNA Ligase 1 (T4RL1) (high concentration of 30,000 units/ml) and ATP (100 mM) were purchased from Sigma Aldrich/Thermofisher Scientific, New England Biolabs, and Promega Corp, respectively. SYBR® Green II RNA Gel Stain (10,000X concentrate in DMSO) was purchased from Thermofisher Scientific. Ribonuclease R (RNase R) was purchased from Lucigen Corporation.
Seed RNAs
Custom RNA oligomers of pre-defined sequences were purchased from Integrated DNA Technologies (http://www.idtdna.com/). All sequences had a 5′ phosphate and a 3′ OH. The following sequences were used:
-
1.
A 20-mer with the sequence GAUACCCUACCCGGUCGGAU.
-
2.
Four 9-mers with the sequences GGGGCUAUA, GCGGUUCGA, UCCCGCUUA, and GCUCCACCA.
-
3.
A completely random RNA 20-mer.
Reaction Design
The original stock of Benzonase/Pierce Nuclease was diluted to 0.5U/µl to be used in the reactions. The final concentrations of the Benzonase, T4RL1, ATP, and the RNA oligo were, 0.025 U/µl, 831U/µl, 2.77 mM, and 89.45 µM, respectively. The reactions were performed at 37 °C in microfuge tubes. Aliquots of 10 ul were collected and added to microfuge tubes (on ice) containing gel loading dye (47.5% formamide, 0.01% SDS, 0.01% bromophenol blue, 0.005% xylene cyanol, 0.5 mM EDTA EDTA), for gel analysis. Aliquots of 10 µl were added to microtubes on ice containing EDTA to inhibit the enzyme action, and immediately transferred to minus 20 °C for subsequent sequence analysis. The aliquots were removed at 30, 60, 90, 120, 150, and 180 min. Samples were run on a 20% polyacrylamide denaturing gel containing (18.5:1.5 acrylamide/bis-acrylamide and 7.7M urea) 200V/20 mA for 3–4 h to allow sufficient resolution of the samples. The gels were placed on 1X TBE buffer containing appropriate dilutions of the SYBR® Green II RNA Gel Stain. The gels were visualized under UV after 45 min of dye exposure. The length distribution in each sample was separately characterized using the Agilent 2100 Bioanalyzer. When required, aliquots of 5 ul were removed and frozen immediately (− 20 °C) for quantifying ATP levels later.
Determination of ATP Levels
A standard curve for ATP levels was derived using the ATP Determination Kit (Thermofisher Scientific) using a Femtomaster-FB12 luminometer (Zylux Corp., Oak Ridge, TN, USA). EDTA is the only reaction component which could have potentially interfered with the ATP assay. However, while one aliquot was treated with EDTA to stop the reaction, the aliquots removed for the ATP assay did not have EDTA added to them. Instead, the samples were either frozen immediately (for ATP assay at a later stage) or assayed for ATP levels immediately.
Checking for Circularization
The entire RNA reaction mixtures that were run for 120–150 min were treated directly with the magnesium-dependent 3′→5 exoribonuclease Ribonuclease R (RNase R) overnight at 37 °C. The RNase R-treated samples were then run on a 20% polyacrylamide denaturing gel to check for RNase R activity and stained as described above. No evidence of circularization was found.
Processing of Reaction Mixture Samples for Sequencing
The concentration of RNA in each sample was determined using a Qubit Fluorometer 2.0 with RNA HS Assay Kit (Invitrogen), and RNA quality was assessed using an Agilent 2100 Bio analyzer with small RNA chips according to the manufacturer’s protocol. A NEXTflex small RNA-Seq Kit V3 (Bioo Scientific) was used to prepare the libraries following the manufacturer’s modified No Size Selection Protocol.
For each small RNA library construction, RNA from the reaction pool was diluted to a final quantity/volume of 1–10 ng in a total volume of 10.5 µl RNAse-free water. The diluted RNA was then used as starting material and treated with the reagents supplied with the NEXTflex® Small RNA Sequencing Kit v3 for Illumina® Platforms (http://www.biooscientific.com/Portals/0/Manuals/NGS/5132-05-NEXTflex-Small-RNA-Seq-v3 -16-06.pdf). The RNAs were first treated for 3′ 4N Adenylated Adapter ligation. Excess adapters were removed and inactivated, followed by 5′ 4N Adapter Ligation. The RNAs containing the Adapters at the 5′ and 3′ ends were then subjected to Reverse Transcription-First Strand Synthesis, on a Thermocycler (30 min 42 °C, 10 min 90 °C). The reverse transcribed products were processed using the NEXTflex™ Small RNA Sequencing Kit v3 No Size Selection Bead Cleanup Protocol which is designed to retain larger (reverse transcribed DNA) products.
The NEXTflex™ Barcode Primers and NEXTflex™ Universal Primer were then added to the reverse transcribed DNA. A different barcoded primer was used for each sample in order to allow multiplex sequencing. The DNA was then subjected to 95 °C for 2 min on a Thermocycler followed by PCR Amplification over 10–15 cycles of (20 s–95 °C, 30 s–60 °C, 15 s–72 °C). The PCR cycles were there followed by a 2-min incubation at 72 °C. Following the PCR amplification step, the libraries were further cleaned up using NEXTflex Cleanup Beads to retain the cDNA while discarding any of the remaining primer(s). The cleaned-up libraries were cleaned up using NEXTflex Cleanup Beads and were validated with an Agilent 2100 Bio analyzer to check size and purity.
The concentration of each cDNA library was determined using a Qubit Fluorometer. Libraries were diluted to 2 nM separately and pooled together. Then the pooled samples were denatured and diluted to 1.8 pM following Illumina’s protocol, and sequenced by a mid-output 150 cycles sequencing kit (Illumina) on Illumina NextSeq 500 platform for 75/75 cycles. RNA sequencing applications often employ RNA spike-in controls (Jiang et al. 2011). We adopted a spike-in control in the form of the well-established ΦX174 (Michel et al. 2010) in order to ensure quality sequencing data at the end of the workflow, (Ledergerber and Dessimoz 2011).
Sequence Data Processing
All high-throughput sequencing reads obtained from the Illumina NextSeq 500 sequencing protocol were processed using a combination of in-house scripts and publicly available utility programs. Raw sequencing data were processed with bcl2fastq Conversion Software (https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2-18.html) to convert to the FASTQ File format and produce reads that had zero mismatches in their barcodes. The four lanes for each barcode were merged with an in-house script. Next, the barcode reads were filtered at 0.01% expected error without N’s with vsearch. If the barcode reads did not pass filter, then the associated forward and reverse reads were also removed. The 3′ and 5′ end adapters were trimmed with fastq-mcf v1.04.807 from ea-utils (Aronesty 2013). This step reduces reads still having adapters to approximately 0.1% of total reads. Residual adapters are searched with the usearch subcommand search_oligodb using the forward read-through sequence (5-‘AACTCCAGTCACATCTCG-3’) and reverse read-through sequence (5′-GTGTAGATCTCGGTGGTCGCCGTATCATT-3′) with a maximum difference of 3. The search results were parsed and associated reads trimmed with an in-house awk script. There were still some forward reads that contained (5′-TGGAATT[A/T/C/G]*-3′) or (5′-AACTCCAGTCAC-3′) and some reverse reads contained (5′-GATCGTCG[A/T/C/G]*-3′) so they were also removed with another set of in-house awk scripts. This step reduced reads still having adapters to approximately 0.000001% of the total reads. Since the NEXTFlex Small RNA sequencing kit introduced 4 random nucleotides to the ends of the reads, these were then clipped off with in-house awk scripts. The read length distributions were then computed at this point using an in-house awk script. About 20% of the reads were the result of a PhiX spike-in that was used. The reads were thus filtered to remove all PhiX sequences using usearch. Then they were filtered at 1% expected error, without N’s, and minimum length of 1 nucleotide through vsearch3 v2.4.4 subcommand fastq_filter. This step produced a mixture of singletons and paired reads that were separated with fastq_pair (https://github.com/linsalrob/fastq-pair). The paired reads were then merged with usearch with minimum merge length of 1 nucleotide, 1% expected error, zero difference, and minimum overlap length of 6 nucleotides. The remaining unmerged pairs from this step were then merged with fastq-join v1.01.759 from ea-utils, filtered for N’s and only reads with less than 1% expected error were kept through the vsearch fastq filter subcommand. The first, second, and unmerged reads from each step were kept separately and their read distribution profiles were computed with vsearch subcommand fastq_stats as well as a set of in-house awk scripts. The resulting first, second, and unmerged reads from each steps were concatenated into one file while discarding any reads that contained stretches of homopolymer of at least 6 nucleotides. The resulting file were then dereplicated through vsearch derep_fulllength subcommand while conserving abundance information and relabeled to contain source information. A similar post-processing approach was applied for the 9-mer sequencing data minus the alignment.
Data Analysis
The dereplicated sequences were converted from FASTA format into a tabular format. From there, they were collated into one large table containing all sequences and their abundance found within each set of experiments. These tables were used to compute the information found in Tables 1, 2, 3, 4, and 5 as well as supplementary Tables S1–S9. The computation of first occurrence was simply the counting of the appearance of a particular sequence at a certain time point such that it did not appear in previous time points. The summation of first occurrences at each time point gave the sequence diversity of the experiment, which indeed equal the non-redundant sequences found in the experiments. The dereplicated sequences for the 20-mer seed-derived sequences were also aligned against the DNA version of the seed sequence (5′-GATACCCTACCCGGTCGGAT-3′) using clustal-omega (Sievers et al. 2011) v1.2.4 at 1000 iterations and muscle v3.8.31 (Edgar 2004). The results from both approaches were compared and collated to obtain a comprehensive picture.
In order to better understand the manner in which sequence space is being explored, consecutive 10-mers were derived from the non-redundant sequences. The seed sequence contains 11 such 10-mers. This was done with an in-house awk script. The total count, non-redundant count (diversity), and first occurrence are shown in Table 3. The un-weighted nucleotide bias was also compared between the 11 10-mers that came from the seed sequence vs all of the 10-mers that were produced from the experiment. The amount of G, C, A, and Ts were counted for the starting oligomers and bias were computed as (base count / total bases) – 25%. The same was done for each position of the 10-mers, i.e., [base count/ total oligomers (11 or 182,473)] – 25%. This information is presented in Table 4. The count of their occurrences in each sequence (frequency) as well as their duplication count, the occurrence multiplied by the sequence counts, were tallied and used to compute the proportions shown in Table 5.
Results
Initially, the activity of each enzyme was verified and a mutually compatible buffer system was established. A dynamic evolution experiment was conducted over a 180-min period beginning with a single defined sequence 20-mer RNA, pGAUACCCUACCCGGUCGGAUOH. Samples were taken at 30-min intervals, divided in half, and either loaded on a 20% polyacrylamide denaturing gel for visualization (Fig. 2) or sequenced using standard RNA-seq protocols. The resulting DNA sequence information was collected for samples at 30, 60, 90, 120, 150, and 180 min. The results at 30 min and at 180 min are summarized in Table 1. Only sequences that are present in at least 1% of the population are shown. Results from all six time points are provided in Supplementary Tables S1–S7.

Denaturing polyacrylamide gel of samples obtained from competition experiment between 0.5 U/µL of Benzonase and T4 RNA ligase with ATP. Control is the seed oligo only, while the remaining lanes correspond to increasing time point in minutes when the reaction was stopped. The asterisk (*) represents the band that corresponded to a single end-to-end ligation of the control to form a 40-mer, which became lighter as the reaction progressed
The most striking feature is the strong intensity of bands in the length 40 region (Fig. 2) after only 30 min. After 90 min, the intensity of this band begins to decline as the available ATP decreases. By 180 min, the Benzonase has become dominant and the larger RNAs are likely being degraded. The percentage of the remaining original 20-mer decreased at each time point, while non-redundant sequences continued to accumulate as a significant fraction of the overall diversity at each time point (Fig. 3 and Supplementary Tables S8 and S9).

The fraction of seed oligo and unique sequences are inversely related with time. As the seed oligo is diminished, the unique sequences increased until the 150-min mark after which unique sequences began to diminish as a result of the activity of Benzonase enzyme overtaking the activity of T4 RNA ligase. The percentage for both seed oligo and unique sequences were calculated with respect to the total sequence count at each time point and total diversity, respectively. The total sequence count was not constant due to sample loading variations during sequencing. The total diversity was computed from the total unique sequences found in all time points
The read size distribution seen in the sequencing results, Fig. 1, at each time point appears to be inconsistent with the distribution of RNA bands seen by gel electrophoresis. This is likely the result of poor dye staining of small oligomers. This interpretation is supported by studies with the Agilent 2100 Bioanalyzer (Supplementary Figure S1), in which the 40-mer peak was minimal. However, T4 RNA ligase is well known to catalyze circularization (Tessier et al. 1986) and it is known that standard RNA-Seq protocols ignore the presence of such circular products (Szabo and Salzman 2016). To eliminate the possibility of significant circularization, the entire RNA pool produced by ligation alone was completely digested by RNAse R which is well known to be unable to digest circular RNAs (Suzuki et al. 2006; Vincent and Deutscher 2006). No surviving RNA was seen thereby indicating that significant circularization did not occur. The self-ligated 40-mer product that would precede circularization was seen in the 30-min pool but its frequency (0.96%) was less than the 1% needed to make the list in Table 1.
The raw sequencing results for all reads were uniformly 75 nucleotides in length with approximately 7–10 million reads at each time point. The pre-processing of sequence data generated reads of very high quality. After merging the forward and reverse reads, filtering for N’s and expected error of 0.01%, approximately 98% of the input reads remained. The read length after this step ranged from 4 through the 120’s with the majority of reads in the 1920s. The stringent filtering step removed some longer reads in the 110’s–120’s range. However, there were very few of these to begin with.
Initially, the most common sequence was the original 20-mer seed. With the progression of time, the quantity of the original sequence decreased from 45.86% at 30 min to 2.45% at 180 min, Fig. 3 and Table 1. It was in fact replaced as the most common sequence beginning at 90 min by the sequence 5′-GGATGATACCCTACCCGGTCGGAT-3′, in which the fragment GGAT was ligated to the original seed. The third most common sequence after 30 min was CCTACCCGGTCGGAT in which the fragment GATAC was removed. This sequence remained fourth after 180 min. A comparison of the most common sequences at 30 and 180 min make it clear that the dominant products have changed considerably. At 30 min, all the dominant sequences end in GGTCGGAT, whereas by 180 min only 8 of 22 dominant sequences have this terminal sequence.
In order for cycles of ligation and degradation to facilitate increased complexity in the prebiotic world, it is essential that larger sequence products are produced. It is not sufficient to be reproducing endless variants of similar size to the original seed. In fact, larger sequences are produced at modest but significant numbers. Thus, an analysis of the data shows that non-redundant sequences in the size range of 40–80 residues continue to accumulate throughout all 180 min, typically representing over 2% of the unique sequences (Table 2). There is a decline near the end, likely as a result of decreased ligase activity.
The cluster alignments produced by clustal-omega (Sievers et al. 2011) and muscle (Edgar 2004) were quite different upon cursory inspections. The clustal-omega alignments centered on the two core sequences 5′-GATACCCTACCC-3′, and 5′-CCCGGTCGGAT-3′, which unsurprisingly are the two halves of the template. These two sequences appeared repeatedly in the many expansions on either ends of the core. The cluster alignments generated by MUSCLE (Edgar 2004) showed distinct zones within the template where sequence expansions were occurring. This took the pattern of 5′-GAT*ACC*CTACCCGG*TCGGAT-3′. The rest of the alignments were structured such that sequence variations and expansions occurred on the ends or in between these core sequences.
The 10-mer analysis allowed a better characterization of how and the extent to which sequence space was being explored in the 20-mer studies. With seven cytosines, the seed sequence had a clear bias for cytosine. This meant that the original eleven 10-mers that were derived from the starting sequence were also heavily biased in cytosine, Table 4. This bias was reduced from 22.27% to just 1.69% over the course of the experiment. This indicates that the competition between ligase and benzonase did mix up the identity of the nuclear bases at each of the ten positions in the 10-mer. Overall, there were 182,473 non-redundant 10-mers at 9,952,856 occurrences from all time points, Table 4. For the most part, each time point had about the same amount of new 10-mers except for the 30- and 150-min mark. The 30-min mark had about double the amount of 10-mer first occurrences, while the 150-min mark had about half that on average. It is not clear what happened at the 150-min mark. Speculatively, this could reflect the loss of ATP activity or simply be an artifact of library loading as indeed there were fewer reads that came from this time point versus the other time points.
With respect to sequence space exploration, the enzyme combination is not so adept. As can be seen in Table 5, the majority of the oligomers, the top 20, are from the original 11 10-mers and the various consecutive 10-mer found around the joint sequences between two end-to-end ligated starting sequences, e.g., TGATACCCTA or ATGATACCCT. After this, there is a precipitous drop in frequency due to the fact that any other oligomers must come from novel mixing and joining of the ligation and digestion fragments. However, this is not to say that this experimental technique is a dead end as novel recombination is definitely achieved and exploration would likely continue if the period of competition were extended.
In an actual prebiotic environment, there would likely be a mixture of many seed oligomers rather than just one. To begin to understand what would happen, four 9-mers (5′-PGGGGCUAUAOH-3′, 5′-PGCGGUUCGAOH-3′, 5′-PUCCCGCUUAOH-3′, and 5′-PGCUCCACCAOH-3′) were mixed together and exposed to the same conditions as the 20-mer. This included exposure to T4 RNA ligase only and T4 RNA ligase and Benzonase together. When the four individual 9-mers are ligated in the indicated order (1-2-3-4), the resulting 36mer corresponds to the alanine tRNA minihelix (Francklyn and Schimmel 1989). This sequence was in fact found, but alternative 36-mers were far more common. After control sequences were removed, the most common sequence with and without Benzonase was 5′-GGGGCUAUAGCGGUUCGAUCCCGCUUA-3′. This is a ligation of the first, second, and third 9-mer. A variety of products were formed containing sequences containing portions of two or more 9-mers. Some representative examples are shown in Fig. 4.

Diagram of the 9-mer reactions. The four 9-mer seed oligomers are represented in the orange box and colored for ease of tracking in the reaction products. The 5′ phosphate is characterized with a small yellow key. Although the figure includes both the T4 RNA ligase and Benzonase enzymes, there were two separate reactions, one with T4 RNA ligase only and one with both enzymes. The bottom right blue box shows a selection of representative reaction products found in deep sequencing of the RNAs. Most RNA products are simply ligations of one or more of the original 9-mers or ligation of partial 9-mers with fully complete 9-mers. However, there were a few examples where the shuffling and re-ligation resulted in sequences with little resemblance to the original 9-mers. An example is highlighted in purple. The count for each sequence are shown on the left of the bounding box
Finally, a completely random 20-mer was used as the initial seed. Following 120 min of exposure to the ligase/Benzonase regimen, the RNA pool was sequenced. No accumulation of specific sequences was observed.
Discussion
The immediate observation is that a system of simultaneous RNA ligation and degradation is in fact achievable. Analysis of the products, Table 2, makes it clear that as time continued, the diversity of the sequences in the pool continued to increase. By the end of the run 245,170 sequences were found to have occurred in one or more samples. Most importantly large numbers of new unique sequences were continuing to accumulate by well over 10% at each time point including 180 min. This indicates that the sequencing had not yet fully captured the number of individual sequences actually encountered during the experiment. To do so would require multiple sequencing runs at each time point until the number of unique sequences in each new sequencing run significantly declined. Although the resulting number would likely be very large, the ultimate total of unique 20-mer variants would be unlikely to approach the 420 examples that would occur if the entire sequence space were explored.
In order to better understand the extent of sequence space exploration, the fate of 10-mers embedded in the original seed and the 10-mers derived from them was examined. It is important to stress that this analysis was done un-weighted, that is without accounting for the duplication level of the oligomers. The reason for this being that each time point could not be scaled to one another because the library loading amount (mass) for each time point were not known. Despite this limitation, overall exploration of sequence space could be assessed and found to be modest in that many oligomers produced early on continued to be major components of the population after 180 min. This suggests that longer run time would be useful. The analysis also showed initial cytosine bias at individual positions in the 10-mers was already dramatically reduced at 180 min.
Of special interest are the products formed. An examination of the primary fragments produced (Table 1) shows that the sequences are not changing by point mutations. This is expected as the experimental plan is devoid of any component, which would be expected to introduce point mutations. The changes are almost completely additions and subtraction of fragments with the result that small K-mers in the original seed (GATAC, CCTACCC, and GGTCGGAT) tend to persist. This may in part reflect the fact that Benzonase does not cleave to monomers but instead leaves fragments of 3–5 residues, which can then be utilized by the ligase. Thus, diversity in the present system is only introduced by cleavage of end–end ligation products. However, in nature novel sequences might also be derived from random cleavage of circularized products. So while circular RNAs were apparently not a significant product in the current system, they would, if present, provide another path to diversity. It is also clear that Benzonase is not an agnostic enzyme as it will likely make it difficult for RNAs containing regions of secondary structure to accumulate.
In a prebiotic world, the seed sequence would almost certainly be a mixture of many sequences. In the present case, we focused primarily on the consequences of the offspring of one initial seed sequence. If, however, there are two or more seed sequences in the starting mixture, there will likely be interactions between them essentially immediately. As a result, the ease of producing a wide diversity of RNAs irrespective of any bias in either the ligation or degradation reactions will be increased. In order to test this hypothesis, the core experiment was repeated with four 9-mers as the seed. It was in fact found that as predicted, large numbers of sequences derived from parts or all of two or more individual 9-mers were found in significant numbers. Some examples are shown in Fig. 4.
One might expect that all possible ligations of the four 9-mers might occur. In fact, 357 unique 36-mers were seen among the reads. The actual numbers are likely higher as only a small portion of the mixture was actually sequenced. Additional sequencing runs would reveal the presence of additional sequences. However, most of the 256 possible ligations of the original four 9-mers are in fact already represented. The most common example was 1-1-2-3 which occurred 1772 times, while the combination 1-2-3-4 occurred just once. It is clear from this result that either or both the Benzonas-ligase system or sequencing system is biased in ways that have not yet been fully characterized.
Further experimental work will be necessary to better understand what the biases are and how sequence space is being explored by the Benzonase/ligase system. It is, however, extremely likely that a true prebiotic system would also explore sequence space in a biased way. Since we do not currently have a good understanding of what that prebiotic system would be, we therefore cannot fully understand what the bias would be. It is, however, unlikely to be the same as that seen in the Benzonase/ligase system. What is clear is that although bias will effect which areas of sequence space initially get explored, large regions of sequence space will nevertheless be explored. This will be especially true when there are multiple seed sequences present as would likely be the prebiotic case. As a result, the effect of bias will likely be quickly minimized.
It has previously been argued that in the hypothetical prebiotic RNA World, high rates of mutation would be expected to negatively impact the emergence of functional sequences (Higgs 1998; Leu et al. 2011; Rajamani et al. 2010). The system described here provides a plausible alternate pathway towards exploration of RNA sequence space that is largely devoid of point mutations. Indeed, from an origins perspective, the present system demonstrates that cycles of ligation and degradation can rapidly produce RNAs of increasing diversity and size despite the absence of a replicase.
A key issue for future studies is the question of an RNA World vs an RNA/Peptide World. It remains to be seen if the composition of the pools produced in response to a single or multiple seed are different when small peptides are present. However, the present experimental system includes two modern proteins and in that respect is not an ideal model of the prebiotic world. It would be preferable to develop a system devoid of such proteins. This might be possible. RNA degradation can be achieved in a variety of ways including exposure to metals such as ferric iron or alkaline hydrolysis. Ligation is more problematic but can occur, for example, with the artificial L1 ligase (Robertson et al. 2001). A challenge will be obtaining the correct termini so that both reactions work.
References
Aronesty E (2013) Comparison of sequencing utility programs. Open Bioinform J 7:1
Atencia EA, Montes M, Gunther Sillero MA, Sillero A (2000) Several dinucleoside polyphosphates are acceptor substrates in the T4 RNA ligase catalyzed reaction. Eur J Biochem 267:1707
Bean HD, Sheng Y, Collins JP, Anet FA, Leszczynski J, Hud NV (2007) Formation of a beta-pyrimidine nucleoside by a free pyrimidine base and ribose in a plausible prebiotic reaction. J Am Chem Soc 129:9556
Benner SA, Jermann TM, Opitz JG, Raillard SA, Zankel TR, Trautwein-Fritz K, Stackhouse J, Ciglic MI, Haugg M, Trabesinger-Ruf N, Weinhold EG (1996) Developing new synthetic catalysts. How nature does it. Acta chem Scandinavica 50:243
Benner SA, Kim HJ, Carrigan MA (2012) Asphalt, water, and the prebiotic synthesis of ribose, ribonucleosides, and RNA. Acc Chem Res 45:2025
Briones C, Stich M, Manrubia SC (2009) The dawn of the RNA World: toward functional complexity through ligation of random RNA oligomers. RNA 15:743
Cafferty BJ, Fialho DM, Khanam J, Krishnamurthy R, Hud NV (2016) Spontaneous formation and base pairing of plausible prebiotic nucleotides in water. Nat commun 7:11328
Cech TR (1993) The efficiency and versatility of catalytic RNA: implications for an RNA world. Gene 135:33
Cech TR (2009) Evolution of biological catalysis: ribozyme to RNP enzyme. Cold Spring Har Symp Quant Biol 74:11
Cech TR (2015) RNA World research-still evolving. RNA 21:474
Decker P, Schweer H, Pohlamnn R (1982) Bioids: X. Identification of formose sugars, presumable prebiotic metabolites, using capillary gas chromatography/gas chromatography—mas spectrometry of n-butoxime trifluoroacetates on OV-225. J Chromatogr A 244:281
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792
Eschenmoser A (2011) Etiology of potentially primordial biomolecular structures: from vitamin B12 to the nucleic acids and an inquiry into the chemistry of life’s origin: a retrospective. Angew Chem 50:12412
Filipowicz W (2017) Traversing the RNA world. J Biol Chem 292:8122
Forsythe JG, Yu SS, Mamajanov I, Grover MA, Krishnamurthy R, Fernandez FM, Hud NV (2015) Ester-mediated amide bond formation driven by wet-dry cycles: a possible path to polypeptides on the prebiotic earth. Angew Chem 54:9871
Fox GE (2010) Origin and evolution of the ribosome. Cold Spring Harb Perspect Biol 2:a003483
Fox G (2016) Origins and early evolution of the ribosome. In: Jagus GHR (ed) Evolution of the protein synthesis machinery and its regulation. Springer, Berlin, pp 31–60
Francklyn C, Schimmel P (1989) Aminoacylation of RNA minihelices with alanine. Nature 337:478
Gilbert W (1986) Origin of life: the RNA world. Nature 319:618
He C, Gallego I, Laughlin B, Grover MA, Hud NV (2017) A viscous solvent enables information transfer from gene-length nucleic acids in a model prebiotic replication cycle. Nat Chem 9:318
Higgs PG (1998) Compensatory neutral mutations and the evolution of RNA. Genetica 102–103:91
Higgs PG, Lehman N (2015) The RNA World: molecular cooperation at the origins of life. Nature reviews Genetics 16:7
Horning DP, Joyce GF (2016) Amplification of RNA by an RNA polymerase ribozyme. Proc Natl Acad Sci U S A 113:9786
Hud NV, Cafferty BJ, Krishnamurthy R, Williams LD (2013) The origin of RNA and “my grandfather’s axe”. Chem Biol 20:466
Jiang L, Schlesinger F, Davis CA, Zhang Y, Li R, Salit M, Gingeras TR, Oliver B (2011) Synthetic spike-in standards for RNA-seq experiments. Genome Res 21:1543
Kawamura K, Maurel MC (2017) Walking over 4 Gya: chemical evolution from photochemistry to mineral and organic chemistries leading to an RNA world. Orig Life Evol Biosph 47(3):281–296
Kim HJ, Ricardo A, Illangkoon HI, Kim MJ, Carrigan MA, Frye F, Benner SA (2011) Synthesis of carbohydrates in mineral-guided prebiotic cycles. J Am Chem Soc 133:9457
Lahav N (1993) The RNA-world and co-evolution hypotheses and the origin of life: implications, research strategies and perspectives. Orig Life Evol Biosph 23:329
Larralde R, Robertson MP, Miller SL (1995) Rates of decomposition of ribose and other sugars: implications for chemical evolution. Proc Natl Acad Sci USA 92:8158
Leal NA, Sukeda M, Benner SA (2006) Dynamic assembly of primers on nucleic acid templates. Nucleic Acids Res 34:4702
Ledergerber C, Dessimoz C (2011) Base-calling for next-generation sequencing platforms. Brief Bioinform 12:489
Leu K, Obermayer B, Rajamani S, Gerland U, Chen IA (2011) The prebiotic evolutionary advantage of transferring genetic information from RNA to DNA. Nucleic Acids Res 39:8135
Ma W (2014) The origin of life: a problem of history, chemistry, and evolution. Chem Biodivers 11:1998
Meiss G, Gast FU, Pingoud AM (1999) The DNA/RNA non-specific Serratia nuclease prefers double-stranded A-form nucleic acids as substrates. J Mol Biol 288:377
Michel A, Clermont O, Denamur E, Tenaillon O (2010) Bacteriophage PhiX174’s ecological niche and the flexibility of its Escherichia coli lipopolysaccharide receptor. Appl Environ Microbiol 76:7310
Nestle M, Roberts WK (1969) An extracellular nuclease from Serratia marcescens. I. Purification and some properties of the enzyme. J Biol Chem 244:5213
Orgel LE, Crick FH (1993) Anticipating an RNA world. Some past speculations on the origin of life: where are they today? FASEB J 7:238
Oró J (1960) Synthesis of adenine from ammonium cyanide. Biochem Biophys Res Commun 2:407
Rajamani S, Ichida JK, Antal T, Treco DA, Leu K, Nowak MA, Szostak JW, Chen IA (2010) Effect of stalling after mismatches on the error catastrophe in nonenzymatic nucleic acid replication. J Am Chem Soc 132:5880
Rich A (1962) On the problems of evolution and biochemical information transfer. In: Kasha M PB (ed) Horizons in Biochemistry. Academic Press, New York, pp 103–126
Robertson MP, Joyce GF (2012) The origins of the RNA world. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a003608
Sagi VN, Punna V, Hu F, Meher G, Krishnamurthy R (2012) Exploratory experiments on the chemistry of the “glyoxylate scenario”: formation of ketosugars from dihydroxyfumarate. J Am Chem Soc 134:3577
Saladino R, Crestini C, Ciciriello F, Costanzo G, Di Mauro E (2007) Formamide chemistry and the origin of informational polymers. Chem Biodivers 4:694
Sczepanski JT, Joyce GF (2014) A cross-chiral RNA polymerase ribozyme. Nature 515:440
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
Suzuki H, Zuo Y, Wang J, Zhang MQ, Malhotra A, Mayeda A (2006) Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 34:e63
Szabo L, Salzman J (2016) Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet 17:679
Tessier DC, Brousseau R, Vernet T (1986) Ligation of single-stranded oligodeoxyribonucleotides by T4 RNA ligase. Anal Biochem 158:171
Vaidya N, Manapat ML, Chen IA, Xulvi-Brunet R, Hayden EJ, Lehman N (2012) Spontaneous network formation among cooperative RNA replicators. Nature 491:72
Vincent HA, Deutscher MP (2006) Substrate recognition and catalysis by the exoribonuclease RNase R. J Biol Chem 281:29769
Weber AL (1992) Prebiotic sugar synthesis: hexose and hydroxy acid synthesis from glyceraldehyde catalyzed by iron(III) hydroxide oxide. J Mol Evol 35:1
Yeates JA, Nghe P, Lehman N (2017) Topological and thermodynamic factors that influence the evolution of small networks of catalytic RNA species. RNA 23(7):1088–1096
Acknowledgements
This project/publication was made possible through the support of a Grant from the John Templeton Foundation. The opinions expressed in this publication are those of the authors and do not necessarily reflect the views of the John Templeton Foundation.
Author information
Authors and Affiliations
Contributions
MRT and QT contributed equally. MRT, AM, DC, and GEF designed experimental work which was carried out by MRT, AM, and DC. QT designed and implemented the data analysis procedures and prepared tables and figures. MP and GEF conceived the project. GEF and MP contributed intellectually and assisted with manuscript preparation and proofreading. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tirumalai, M.R., Tran, Q., Paci, M. et al. Exploration of RNA Sequence Space in the Absence of a Replicase. J Mol Evol 86, 264–276 (2018). https://doi.org/10.1007/s00239-018-9846-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-018-9846-8