
Abstract
The AdpA protein from a streptomycin producer Streptomyces griseus is a founding member of the AdpA family of pleiotropic regulators, known to be ubiquitously present in streptomycetes. Functional genomic approaches revealed a huge number of AdpA targets, leading to the claim that the AdpA regulon is the largest one in bacteria. The expression of adpA is limited at the level of translation of the rare leucyl UUA codon. All known properties of AdpA regulators were discovered on a few streptomycete strains. There are open questions about the true abundance and diversity of AdpA across actinobacterial taxa (and beyond) and about the possible evolutionary forces that shape the AdpA orthologous group in Streptomyces. Here we show that, with respect to the TTA codon, streptomycete adpA is more diverse than has been previously thought, as the genes differ in presence/position of this codon. Reciprocal best hits to AdpA can be found in many actinobacterial orders, with a domain organization resembling that of the prototypical AdpA, but other configurations also exist. Diversifying positive selection was detected within the DNA-binding (AraC) domain in adpA of Streptomyces origin, most likely affecting residues enabling AdpA to recognize a degenerate operator. Sequence coding for putative glutamine amidotransferase (GATase-1) domain also shows signs of positive selection. The two-domain organization of AdpA most likely arose from a fusion of genes encoding separate GATase-1 and AraC domains. Indeed, we show that the AraC domain retains a biological function in the absence of the GATase-1 part. We suggest that acquisition of the regulatory role by TTA codon is a relatively recent event in the evolution of AdpA, which coincided with the rise of the Streptomycetales clade and, at present, is under relaxed selective constraints. Further experimental scrutiny of our findings is invited, which should provide new insights into the evolution and prospects for engineering of an AdpA-centered regulatory network.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The adpA gene was first shown in Streptomyces griseus to be a pleiotropic regulator influencing processes as diverse as morphological differentiation and streptomycin production (Ohnishi et al. 1999). In S. griseus the expression of adpA is dependent on small signaling molecule, γ-butyrolactone, referred to as A-factor. The latter is produced in a growth-dependent manner and turns on the transcription of adpA by binding to the A-factor receptor protein (ArpA), which has bound the adpA promoter, and dissociating the ArpA from the DNA (Yamazaki et al. 2004; Ohnishi et al. 2005; Horinouchi 2007). In S. coelicolor adpA expression it has been shown to be controlled by cis-antisense RNA (Šetinová et al. 2018). Recently the AdpA regulon was demonstrated to encompass from 100 to 500 genes, depending on streptomycete species. (Higo et al. 2011, 2012; Guyet et al. 2014). AdpA was also shown to regulate replication (Wolański et al. 2011, 2014). The adpA deletion in streptomycetes affects the aerial mycelium formation and, often, antibiotic production as well (Ohnishi et al. 1999; Nguyen et al. 2003; Takano et al. 2003; Zhao et al. 2007; Pan et al. 2009; Lopez-García et al. 2010; Xu et al. 2017). All available experimental and genomic data attest to the focal role of AdpA in Streptomyces biology.
In addition to being controlled by the hormone-like molecule, asRNA and the repressor protein ArpA, the AdpA expression is subject to a remarkable regulation at the level of translation of UUA codon within the adpA transcript. The UUA codon is the rarest one within GC-rich genomes of Actinobacteria (Chater and Chandra 2008; Zaburannyy et al. 2009). Gene bldA encodes the only tRNALeuUAA that is able to read the UUA codon efficiently, and its delayed occurrence during a cell cycle limits the expression of UUA-containing genes (Lawlor et al. 1987; Chater and Chandra 2008; Hackl and Bechthold 2015). In the latter, TTA codon(s) are usually located close to a start codon of a gene. It is thought to lower the cost of abortive translation until the tRNALeuUAA concentration in cells exceeds a certain threshold level (Chater and Chandra 2008). So far, a biased intragenic position of TTA was documented for Streptomyces genes (Ventura et al. 2007). The TTA-mediated regulation of gene expression is well documented in a number of streptomycetes, but the role for this codon has not been investigated beyond the genus Streptomyces. Only recently, the TTA codon-containing genes in Frankia were analyzed (Sen et al. 2012). Frankia genomes were shown to contain a higher number of TTA+ genes compared to most of the other actinomycetes. However, this codon does not seem to play a regulatory role in Frankia (Sen et al. 2012).
Aforementioned observations suggest that the AdpA protein and its codon sequence have undergone adaptive evolution in Streptomyces, although no attempts were undertaken to address this question directly. Our understanding of the abundance, structural, and functional diversity of members of the AdpA orthologous family within and beyond Streptomyces is currently based on rather modest number of genomes (Chandra and Chater 2014), as compared to what is currently available. The present work is an attempt to address aforementioned questions via a bioinformatics analysis of available genomic data. We demonstrate that the AdpA orthologous group is diverse (regarding its genetic architecture) and abundant within Actinobacteria; significant structural diversity was found even for the best studied (and previously thought to be homogeneous) group of streptomycete adpA sequences. Several AdpA-like sequences were identified beyond actinobacterial taxa, hinting at the ancient evolutionary origin of this fascinating group of regulators. Here, for the first time, through the analysis of AdpA orthologous group of Streptomyces origin we present evidence that certain sites within DNA-binding helix-turn-helix motif are subject to positive selection. In contrast to previous reports, we revealed no signals of selection on TTA codon. Finally, we suggest that a typical two-domain AdpA arose as a result of a fusion of genes that originally encoded these domains.
Materials and Methods
Gene Sequences
A set of all probable AdpA orthologs was collected from GenBank using a reciprocal best hit (RBH) strategy based on an E value threshold of 10 and BLOSUM62 similarity matrix (Moreno-Hagelsieb and Latimer 2008). S. griseus AdpAgr (Ohnishi et al. 2005), S. coelicolor AdpAsc (Wolański et al. 2011), and S. clavuligerus AdpAcl (Lopez-García et al. 2010) were initially tested as BLASTP queries. When necessary, manual annotation of genes was conducted by examining the database hits of BLASTP with KEGG (Tanabe and Kanehisa 2012) and CDD (Marchler-Bauer et al. 2011) databases, and the results of FRAMEPLOT analysis (Ishikawa and Hotta 1999).
The codon sequences of identified RBHs were aligned using the ProGraphMSA program (Szalkowski 2012) with options -m --codon -f -w -F, for amino acid alignment options were -f -m -A. The codon alignment was further refined by deleting columns in alignment where gaps constituted more than 90% of the content. Alignments of genes that contain ‘classical’ TTA codons and those which do not were extracted from the initial alignment.
Phylogenetic Analysis
A phylogenetic tree was inferred by maximum likelihood using CodonPhyML (Gil et al. 2013). The following models were used and compared: codon models M0 and M0+Г (with among-site rate variation), and amino acid model LG+Г+F (Le and Gascuel 2008) with amino acid frequencies estimated empirically from data. Model M0 assumes constant selection pressure on the protein across the sites, as measured by ω = dN/dS ratio of non-synonymous to synonymous substitution rates (Goldman and Yang 1994; Nielsen and Yang 1998). When sequences evolve neutrally, under no selection, it is considered that ω = 1, whereas ω < 1 indicates purifying selection, and ω > 1 positive selection.
Selection Pressure Analysis
To estimate ω, random-sites models (REL) which allow for across-site variation in dN and dS (Pond and Muse 2005) were applied using the HyPhy software package (Kosakovsky Pond et al. 2005) (version 2.2.4). The phylogenetic trees constructed under the best fitting model in CodonPhyML analysis were used for the analysis of selection pressure. Most REL models pre-specify a number of ω classes and fit the values for each class as well as the fraction of sites belonging to each class (Nielsen and Yang 1998; Yang et al. 2000; Kosakovsky Pond and Frost 2005). Probabilities of each individual site to fall into one of the classes are inferred via an empirical Bayes approach. Traditional codon models with ω parameter estimate dN and dS as a ratio and assume a constant dS (Nielsen and Yang 1998; Suzuki and Gojobori 1999; Yang et al. 2000; Yang and Bielawski 2000). However, in some cases, synonymous substitutions could be under selection (Sharp et al. 1995). To account for the effect of variation of synonymous substitution rates across sites, we performed codon selection analysis using the dNdSRateAnalysis program from HyPhy. The ‘Nonsynonymous’ model assumes a constant dS (= 1), but samples dN values from a given rate distribution. The ‘Dual’ model estimates dS and dN independently, sampling both from a given rate distribution (Pond and Muse 2005). In this study, both models were fitted using the MG94 × REV core rate matrix with rates from the general discrete distribution (GDD) and three classes. All models were fitted several times using randomized initial values to find the global optimum. To test for site-to-site variation in dS across both AdpA datasets, Dual and Non-synonymous models were compared using a likelihood ratio test (LRT).
To detect amino acid sites under positive selection, we used the Bayesian approach (Pond and Muse 2005) as implemented in dNdSResultProcessor of the HyPhy software. We selected every site that had a Bayes factor (BF) for positive selection greater than 100 (Kass and Raftery 1995).
Plasmid Construction and Complementation Studies for Functional Experiments
The coding sequence of Streptomyces albus J1074 adpA orthologue (XNR_4181) together with its putative ribosome-binding site was amplified using primers xnr4181_XbaIup and xnr4181_EcoRIrp yielding 1250 bp amplicon (these and other primer sequences mentioned throughout the work are listed in Table 1). This PCR product was digested with restriction endonucleases XbaI and EcoRI, and cloned into respective sites of integrative, cumate-inducible expression vector pGCymRP21 (Horbal et al. 2014) to give pGC4181. The adpA genes from S. coelicolor M145 (adpA sc ), S. clavuligerus ATCC27064 (adpA cl ), S. ghanaensis ATCC14672 (adpA gh ), as well as XNR_1761 gene, were cloned likewise using appropriate primer pairs listed in Table 1, thus leading to plasmids pGCSCO, pGCSCLA, pOOB103c, and pGC1761, respectively. Part of the XNR_4181 encoding the AraC–HTH DNA-binding domain (700–1245 bp) was amplified with xnr4181araC_up and xnr4181_EcoRIrp primers yielding 601 bp amplicon. The forward primer contained artificially introduced start codon and the ribosome-binding site. The PCR product was digested with XbaI and EcoRI, and cloned into respective sites of pGCymRP21 giving pGC4181d. The same approach was used to clone coding sequences of DNA-binding domains of adpA sc , adpA cl , and adpA gh , generating pGCSCOd, pGCSCLAd, pGCSGHd.
All plasmids (together with pGCymRP21) were transferred conjugally from E. coli ET12567 (pUZ8002) into S. coelicolor M851 (∆adpA mutant; Takano et al. 2003), as described elsewhere (Ostash et al. 2015). Apramycin-resistant transconjugants were verified with PCR, using primers specific to the vector marker gene aac3(IV). The transconjugants and control strains (M851 pGCymRP21+, its initial M145 strain) were grown for 3–5 days on R5 agar plates (Kieser et al. 2000) supplemented with 50 µM cumate to induce the expression of the cloned genes.
Results and Discussion
Abundance and Diversity of AdpA Orthologous Family Within Phylum Actinobacteria
We explored amino acid sequences of three experimentally studied AdpA proteins—AdpAgr, AdpAsc, and AdpAcl—as BLASTP queries to identify putative orthologs across Actinobacteria. Our initial searches (in 2013) yielded 323 unique protein sequences in total. The number of reciprocal best hits (RBHs) retrieved for each of the three queries ranged from 114 to 138 and to 323 (Fig. 1), respectively, and included representatives of most actinobacterial orders (Streptomycetales, Corynebacteriales, Bifidobacteriales etc) (Fig. S1, Electronic Supplementary Materials; ESM). For example, AdpAcl has led to 177 RBHs unidentified when using either AdpAgr or AdpAsc as queries. (Here and further down in this paper we label the 323 AdpA RBHs identified by AdpAcl as “initial dataset”).

Venn diagram for orthologs detected against GenBank database using AdpA queries from three Streptomyces species shown on the figure. BLAST searches were carried out against GenBank for all proteins of 323 species using BLOSUM62 matrices at default E value cut-off
As mentioned above, AdpA consists of ThiJ/PfpI/DJ-1-like (also known as GATase-1) dimerization domain and an AraC/XylS-type DNA-binding domain at its N- and C-termini, respectively (Ohnishi et al. 2005). Searching across the Actinobacteria class, we discovered that certain species have no full-length AdpA RBHs. Rather, orthology was revealed between the query and proteins featuring either single AraC or GATase-1 domains. This prompted us to analyze a more extensive set of actinobacterial genomes in order to study a diversity of domain organization of AdpA regulators within the class. As a source of genomes we used the PATRIC database (Wattam et al. 2014) and the full-length AdpAcl, GATase-1cl, or AraCcl domains of AdpAcl as the queries. Our updated search yielded 1394 unique RBHs, which spanned 17 orders of class Actinobacteria class (Fig. 2). With respect to available genome sequences, streptomycetes constitute the most deeply covered single genus within the phylum Actinobacteria, with the largest (390) set of probable AdpA orthologs (Fig. 2).

The unrooted tree of Actinobacteria and Cyanobacteria phyla reconstructed using PhyloPhlAn phylogenetic pipeline. Collapsed clades (at the level of orders) are color-highlighted and labeled with the order name. The latter is followed by round brackets showing two numbers: first one is for quantity of retrieved AdpA orthologs, and second one—number of genomes where no AdpAcl ortholog was found. The types of domain organization of AdpA orthologs are shown next to corresponding orders. Pink rectangular denotes GATse domain; violet hexagon—AraC HTH domain; light pink rectangular—truncated GATse domain; blue octagon—arabinose-binding domain of AraC transcription regulator; gray oval—domain of unknown function. (Color figure online)
In terms of domain organization, the most diverse AdpA RBHs were revealed in orders Corynebacteriales, Bifidobacteriales, and Micrococcales (Fig. 2). For example, in Corynebacteriales, the 3-Mbp genome of Corynebacterium freneyi DNF00450 carries one gene HMPREF1650_RS07820-encoding ThiJ/PfpI protein orthologous to AdpAcl/GATase-1cl, and no genes for AraC family regulators. Corynebacterium humireducens NBRC 106098 (2.68 Mbp) encodes only three AraC-family transcriptional regulators (one of them—AdpA RBH) and no GATase-1 proteins. Other orders usually possessed AdpA RBHs having typical two-domain architecture. Our results suggest that adpA could originate from the gene fusion. Indeed, pairs of adjacent genes encoding ThiJ- and AraC domain-containing proteins are found in numerous bacterial genomes. Particularly, such gene pairs (paralogous to adpA) were found in S. coelicolor (SCO6746/6747), S. griseus (SGR_981/982), Saccharopolyspora erythraea (SACE_6420/SACE_6421, SACE_4268/4269), Actinosynnema mirum (Amir_4315/4316), Salinispora arenicola (Sare_0089/0090), Kribbella flavida (Kfla_5799/Kfla_5798), and even in chloroflexi bacterium Herpetosiphon aurantiacus (Haur_2399/Haur_2400).
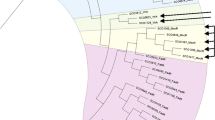
Streptomycete adpA genes fell into several groups with regard to the position of TTA codon (Fig. 3). The absolute majority had TTA codon in the ‘classical’ position, as it is in S. griseus adpA gene, between GATase-1 and HTH domains. In a few adpA orthologs TTA codon was found within the GATase-1 domain, closer to the start of the gene; in S. sp. HmicA12 TTA was near the beginning of AraC domain; and in S. neyagawaensis TTA codon was located close to the stop codon of adpA. Finally, we identified adpA genes (from S. sp. C, S. sp. Mg1, S. sp. ScaeMP-e10, and S. lysosuperificus ATCC 31396) that have no TTA at all.

Amino acid alignment of streptomycetes AdpA proteins from S. griseus, S. somaliensis, S. sp. HmicA12, S. lysosuperificus, and S. neyagawaensis showing the position of the TTA codon-encoded leucine amino acid (yellow box) along with the position of the HTH motifs (box) of DNA-binding domain. Conserved (asterisk) and homologous (colon) amino acids are marked underneath. (Color figure online)
An average actinobacterial AdpA protein had the length of 320–345 aa. The largest protein of 705 aa was from Kitasatospora cheerisanensis KCTC 2395; the increased size of the protein appears to result from incorporation of low complexity sequences of unknown function at the C- and N-termini (Fig. S3, ESM). As compared to the other actinobacterial AdpA RBHs, streptomycete AdpA proteins had a longer C-terminal part, and its length was within the 252–496 aa range.
In our first dataset we revealed probable AdpA orthologs in 27 mycobacterial species. However, Mycobacterium tuberculosis H37Rv appeared to lack full-length two-domain AdpA counterpart. The only strain from Mycobacterium tuberculosis complex that returned AdpA RBH—MarA2—was M. tuberculosis 401416. Using the latter protein as a query against H37Rv proteome, protein Rv1931c (AdpA RBH) was identified as a MarA2 ortholog. Interestingly, its gene is adjacent to the one encoding a ThiJ-like protein Rv1930c. Although Rv1931 was not among RBHs for AdpAcl or any other streptomycete AdpA, we decided to take a closer look at mycobacterial AdpA-like proteins given the medical importance of mycobacteria and one report (Frota et al. 2004) showing that rv1931c plays a role in the virulence of M. tuberculosis.
We constructed position-specific weight matrix from known AdpA operator sequences (Fig. S4, ESM) and searched for the presence of such operators in the promoters of genes for mycobacterial AdpA orthologs. PREDetector (Hiard et al. 2007) and MEME (Bailey et al. 2009) identified putative AdpA operators upstream of 18 mycobacterial genes for AdpA RBHs. These operators had E values (at least 10−10) and p values (see Table S1 for examples, ESM) low enough to consider discovered motifs significant. Also, we revealed a high degree of gene conservation around adpA in these Mycobacterium species (Fig. S5, ESM).
We carried out genome-wide screening of mycobacterial genomes for AdpA operators using PreDetector. The operators have been identified in the promoters of whiB genes from genomes of M. smegmatis JS623, M. rhodesiae NBB3, M. abscessus subsp. bolletii 50594, and M. tuberculosis H37Rv (Table S2, ESM). Some of the WhiB-encoding genes are regulated by AdpA in S. griseus (Higo et al. 2012). WhiB proteins are well known for their diverse roles in actinobacterial morphogenesis, cell division, virulence, primary and secondary metabolism, and intrinsic antibiotic resistance. Our findings suggest that AdpA-like regulators in mycobacteria may play an important function in regulating different cell processes. It is of interest to investigate whether AdpA-like proteins can be pleiotropic regulators of diverse cellular processes in mycobacteria, or whether such regulatory model has evolved only in certain genera, such as Streptomyces.
Distribution of Probable AdpA Orthologs Beyond Phylum Actinobacteria
We decided to analyze the closest neighbors of Actinobacteria on the bacterial tree. Transcriptional regulators of AraC family constitute one of the most abundant functional classes encoded in genomes from all domains of life (Aziz et al. 2010). However, AraC domain is not listed as having one of “the 100 most ubiquitous functional roles in genomes.” Abundance without ubiquity is believed to be an indicator of adaptive, organism-specific, or habitat-specific functionality. Different approaches towards phylogenetic reconstruction (Ciccarelli et al. 2006; Jolley et al. 2012; Segata et al. 2013) suggest that cyano- and actinobacteria are one of the most deeply branched phyla that share the closest last common ancestor. According to literature-based evolutionary timescale server TimeTree (http://www.timetree.org/), cyano- and actinobacteria diverged around 2850 Mya, which is less than divergence time between actinobacteria and Firmicutes, such as Bacillus (3070 Mya). Indeed, using the PhyloPhlAn method (Segata et al. 2013), we confirmed previous results for our extended set of actinobacterial genomes (Fig. 2). PhyloPhlAn is a computational pipeline for reconstructing highly accurate and resolved phylogenetic trees based on whole-genome sequence information. This method assigns microbial phylogeny and putative taxonomy using > 400 proteins optimized from among 3737 genomes. Also the method measures the sequence diversity of all clades, classifies genomes from deep-branching candidate divisions through closely related subspecies, and improves consistency between phylogenetic and taxonomic groupings. We used PhyloPhlAn because it implements insertion operations, so we could incorporate new genomes of interest into pre-built microbial tree of life.
After an analysis of 198 genomes, we identified nine AdpAcl RBHs within cyanobacterial orders of Nostocales, Synechococcales, and Oscillatoriales. Interestingly, only three of them are full-length orthologs with two-domain organization: those were from Acaryochloris sp. CCMEE 5410, Oscillatoriales cyanobacterium ‘hensonii’ and Hassallia byssoidea VB512170. Others contained only C-terminal AraC domain. Thus, our genome mining efforts yielded a great diversity of genetic architectures within a group of AdpA-like proteins of Actinobacteria and Cyanobacteria, as summarized in Fig. 2.
Estimation of Selective Pressure on the Protein
To infer adpA codon positions under selection on the protein level, we choose to examine the initial (smaller) dataset because it contains the most extensively studied and validated sequences. We analyzed the entire 323 adpA gene dataset, only TTA+ genes (91; mostly Streptomyces), and 232 TTA− genes. Here TTA+ genes were those having TTA codon within the interdomain region (as in adpA cl ); the TTA− dataset consisted of genes that either lack TTA or carry it in positions other than in adpA cl .
After performing the analyses with CodonPhyML program, we compared the fit of three different models for three datasets using AIC (Akaike 1973). Table 2 indicates that the codon model M0+Γ gives a better fit to adpA datasets compared to other models. Thus, phylogenetic trees constructed under M0+Γ model were used for further selection pressure analysis. We evaluated the selection pressure at the protein level and examined the effect of among-site variation in dS on the estimation of ω in our datasets. For all three datasets the site-to-site variation in dS was significant (p value ≪ 0.01; Table 3).
For the group of 91 TTA+adpA genes, highest synonymous substitutions rates (sites with fast inferred dS rate categories) can be observed at the C-terminus downstream of a DNA-binding motif (Fig. 4). Two small regions with slowest a posteriori dS rates locate in GATase domain and another two in the interdomain part and HTH domain of the protein. Interestingly, one of these regions contains TTA codon, whose adjacent positions correspond to the inferred lowest dS rate categories. At the same time, three regions with sites having highest values of dN/dS can be found in GATase domain and one in HTH domain (Fig. 4). We identified 93 positively selected sites in this dataset, 66 of them were located in the region involved in dimerization and 21—in the DNA-binding motif. Two amino acids under positive selection were located next to TTA codons:proline downstream L224 (‘classical’ TTA codon in S. griseus), and glycine upstream of Leu (which is encoded by TTA codon in S. aurantiacus JA 4570, S. lysosuperificus ATCC 31396, and S. violaceusniger SPC6). However, according to our analysis TTA codon is under purifying selection.

Posterior dS and ω at each amino acid site across the adpA gene for dual rate variation model: a TTA+ set; b TTA− set; c all AdpA set. TTA codon is shown as the red box on the x axis of the plot. A schematic of AdpA domain structure is shown above the plot. (Color figure online)
In the DNA-binding motif sites under positive selection included the following amino acid residues: A255, R261, R262, R267, R269, A275, L277, R320 (Fig. S6, ESM). From the crystal structure of AdpA DNA-binding domain it could be inferred that the guanidinium groups of Arg-261, Arg-267, and Arg-269 form salt bridges with the phosphate groups of C4′, T1, and C6′ nucleotides, respectively, of the 10-bp consensus AdpA-binding sequence (5′-TGGCSNGWWY; S:G or C; W:A T; Y:T or C; and N:any nucleotide). C4′ is one of the most highly conserved nucleotides in the AdpA operator. G2 is recognized by R262—the residue essential for the recognition of the consensus sequence (Yao et al. 2013). The Cβ atom of Ala-275, via van der Waals forces, interacts with the 2′-deoxyriboses of C5′ nucleotide. In the HTH2 motif, the side-chain guanidinium of Arg-320 forms electrostatic interactions with the phosphate groups of C10 nucleotide (Yao et al. 2013). Thus, the ability of AdpA to recognize degenerate sequence appears to be due to the ongoing evolutionary pressure for innovation that is taking place in the Streptomyces clade.
In the 232 (TTA−) and 323 (all AdpA) datasets no sites under positive selection were identified. Sites with elevated dS and ω were spread evenly along the gene length in both datasets (Fig. 4). Overall, more sites with elevated ω values were observed in the TTA+ set compared to the 232 and 323 sets.
Functional Significance of AraC Domain: Experimental Test
The aforementioned genomic analysis pointed to the possibility that two-domain architecture of AdpA (as well as other HTH_18 superfamily proteins) could be a result of fortuitous gene fusion. We tested the hypothesis that isolated AraC domain of AdpA should still exhibit biological function in the absence of GATase-1. In the two-domain organization of AdpA, AraC domain is thought to play a principal role in transcriptional regulation, whereas GATase-1 mediates protein dimerization (Yamazaki et al. 2004; Kato et al. 2005). The importance (structural or functional) of dimerization is not supported experimentally.
We cloned the entire sequences of adpA-encoding genes from Streptomyces albus J1074 (XNR_4181), S. ghanaensis ATCC14672 (adpA gh ), S. clavuligerus ATCC7064 (adpA cl ), S. coelicolor M145 (adpA sc ) as well as their AraC domain-encoding segments (XNR_4181_dbd, adpA gh _dbd, adpA cl _dbd, adpA sc _dbd) into cumate-inducible expression vector pGCymRP21. The resulting plasmids, referred to as pGC4181, pOOB103c, pGCSCLA, pGCSCO, pGC4181d, pGCSGHd, pGCSCLAd, and pGCSCOd (see Materials and Methods), were introduced into adpA sc knockout strain S. coelicolor M851 (Takano et al. 2003). We have also generated and transferred into M851 plasmid pGC1761, which is pGCymRP21 carrying XNR_1761, the closest paralog of XNR_4181 in S. albus genome. Xnr_1761 also belongs to HTH_18 superfamily of transcriptional regulators, possesses the same domain architecture as AdpA proteins do, and shares 60% aa sequence identity with Xnr_4181. It was interesting to see whether XNR_1761 is able to complement Bld phenotype of S. coelicolor M851.
As could be seen from Fig. 5, M851 pGCymRP21+ strains failed to produce spores (a hallmark of adpA deficiency) on rich R5 medium, while introduction of full XNR_4181, adpA gh , adpA cl , and adpA sc restored spore formation to M851. The introduction of XNR_4181_dbd, adpA gh _dbd, adpA cl _dbd, adpA sc _dbd also led to sporulation of M851. In contrast, introduction of XNR_1761 did not influence M851 morphogenesis, suggesting that effects of adpA genes and their DNA-binding domains on the adpA mutant morphology are rather specific.

Three-day lawns of control [S. coelicolor M145 (1) and S. coelicolor M851 pGCymRP21+(2)] and complemented S. coelicolor strains [S. coelicolor M851 pGCSCOd+ (3), pGCSCO+(4), pGCSCLAd+ (5), pGCSCLA+ (6), pGC4181d+ (7), pGC4181+ (8), pGCSGHd+ (9), pOOB103c+ (10), and pGC1761+ (11)] on R5 agar plates. The adpA mutant-carrying pGCymRP21 (M851 pGCymRP21+) did not produce aerial mycelium. Formation of aerial mycelium and spores could be observed for all complemented strains except S. coelicolor M851 pGC1761+
Discussion
In this work we combined computational and experimental approaches to investigate the diversity, evolution, and function of AdpA family regulatory proteins in Streptomyces and class Actinobacteria. The obtained data expand and complement our understanding of this family, which was previously based on a few model streptomycetes.
It remained an open question whether AdpA, as a two-domain protein of high pleiotropicity, was “invented” by Streptomyces, or whether it has originated before the Streptomyces branched off the actinobacterial phylogenetic tree. Chandra and Chater (2014) addressed this issue on the basis of 100 actinobacterial genomes. Their work suggested that AdpA orthologs are ubiquitously present in Streptomyces, and corresponding adpA genes carry a TTA codon in the “classical” region. Importantly, probable AdpA orthologs were sparsely found in some other actinobacterial taxa, suggesting AdpA origin before the speciation of Streptomyces. After mining 2215 actinobacterial genomes, we came to similar conclusions, albeit with a few important amendments. The first one concerns the Streptomyces clade (see Fig. 2). The latter invariably features AdpA orthologs, yet there is diversity with regard to codon TTA. In addition to the “classic” location of TTA between the two domains, we found a few “TTA-free” adpA, as well as orthologs in which this codon is located within the GATse domain or close to the stop codon. The second amendment concerns the presence of AdpA orthologs on a substantially larger phylogenetic scale. Here we show that AdpAcl RBHs exist in many actinobacteria and GATase-1-AraC two-domain configuration prevails. Other variants (such as single GATase-1/AraC domains, or presence of other domains) could be found, mostly in orders Corynebacteriales and Bifidobacteriales. We also identified AdpAcl orthologs in a few cyanobacteria. In our work ortholog identification was based on BLASTP RBHs assuming that protein domains are units of orthology (Gabaldon and Koonin 2013). These criteria are less strict as compared to those used by Chandra and Chater (2014), and so might lead to ortholog misidentification in case of multiparalogous protein families, such as AdpA. Nevertheless, our approach agrees with previous estimates for a number of actinobacterial taxa; greater abundance and diversity of AdpAs could be a natural result of deeper sampling of all taxonomic groups. We believe that our dataset outlines a general picture of AdpA counterparts across long evolutionary distances, creating a starting point for the identification of genuine orthologs beyond Streptomyces and their functional characterization.
The ability to recognize a degenerate operator is the most salient trait of the AdpA function in Streptomycetes. In contrast, nothing is known about the mechanism and the role of probable AdpA orthologues in non-streptomycete species. Here we reveal that Streptomyces AdpA is under positive selection. Positively selected sites are grouped in regions involved in DNA-binding and protein dimerization. The precise location of codon sites under selection could not be identified reliably within other adpA genes. Thus, AdpA, as it is known from the studies of a few model cases, is most likely the Streptomyces-specific innovation, and this knowledge could not be directly extrapolated onto the other genera. Our recent studies of the AdpA-like protein from uncommon actinomycete Actinoplanes teichomyceticus support this suggestion (Ostash et al. 2015).
Our work reaffirms previous finding that only streptomycete adpA genes are associated with the rare TTA codon; nevertheless, a few TTA-free adpA sequences from Streptomyces were revealed. Although previously implicitly assumed (Chater and Chandra 2008; Chandra and Chater 2014), our data revealed no evidence of strong selection for TTA codon in adpA. The TTA-containing genes in streptomycetes are characterized by an extreme positional bias of TTA codon towards the start of genes (Zaburannyy et al. 2009). This is speculated to decrease the energy waste that is proportional to the number of codons that will be translated until the UUA is encountered (Fuglsang 2005). In Streptomyces adpA genes TTA is usually located in the middle of coding sequences, clearly violating the above-mentioned rule. However, such a TTA codon placement is easier to understand if two-domain AdpA arose from gene fusion; in this case TTA would be at the beginning of AraC-encoding gene. Although aforementioned explanation looks theoretically plausible, it is not supported by available evidence. First, there are no separate GATase-1 or AraC-encoding genes across actinobacteria carrying TTA at C- or N-termini, respectively; second, we have found several streptomycete TTA-free adpA as well as several adpA carrying TTA outside of linker sequence; third, there are no signs of positive selection on codon position where TTA is usually located. It is therefore likely that TTA emerged in adpA after two-domain-encoding sequence has been formed, although evolutionary and functional reasons behind this codon remain obscure.
The gene fusion scenario of AdpA evolution is supported by the presence of the adjacent GATase-1 and AraC-encoding genes (adpA paralogues) in actinomycete genomes and heterologous complementation of adpA mutant S. coelicolor M851 with AraC domains derived from native and several heterologous AdpA-encoding genes. In the latter experiment we tested the ability of the isolated AraC domain to complement the most obvious defect of adpA sc mutant, e.g., its inability to erect aerial hyphae. The AdpA is proposed to bind its operator sequences in either monovalent or bivalent mode, namely either one or two AraC domains of the AdpA dimer contact the operators (Yamazaki et al. 2004). Hence a separate AraC domain of AdpA would still be able to function in a monovalent fashion; only targets requiring bivalent AdpA binding will be affected. At present we assume that GATase-1 domain is not absolutely required for AdpA to control morphogenesis. Notably, none of the adpA-/adpA_dbd-expressing constructs fully complemented the loss of an actinorhodin production by the S. coelicolor ∆adpA (M851) strain. Although reasons for that can be different for different genes, we observed that vector-expressed cumate repressor CymR affected actinorhodin biosynthesis. Also, recent report (Xu et al. 2017) showed that full-length AdpA could interfere with antibiotic production. It will be of interest to examine the effect of AraC domain on the other traits known to be influenced by the full-length AdpA (Higo et al. 2012). This should deepen the understanding of the dimerization domain in an overall function of AdpA proteins, which, in turn, will further illuminate the evolutionary trajectory of this fascinating family of regulators.
References
Akaike H (1973) Information theory and an extension of the maximum likelihood principle. In: Petrov BN, Csaki F (eds) Second international symposium on information theory. Akademiai Kiado, Budapest, pp 267–281
Aziz RK, Breitbart M, Edwards RA (2010) Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Res 38(13):4207–4217
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME suite: tools for motif discovery and searching. Nucleic Acids Res 37(Suppl 2):202–208
Chandra G, Chater KF (2014) Developmental biology of Streptomyces from the perspective of 100 actinobacterial genome sequences. FEMS Microbiol Rev 38(3):345–379
Chater KF, Chandra G (2008) The use of the rare UUA codon to define “expression space” for genes involved in secondary metabolism, development and environmental adaptation in Streptomyces. J Microbiol 46(1):1–11
Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B, Bork P (2006) Toward automatic reconstruction of a highly resolved tree of life. Science 311(5765):1283–1287
Frota CC, Papavinasasundaram KG, Davis EO, Colston MJ (2004) The AraC family transcriptional regulator Rv1931c plays a role in the virulence of Mycobacterium tuberculosis. Infect Immun 72(9):5483–5486
Fuglsang A (2005) Intragenic position of UUA codons in streptomycetes. Microbiology 151(Pt 10):3150–3152
Gabaldon T, Koonin EV (2013) Functional and evolutionary implications of gene orthology. Nat Rev Genet 14(5):360–366
Gil M, Zanetti MS, Zoller S, Anisimova M (2013) CodonPhyML: fast maximum likelihood phylogeny estimation under codon substitution models. Mol Biol Evol 30(6):1270–1280
Goldman N, Yang Z (1994) A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol 11:725–736
Guyet A, Benaroudj N, Proux C, Gominet M, Coppée JY, Mazodier P (2014) Identified members of the Streptomyces lividans AdpA regulon involved in differentiation and secondary metabolism. BMC Microbiol 14:81
Hackl S, Bechthold A (2015) The Gene bldA, a regulator of morphological differentiation and antibiotic production in Streptomyces. Arch Pharm 348(7):455–462
Hiard S, Maree R, Colson S, Hoskisson PA, Titgemeyer F, van Wezel GP, Joris B, Wehenkel L, Rigali S (2007) PREDetector: a new tool to identify regulatory elements in bacterial genomes. Biochem Biophys Res Commun 357(4):861–864
Higo A, Horinouchi S, Ohnishi Y (2011) Strict regulation of morphological differentiation and secondary metabolism by a positive feedback loop between two global regulators AdpA and BldA in Streptomyces griseus. Mol Microbiol 81(6):1607–1622
Higo A, Hara H, Horinouchi S, Ohnishi Y (2012) Genome-wide distribution of AdpA, a global regulator for secondary metabolism and morphological differentiation in streptomyces, revealed the extent and complexity of the AdpA regulatory network. DNA Res 19(3):259–273
Horbal L, Fedorenko V, Luzhetskyy A (2014) Novel and tightly regulated resorcinol and cumate-inducible expression systems for Streptomyces and other actinobacteria. Appl Microbiol Biotechnol 98(20):8641–8655
Horinouchi S (2007) Mining and polishing of the treasure trove in the bacterial genus streptomyces. Biosci Biotechnol Biochem 71(2):283–299
Ishikawa J, Hotta K (1999) FramePlot: a new implementation of the Frame analysis for predicting protein-coding regions in bacterial DNA with a high G+C content. FEMS Microbiol Rev 174(2):251–253
Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, Wimalarathna H, Harrison OB, Sheppard SK, Cody AJ, Maiden MCJ (2012) Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology 158(4):1005–1015
Kass RE, Raftery AE (1995) Bayes factors. J Am Stat Assoc 90(430):773–795
Kato JY, Ohnishi Y, Horinouchi S (2005) Autorepression of AdpA of the AraC/XylS family, a key transcriptional activator in the A-factor regulatory cascade in Streptomyces griseus. J Mol Biol 350(1):12–26
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical streptomyces genetics. John Innes Foundation, Norwich
Kosakovsky Pond SL, Frost SDW (2005) Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22(5):1208–1222
Kosakovsky Pond SL, Frost SDW, Muse SV (2005) HyPhy: hypothesis testing using phylogenies. Bioinformatics 21(5):676–679
Lawlor EJ, Baylis HA, Chater KF (1987) Pleiotropic morphological and antibiotic deficiencies result from mutations in a gene encoding a tRNA-like product in Streptomyces coelicolor A3(2). Genes Dev 1(10):1305–1310
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320
López-García MT, Santamarta I, Liras P (2010) Morphological differentiation and clavulanic acid formation are affected in a Streptomyces clavuligerus adpA-deleted mutant. Microbiology 156(8):2354–2365
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH (2011) CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res 39(Suppl 1):225–229
Moreno-Hagelsieb G, Latimer K (2008) Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 24(3):319–324
Nguyen KT, Tenor J, Stettler H, Nguyen LT, Nguyen LD, Thompson CJ (2003) Colonial differentiation in Streptomyces coelicolor depends on translation of a specific codon within the adpA Gene. J Bacteriol 185(24):7291–7296
Nielsen R, Yang Z (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148(3):929–936
Ohnishi Y, Kameyama S, Onaka H, Horinouchi S (1999) The A-factor regulatory cascade leading to streptomycin biosynthesis in Streptomyces griseus: identification of a target gene of the A-factor receptor. Mol Microbiol 34(1):102–111
Ohnishi Y, Yamazaki H, Kato JY, Tomono A, Horinouchi S (2005) AdpA, a central transcriptional regulator in the A-factor regulatory cascade that leads to morphological development and secondary metabolism in Streptomyces griseus. Biosci Biotechnol Biochem 69(3):431–439
Ostash B, Yushchuk O, Tistechok S, Mutenko H, Horbal L, Muryn A, Dacyuk Y, Kalinowski J, Luzhetskyy A, Fedorenko V (2015) The adpA-like regulatory gene from Actinoplanes teichomyceticus: in silico analysis and heterologous expression. World J Microbiol Biotechnol 31(8):1297–1301
Pan Y, Liu G, Yang H, Tian Y, Tan H (2009) The pleiotropic regulator AdpA-L directly controls the pathway-specific activator of nikkomycin biosynthesis in Streptomyces ansochromogenes. Mol Microbiol 72(3):710–723
Pond SK, Muse SV (2005) Site-to-site variation of synonymous substitution rates. Mol Biol Evol 22(12):2375–2385
Rodrigue N, Lartillot N, Philippe H (2008) Bayesian comparisons of codon substitution models. Genetics 180(3):1579–1591
Segata N, Börnigen D, Morgan XC, Huttenhower C (2013) PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat Commun 4:2304
Sen A, Thakur S, Bothra AK, Sur S, Tisa LS (2012) Identification of TTA codon containing genes in Frankia and exploration of the role of tRNA in regulating these genes. Arch Microbiol 194(1):35–45
Šetinová D, Šmídová K, Pohl P, Musić I, Bobek J (2018) RNase III-binding-mRNAs revealed novel complementary transcripts in Streptomyces. Front Microbiol 8:2693
Sharp PM, Averof M, Lloyd AT, Matassi G, Peden JF (1995) DNA sequence evolution: the sounds of silence. Philos Trans R Soc Lond B 349(1329):241–247
Suzuki Y, Gojobori T (1999) A method for detecting positive selection at single amino acid sites. Mol Biol Evol 16(10):1315–1328
Szalkowski AM (2012) Fast and robust multiple sequence alignment with phylogeny-aware gap placement. BMC Bioinform 13:129
Takano E, Tao M, Long F, Bibb MJ, Wang L, Li W, Buttner MJ, Bibb MJ, Deng ZX, Chater KF (2003) A rare leucine codon in adpA is implicated in the morphological defect of bldA mutants of Streptomyces coelicolor. Mol Microbiol 50(2):475–486
Tanabe M, Kanehisa M (2012) Using the KEGG database resource. Curr Protoc Bioinform 38:1.12:1.12.1–1.12.43
Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, van Sinderen D (2007) Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev 71(3):495–548
Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, Gillespie JJ, Gough R, Hix D, Kenyon R, Machi D, Mao C, Nordberg EK, Olson R, Overbeek R, Pusch GD, Shukla M, Schulman J, Stevens RL, Sullivan DE, Vonstein V, Warren A, Will R, Wilson MJ, Yoo HS, Zhang C, Zhang Y, Sobral BW (2014) PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res 42(D1):581–591
Wolański M, Donczew R, Kois-Ostrowska A, Masiewicz P, Jakimowicz D, Zakrzewska-Czerwińska J (2011) The level of AdpA directly affects expression of developmental genes in Streptomyces coelicolor. J Bacteriol 193(22):6358–6365
Wolański M, Jakimowicz D, Zakrzewska-Czerwińska J (2014) Fifty years after the replicon hypothesis: cell-specific master regulators as new players in chromosome replication control. J Bacteriol 196(16):2901–2911
Xu J, Zhang J, Zhuo J, Li Y, Tian Y, Tan H (2017) Activation and molecular mechanism of a cryptic oviedomycin biosynthetic gene cluster via the disruption of a global regulatory gene adpA in Streptomyces ansochromogenes. J Biol Chem. https://doi.org/10.1074/jbc.M117.809145
Yamazaki H, Tomono A, Ohnishi Y, Horinouchi S (2004) DNA-binding specificity of AdpA, a transcriptional activator in the A-factor regulatory cascade in Streptomyces griseus. Mol Microbiol 53(2):555–572
Yang Z, Bielawski JP (2000) Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15(12):496–503
Yang Z, Nielsen R, Goldman N, Pedersen AM (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155(1):431–449
Yao MD, Ohtsuka J, Nagata K, Miyazono KI, Zhi Y, Ohnishi Y, Tanokura M (2013) Complex structure of the DNA-binding domain of AdpA, the global transcription factor in Streptomyces griseus, and a target duplex DNA reveals the structural basis of its tolerant DNA sequence specificity. J Biol Chem 288(43):31019–31029
Zaburannyy N, Ostash B, Fedorenko V (2009) TTA Lynx: a web-based service for analysis of actinomycete genes containing rare TTA codon. Bioinformatics 25(18):2432–2433
Zhao J, Wen Y, Chen Z, Song Y, Li J (2007) An adpA homologue in Streptomyces avermitilis is involved in regulation of morphogenesis and melanogenesis. Chinese Sci Bull 52(5):623–630
Acknowledgements
The work was supported by grant BG-41Nr from the Ministry of Education and Science of Ukraine (to B.O.).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rabyk, M., Yushchuk, O., Rokytskyy, I. et al. Genomic Insights into Evolution of AdpA Family Master Regulators of Morphological Differentiation and Secondary Metabolism in Streptomyces. J Mol Evol 86, 204–215 (2018). https://doi.org/10.1007/s00239-018-9834-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-018-9834-z