
Abstract
Pattern recognition receptors (PRRs) are specialized receptors that represent a key component of the host innate immune system. Whether molecular evolutionary history of different PRR classes have involved different genetic mechanisms underlying diverse pathogen environment in mammals, and whether distinct ecology of mammals may have imposed divergent selective pressures on the evolution of the PRRs, remained unknown. To test these hypotheses, we investigated the characterization of 20 genes belonging to four PRR classes in mammals. Evidence of positive selection was found in most (17 of 20) PRR genes examined, and most positively selected sites (84%) undergoing radical changes were found to fall in important functional regions, consistent with the co-evolutionary dynamics between the hosts and their microbial counterparts. We found different evolutionary patterns in different PRR classes, with the highest level of positive selection in C-type lectin receptor (CLR) family, suggesting that the capability of CLRs in response to a wide variety of ligands might explain their malleability to selection pressures. Tests using branch models that partitioned the data along habitat and social behavior found significant evidence of divergent selective pressures of PRRs among mammalian groups. Interestingly, species-specific evolution was detected on RIG-I-like helicase genes (RLRs) in cetaceans, suggesting that RLRs might play a critical role in the defense against widespread marine RNA viruses during their divergence and radiation into marine habitats. This study provides a comprehensive look at the evolutionary patterns and implications of mammalian PRRs, and highlights the importance of ecological influences in molecular adaptation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
The innate immune system is the first line of host defense in all mammals, where it plays a crucial role in the body’s protective response to ensure the removal of various microbial infections. The function of the innate immune is to recognize pathogen-associated molecular patterns (PAMPs) using a variety of host receptors called pattern recognition receptors (PRRs) and to activate the innate immune system through changes in cytokine expression, including pro-inflammatory cytokines and type I interferons (IFNs) (Medzhitov 2001). There are four main classes of germline-encoded PRRs implicated in the innate immune response, including Toll-like receptors (TLRs) and non-TLRs such as C-type lectin receptors (CLRs), RIG-I-like helicases (RLRs), and the NOD-like receptor (NLR) family (Takeuchi and Akira 2010). TLRs, a major class of PRRs, are type I transmembrane glycoproteins and can be expressed at the cell surface or in membrane compartments of specialized immune cells (Iwasaki and Medzhitov 2010). The immunity response of TLRs involves detecting invading pathogens outside cells and in intracellular endosomes and lysosomes via extracellular leucine-rich repeat (LRR) sequences and transmitting signals through the cytoplasmic Toll–interleukin (IL)-1 receptor (TIR) domain (Akira et al. 2006). The CLRs are another type of membrane-bound receptors that are characterized by the presence of a carbohydrate-binding domain and expressed by dendritic cells. CLRs are involved in the induction expression of specific cytokines by either modulating TLR signaling or directly activating nuclear factor-κB (Geijtenbeek and Gringhuis 2009). The RLR family specifically detects viral RNA in the cytoplasm of cells and induces IFNs, which are composed of two N-terminal caspase activation and recruitment domain (CARD)-related motifs, a central DEAD box helicase/ATPase domain, and a C-terminal repressor domain (Yoneyama and Fujita 2009). In addition, NLRs have a characteristic central nucleotide-binding domain and C-terminal leucine-rich repeats that also detect cytoplasmic pathogens (Takeuchi and Akira 2010).
The evolution of PRRs has received considerable attention because they have the potential to be subjected to a variety of selection pressures from a wide diversity of coevolving pathogens (Sironi et al. 2015). For example, recent studies performed in avians (Yilmaz et al. 2005; Alcaide and Edwards 2011; Grueber et al. 2014), ungulates (Jann et al. 2008; Quéméré et al. 2015; Ishengoma and Agaba 2017), cetaceans (Shen et al. 2012; Ishengoma and Agaba 2017), rodents (Tschirren et al. 2011; Fornůsková et al. 2013), and primates (Nakajima et al. 2008; Wlasiuk and Nachman 2010) revealed different degrees of positive selection acting on TLR members during their evolutionary history. In contrast, general patterns of selection on non-TLR immune defense genes are just beginning to emerge, with only very few reports available. For instance, recently, de Matos et al. (2013) reported that three RLR genes, RIG-I, MDA5, and LGP2, have all been subject to long-term selective pressures during mammalian evolution. Despite several studies on the evolution of TLRs and RLRs in mammals, a clear picture of the evolution of all PRR classes has not emerged. Studies to date have not incorporated analyses of all mammalian PRRs and have not assessed their different patterns of molecular adaptation, which potentially limit our understanding of evolutionary mechanisms responsible for diverse pathogen recognition.
Mammals are the most complex group among vertebrates and include diverse species that occupy aerial, aquatic, and terrestrial niches (Werdelin 2007). Slade and McCallum (1992) reported that the marine mammals (i.e., cetaceans), which evolved from terrestrial lineages (i.e., ungulate), have lost the histocompatibility complex (MHC) diversity due to a decrease in exposure to microparasitic diversity in the marine environment (Slade and McCallum 1992). This report suggests that mammals must have been confronted with different pathogenic challenges from distinct ecology, which might link to the evolution of immune-related genes. In addition, mammals also exhibit stark contrasts in social behavior with grouping living and solitary living that results in differential degrees of parasite transmission (Alexander 1974). In particular, mammalian with large different feeding habits, e.g., carnivores, omnivores, herbivores, and insectivorous, also affect infectious disease dynamic because parasites depend on their ecological network of living (Lafferty et al. 2008). Consequently, we hypothesized that ecological differences among mammals occupying different ecosystems, social behavior, and diets, may be driving divergent evolution of mammalian PRRs.
Given the ever-changing environmental pathogens, the adaptation of innate immunity must have made a particularly great contribution to mammals’ resistance to complex microbial milieu. Therefore, understanding the molecular mechanisms underlying immune receptors is extremely important for developing insights into host defense processes against infections in different mammalian groups during their adaption to various habitats. In this study, we performed comparative evolutionary analyses of 20 PRR-related genes from most representative lineages of mammals, and sought to (1) comprehensively reveal the evolutionary patterns for mammalian PRRs, (2) investigate different classes of PRRs that might display different patterns of molecular evolution, (3) test whether ecological differences between mammals have resulted in divergent selective pressures on PRR genes.
Methods
Species Coverage and Sequence Retrieval
We screened a list of 20 PRR-related genes, including 10 TLRs (TLRs 1–10), 4 NLRs (NLRP3, NLRX1, NOD1, NOD2), 3 RLRs (LGP2, MDA5, RIGI), and 3 CLRs (Dectin-1, Dectin-2, MINCLE) by searching research articles and recent review papers (Takeuchi and Akira 2010). The gene sequences derived from the genomes of baiji (Lipotes vexillifer), minke whale (Balaenoptera acutorostrata), killer whale (Orcinus orca), sperm whale (Physeter macrocephalus), bowhead whale (Balaena mysticetus), Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis, unpublished), seal (Leptonychotes weddellii), walrus (Odobenus rosmarus), manatee (Trichechus manatus), polar bear (Ursus maritimus), Brandt’s bat (Myotis brandtii), David’s myotis (Myotis davidii), black flying fox (Pteropus alecto), long-winged bat (Taphozous longimanus), and big brown bat (Eptesicus fuscus) were retrieved by BLAST (NCBI) using the published human genes as queries (Johnson et al. 2008). Additionally, the coding sequences of the remaining species were obtained from the Ensembl genome browser (http://www.ensembl.org/) with the accession numbers listed in supplementary Table S1. The species composition of each orthologous PRR gene varied from 35 to 48 species from the most representative mammalian groups: Cetartiodactyla, Carnivora, Chiroptera, Rodents, and Primates. The nucleotide and deduced amino acid sequences were aligned using MEGA 6.0 (Tamura et al. 2013) and were checked by eye.
Test of Evolutionary Selective Pressure
To assess the molecular evolution of PRR-related genes, we compared the rate per site of non-synonymous substitutions (dN) to the rate per site of synonymous substitutions (dS) using the codon-based maximum likelihood (CodeML) implemented in PMAL 4.7 (Yang 2007). Ratios of dN/dS (ω) < 1, = 1, > 1 are interpreted as evidence of purifying selection, neutral selection, and positive selection, respectively. The well-supported phylogeny of mammals was used as the input tree in all analyses (Fig. S1) (Ranwez et al. 2007).
To estimate ω for every codon in the alignment, two alternative models, M8 and M8a, were performed. Model M8a restricted sites with ω ≤ 1, whereas model M8 included a class of sites with ω > 1 (Swanson et al. 2003). A likelihood ratio test (LRT) with χ2 distribution was conducted to determine the nested model comparison at a threshold with p < 0.05, and Bayes empirical Bayes (BEB) analysis was used to identify sites under positive selection with posterior probabilities > 0.95 (Yang et al. 2005).
Simultaneously, we applied alternative approaches based on maximum likelihood in the Datamonkey web server (http://www.datamonkey.org) to further examine the extent of evolutionary pressure occurring at every codon, the advantage of which is incorporated variation in the rate of synonymous substitution (Pond and Frost 2005). The single likelihood ancestor counting (SLAC) model is based on the reconstruction of the ancestral sequences and the counts of synonymous and nonsynonymous changes at each codon position in a phylogeny. The fixed-effect likelihood (FEL) model estimates the ratio of nonsynonymous to synonymous substitutions on a site-by-site basis, without assuming an a priori distribution of rates across sites. The random effect likelihood (REL) model first fits a distribution of rates across sites and then infers the substitution rates for individual sites. We accepted sites with p values < 0.1 for SLAC and FEL and Bayes Factors > 50 for REL as candidates for selection.
Given that the aforementioned ML methods do not take into account the magnitude of changes in the physicochemical properties of amino acids resulting from nonsynonymous substitutions, TreeSAAP 3.2 software (Woolley et al. 2003) was used to detect significant physicochemical amino acid changes among residues. A total of 31 structural and biochemical amino acid property changes were considered, and these changes, whether radical or affecting the whole protein, were indicated by z-score categories produced via goodness-of-fit testing. The number of radical changes (z-score categories: 6–8) per codon in amino acid properties was regarded as a proxy of positive selection; more radical changes might be a suggestive of adaptive evolution. The sites identified under selection in at least two of the ML methods were considered for further analyses.
To assess positively selected sites along a specific lineage of mammalian phylogeny, we employed the branch-site model A. The branch-site model A allows ω to vary among not only sites but also ‘foreground’ (lineages tested to be under positive selection) and ‘background’ (the remaining lineages) branches specified by the user (Zhang et al. 2005). Each branch of mammals was specified as foreground branches according to the species tree in independent branch-site model test. The branch-site model A assumes four classes of sites, especially allowing codons under positive selection along foreground lineage with ω2 > 1 and compared with the null hypothesis with fixed ω2 = 1 in all-mammal dataset.
To assess the potential influence of ecological variables on PRRs divergence, the branch models were conducted where ω could vary among different branch groups. Specifically, a one-ratio model with fixed ω across the tree was compared with various intermediate models (e.g., 2-, 3-, 4-ratio) in which ω was allowed to differ between the background and focal branches. Based on a priori knowledge of mammalian ecological variables for living taxa, three classes were considered, i.e., diet (herbivorous, carnivorous, insectivorous, omnivorous), habitat (aerial, aquatic, terrestrial), and social behavior (gregariously terrestrial mammals, gregariously aquatic mammals, solitary), and analyses were conducted along each clade partition of each divergence clade (Fig. S2). For example, the one-ratio model was compared with a two-ratio (2ω) model that allowed one ω for aerial (e.g., bats) and aquatic (e.g., cetacean, pinnipeds, sirenian) mammalian branches and another for the remaining branches (terrestrial mammals), aiming to test whether different selective pressures acting on the lineages led to specific habits (i.e., aerial and aquatic lifestyle). Furthermore, to explore the rate variation among aerial and aquatic mammalian clades with different habitats, the three-ratio (3ω) model assumes that the branches of aquatic mammals and aerial mammals have independent rates and provided a third ω value for the remaining mammalian branches (terrestrial mammals), which was compared with the 2ω model. In all cases, the nested models were compared using the LRTs, and three starting values of ω of 0.5, 1, and 1.5 were conducted to ensure convergence. Multiple testing for positive selection on genes was corrected by performing a false discovery rate (FDR) test at a cutoff of 0.05 (Storey and Tibshirani 2003) for all analyses.
Identification of Functional Domains
To determine the delimitation of each domain of PRR molecules, the SMART webserver (http://smart.embl-heidelberg.de/) was used. The human delimitations were used as a reference for the remaining species. To gain insights into the functional significance of the putative selected sites, we mapped the sites under positive selection to crystal structures using PyMOL (http://pymol.org). The three-dimensional (3D) structures of genes were predicted using the homology modeling software provided by the I-TASSER server (Zhang 2008). The protein sequences of these genes were derived from the human reference.
Results
Site Selection of PRR-Related Genes in All Mammals
The one-ratio model analyses showed that the ω values for all 20 PRR-related genes were significantly less than 1 (Table S2), ranging from 0.122 (NLRX1) to 0.517 (Dectin-2), suggesting that these genes do have functions and that strong purifying selection plays a central role in maintaining their important roles in innate immunity. The mean dN/dS values of NLRs were significantly lower than those of CLRs (p < 0.001), viral TLRs (p < 0.001), and RLRs (p < 0.05), suggesting that NLRs may have been subjected to a relatively stronger functional constraint (Fig. S3).
Despite evidence of the overall purifying selection, likelihood ratio test (LRT) showed that the M8 model, including sites with ω > 1, fitted the data significantly better than the neutral model M8a for 17 PRR-related genes, including 10 TLRs (TLRs 1–10), 3 RLRs (LGP2, MDA5, RIGI), 3 CLRs (Dectin-1, Dectin-2, MINCLE), and 1 NLR (NOD1, except for NLRP3, NLRX1, NOD2) (Table 1). The M8 model detected a total of 91, 34, 9, and 6 sites to be under positive selection in the TLR, RLR, CLR, and NLR families, respectively. The significant evidence of positive selection acting on these 17 PRR-related genes was also identified using other maximum likelihood (ML) methods implemented in Datamonkey, many of which coincided with the codons identified by M8 (Table 1). When all ML analyses were considered together, 75 codons from 17 genes (TLR1: 6, TLR2: 3, TLR3: 5, TLR4: 11, TLR5: 3, TLR6: 2, TLR7: 6, TLR8: 10, TLR9: 3, TLR10: 2, NOD1: 2, LGP2: 3, MDA5: 4, RIGI: 8, Dectin-1: 2, Dectin-2: 3, MINCLE: 2) were detected to be under positive selection in the all-mammal dataset by at least 3 ML methods, which corresponded to 0.21–1.42% of positively selected codons for each gene. Of these 75 codons, 68% (51/75) were positively selected in TLRs, 20% (15/75) were positively selected in RLRs, 9.3% (7/75) were detected in CLRs, and only 2.7% (2/75) were identified in NLRs. Within the TLR family, the proportion of positively selected codons ranged from 0.25% (TLR10, 2 sites) to 1.33% (TLR4, 11 sites); TLR8 stood out within viral TLRs because 0.97% (10 sites) of positively selected codons were identified, followed by the non-viral TLR4 (Fig. S4). For the RLR and CLR families, the highest proportions of positively selected codons were detected at RIGI (8 positions, 0.87%) and Dectin-2 (3 positions, 1.42%), respectively (Table 1). It is interesting to note that the CLR family had the highest proportion of positively selected sites (1.12%), whereas the NLR family had the lowest, but still significantly positive value (0.053%). Remarkably, 84% (63/75) (46 for TLRs, 14 for RLRs, 1 for NLRs, and 2 for CLRs) of positively selected sites (PSSs) were also detected to be under radical changes in their physicochemical properties by protein-level approaches implemented in TreeSAAP (Table S3).
Species-Specific Selection
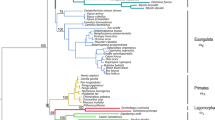
After performing Bonferroni correction for multiple testing, it was suggested that 17 (TLR1, TLR3, TLR4, TLR5, TLR6, TLR8, TLR9, LGP2, MDA5, RIGI, NOD1, NOD2, NLRP3, NLRX1, Dectin1, Dectin2, MINCLE) of 20 genes best fit the alternative model in branch-site analyses along 99 mammalian lineages in total (Table 2; Table S4). In particular, 9 out of 20 genes were detected to be under positive selection along 11 rodentia. Moreover, evidence for positive selection was identified along the lineages leading to 17 chiroptera, 13 primates, 12 carnivora, and 8 artiodactyla branches as foreground at 7, 7, 8, and 7 PRR-related genes, respectively. In cetaceans, 6 out of 20 genes best fit the alternative model along 12 cetacean branches, whereas for the remaining mammalian lineages, the numbers were smaller (e.g., only 0 to 4 genes in perissodactyla, lagomorpha) (Fig. 1). Of these positively selected genes (PSGs), the higher proportions of genes under positive selection were mainly involved in non-TLR families, such as 67% (6/9) for rodentia, 57% (4/7) for chiroptera, 75% (6/8) for carnivora, 83% (5/6) for cetacean, and 100% (7/7) for primate, respectively (Table 2). To better understand the distribution of positive selection in different mammalian lineages, the above inferred positively selected genes were visually mapped onto the mammalian phylogeny (Fig. 1).

Positive selected genes related to PRRs in mammals are mapped to a phylogenetic tree. Colored bars indicated the four types of PRR classes identified under positive selection in the present study. The detailed information of cetacean and chiroptera are shown in the right part. TLRs Toll-like receptors, CLRs C-type lectin receptors, RLRs RIG-I-like helicases, and NLR NOD-like receptor family
Selective Regime in Mammals with Variable Ecology
We used a branch model with multiple ratios to test for divergent selection by partitioning the data into three ecological groupings, including diet, habitat, and social behavior (Fig. S2). For the LRTs among nested models, the two-ratio model that grouped aerial and aquatic mammals as foreground clades showed significance against the simplest model (one-ratio) for the TLR2, TLR3, NLRP3, and NOD2 genes in the data subset of divergent habitat after FDR (Table 3). Additionally, the three-ratio model that placed aerial and aquatic mammals in the foreground clades separately also performed better than the null (two-ratio) model at the four genes aforementioned in the habitat, confirming that there is significant divergent selective pressure between aerial, aquatic, and terrestrial mammals. For the divergent social behaviors, the best-fitted models were to be found in those that partitioned social (aquatic/terrestrial) mammals from solitary taxa (Table 3) at the TLR2, NLRX1, and NOD2 genes after FDR. Interestingly, the four-ratio model, making a division between herbivorous, carnivorous, insectivorous, and outgroups, was a significantly better fit than the three- and two-ratio models at the TLR7 gene in diet pattern (Table 3). However, no significant divergent selective pressure was detected in other genes.
Mapping of PSSs onto Protein Structures
The PSSs were mapped onto the 3D protein structures to further assess their functional significance. For transmembrane proteins (e.g., TLRs, CLRs), the most identified PSSs (82.4% for TLRs and 42.9% for CLRs) were localized in the extracellular domain (ECD) regions, whereas fewer instances of positive selection were detected in the remaining domains (Table S5; Fig. S5). When the RLR family was considered, all sites under positive selection were widely distributed in important functional domains (Table S5; Fig. S5), such as the caspase recruitment domain, the helicase ATP-binding domain, and the helicase superfamily c-terminal domain (HELICc), suggesting obligatory roles during the evolutionary process.
Discussion
Pervasive Positive Selection in Mammalian PRRs
In the innate immune system, PRRs are the major contributor in response to sensing the presence of microorganisms. As the best characterized components among the PRRs, TLRs have been considered selectively constrained for a long time, yet recent studies on the TLR genes of vertebrates (e.g., primates, cetaceans, birds, reptiles, teleosts) have shown that adaptive evolution played a critical role in response to the co-evolutionary arms races with their microbial ligands (Wlasiuk and Nachman 2010; Shen et al. 2012; Voogdt et al. 2016). However, an overall and comprehensive investigation on mammalian PRR evolution and their association with ecological adaptation across mammalian phylogeny has not been conducted before. Here, we presented the first characterization of all PRRs in different mammalian lineages, which could provide some novel insights into the evolution and adaptive implications of the mammalian immune system at the molecular level.
The present analyses found clear signatures of positive selection in most PRR genes (10 TLRs, 3 RLRs, 3 CLRs, and 1 NTR). Neutral site-specific models of evolution were rejected for 17 genes (TLR1, TLR3, TLR4, TLR5, TLR6, TLR8, TLR9, LGP2, MDA5, RIGI, NOD1, NOD2, NLRP3, NLRX1, Dectin1, Dectin2, MINCLE), and a total of 75 robust candidate sites under selection were identified using several ML methods. Of these sites, 84% (63/75) were categorized as radical amino acid changes at the protein level. Particularly, almost all of the radical amino acid changes subjected to positive selection were localized within or near the functional region based on the predicted structural information. For example, up to 82.4% (42/51) of PSSs of TLRs were scattered in the ECD regions (Table S5), which are responsible for ligand binding and auto-regulation and contain 19–25 leucine-rich repeats (LRRs) (Bell et al. 2003). It has been reported that mutations in the ECD domain would increase susceptibility to pathogen infection, such as cytomegalovirus infection in mice (Tabeta et al. 2004) and Mycobacterium avium subsp. paratuberculosis (MAP) infection in cattle (Mucha et al. 2009). Positively selected sites of RLRs were located over CARD and helicase domain, which are also known to play roles in the recognition of viral infection in various cells (Creagh and O’Neill 2006). Overall, the extensive positive selection identified in PRRs across the mammalian phylogeny suggests an enhancement of their host’s defense capacity against invading pathogens during mammalian evolution.
Difference in Selection Between Mammalian PRR Classes
In our study, we noted obvious evidence of positive selection in different TLRs. The strongest signal of positive selection came from the bacterial-sensing TLR4, with 11 codons (1.33%, Table 1) identified as robust candidate sites under positive selection by at least two ML methods. The most accepted explanation for this evidence is that TLR4, aided by myeloid differentiation factor 2 (MD2), can respond to a wide variety of ligands, including those from bacteria lipopolysaccharides, components of yeast, Trypanosoma, and even viruses (Jiménez-Dalmaroni et al. 2016). In addition to the selection detected in TLR4, PSSs in the extracellular domains of TLR1 (174 in LRR5, 205 in LRR7, 488 in LRR18) and TLR2 (174 in LRR3, 211 in LRR7) and in the cytoplasmic domain of TLR1 (621 and 626) (Table S5) were also identified, suggesting that changes in the ligand-binding activity of TLR1/2 heterodimers may be modified through positive selection in mammals (Takeuchi et al. 2002) and further supporting the importance of LRRs in TLR ligand recognition. Not surprisingly, as revealed in previous studies on primates (Wlasiuk and Nachman 2010), birds (Alcaide and Edwards 2011), mammals (Areal et al. 2011), rodents (Fornůsková et al. 2013), terrestrial ungulates, and cetaceans (Ishengoma and Agaba 2017), the present study also detected higher levels of positive selection acting on bacterial-sensing TLRs than on viral-sensing counterparts. Bacterial-sensing TLRs are expressed on the cell surface, which possesses a higher redundancy (i.e., tolerance of damaging mutations) that might effectively recognize bacterial evasion by the host (Barreiro et al. 2009). In contrast, viral TLRs, which sense ancient and conserved viral PAMPs and activate the autophagy response (Lewis and Obbard 2014), are considered to undergo stronger evolutionary constraint than to non-viral TLRs to maintain their dual functions of recognizing nucleic acids from viruses and avoiding self-reactivity.
In contrast to TLRs, patterns of selection acting on mammalian non-TLR innate immune genes are much less clear. In our study, the highest positive selection frequencies were detected at CLRs, with 1.01% in Dectin1, 1.42% in Dectin2, and 0.94% in MINCLE of codons under positive selection (Table 1; Fig. S3). CLRs are expressed by dendritic cells (DCs) and are mainly responsible for the recognition of mannose, fucose, and glucan carbohydrate structures, resulting in interacting with most classes of human pathogens: viruses, fungi, mycobacteria, bacteria, and helminths (Geijtenbeek and Gringhuis 2009). This capability in response to a wide variety of ligands might explain their malleability to selection pressure. Additionally, CLRs not only can act as PRRs but also recognize endogenous ligands to facilitate adhesion between cells (Graham and Brown 2009); thus, the positive selection on CLRs is potentially associated with immune and non-immune functions in mammals. It is worthy to note that Dectin2 stood out among CLRs as the gene having highest proportion of PSSs (1.42%). Dectin2 is known to play important roles in mannose binding, mediating UV-induced immunosuppression, targeting antigen, recognition of fungi, and response to allergens (Graham and Brown 2009). The significant signature of positive selection detected on Dectin2 might suggest its importance in the maintenance of diverse functions in immunity and homeostasis for mammals.
A significant signature of positive selection was also detected on RIGI of the RLR family, but lowest number of codons under selection was observed for MDA5, inconsistent with previous studies (de Matos et al. 2013). This is not particularly surprising given that the power to detect selection with codon-based methods depends on the number of taxa, and the previous study focused on fewer species. Considering that virus-induced type I interferon (IFN) production was completely abolished in fibroblasts and conventional dendritic cells (cDCs) from RIG-I-deficient mice (Kato et al. 2005), positive selection in RIGI seems to help enhance the induction of IFNs after infection with RNA viruses to provoke antiviral immune responses in mammals. In addition, NLRs that also recognize bacterial peptidoglycan components seem to being the least prone to evolutionary change, since only one gene with the lowest number of codons under selection was observed (Table 1; Fig. S3), unlike bacterial-sensing TLRs. It should be noted that mutations in NLRs gene (e.g., NOD2) generate an inflammatory bowel disorder (Takeuchi and Akira 2010). Therefore, imposition of functional constraints in NLRs might be an evolutionary strategy to minimize the dangerous encounter in inflammatory disorder. Taking all of these lines of evidence collectively, positive selection acting on the different PRRs may reflect their particular and critical contributions to host defense during mammalian evolution.
Ecology-Driven Evolution of Mammalian PRRs?
Mammals possess tremendous ecological diversity, from the tiny shrew to the gargantuan blue whale, from swimming pinnipeds to flying bats. As a consequence, the complex evolutionary history associated with the radiation of mammals must pose many distinctly pathogenic challenges from their habitats. Our data support the hypothesis that divergent patterns of positively selected PRR genes in mammals, and differences across habitats and social behaviors may have driven this divergent evolution in mammalian PRRs. For example, the strongest positive selection on rodents was expected, given that their social behaviors could facilitate parasite transmission and sustain acute-immunizing infections (Luis et al. 2015). Most importantly, nine PSGs identified in rodents are only enriched in NLRs (NOD1, NOD2, NLRP3, NLRX1), TLRs (TLR3, TLR4, TLR9), and RLRs (MDA5, RIGI), but no CLR genes were inferred as being under positive selection in rodents. NLRs and RLRs can trigger a subset of responses similar to TLRs and likely act coordinately in innate immunity (Creagh and O’Neill 2006). Our result is suggestive of an important cooperation between these families in rodents, providing a tightly controlled combinatorial repertoire for triggering rodents’ defenses. A particularly interesting finding involves the divergent selective pressures detected on RLR genes between cetaceans and ungulates (Table 2), with cetacean RLRs (MDA5 and RIGI) showing positive selection while ungulates do not. The degree of connectivity and the modes of dispersal are very different in terrestrial and marine ecosystems (McCallum et al. 2003). Cetaceans encountered great changes in the pathogen environment while they moved from land to aquatic ecosystems. Very different pathogens, especially the widespread marine RNA viruses, can be major sources of disease and mortality for marine life (Lang et al. 2009). Therefore, our results implied that RLRs, cytoplasmic virus sensors, may have played an adaptive role in the water re-colonization of cetaceans. Bats are receiving increasing attention as the host reservoirs of a number of zoonotic viral pathogens (Brook and Dobson 2015). Some specific traits, such as their long life spans, flight capabilities, gregarious nature, and roosting sites, may make bats suited to host more viruses (Luis et al. 2013). Various studies have comprehensively documented the evolution of immunity in bats (Zhang et al. 2013; Escalera-Zamudio et al. 2015; Schad and Voigt 2016). For instance, Zhang et al. (2013) reported that the evidence of genetic changes, such as a loss of positive selection in the PYHIN gene family and immunoglobulin superfamily duplication, may contribute to their evolution of innate and adaptive immunity. Similarly, our analyses provide evidence that several PRR genes have been subjected to adaptive evolution in the bat, which in turn suggests that the diversity of ecological specializations among bats has been combined with PRR-inherent factors to accelerate the adaptive evolution of PRRs in these species. Of note, NLRP3 has previously been identified to show positive selection in the bat genome (Zhang et al. 2013); thus, the present observation of positive selection on this gene further suggests its importance to enhance the capacity for inflammasome assembly during viral infection. Finally, we found a stronger signal for positive selection in PRR genes across lineages of laurasiatheria and euarchontoglires than among other lineages such as Afrotheria, and NLR genes were exclusively positively selected along the laurasiatherian and euarchontoglires lineages (Table 2). It is reasonable to suggest that evolution of PRR genes may have been attributable to the rapid radiation and high diversification in taxa such as laurasiatheria and euarchontoglires (Doronina et al. 2017). In summary, these species-specific evolutionary patterns of PRRs might provide useful pointers to the divergent evolution of mammals driven by ecological factors.
References
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 1244:783–801
Alcaide M, Edwards SV (2011) Molecular evolution of the Toll-like receptor multigene 1 family in birds. Mol Biol Evol 28:1703–1715
Alexander RD (1974) The evolution of social behavior. Annu Rev Ecol Syst 5:325–383
Areal H, Abrantes J, Esteves PJ (2011) Signatures of positive selection in Toll-like receptor TLR genes in mammals. BMC Evol Boil 111:368
Barreiro LB, Ben-Ali M, Quach H, Laval G, Patin E, Pickrell JK, Kidd JR (2009) Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. Plos Genet 57:e1000562
Bell JK, Mullen GE, Leifer CA, Mazzoni A, Davies DR, Segal DM (2003) Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol 2410:528–533
Brook CE, Dobson AP (2015) Bats as ‘special’ reservoirs for emerging zoonotic pathogens. Trends Microbiol 233:172–180
Creagh EM, O’Neill LA (2006) TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol 278:352–357
de Matos AL, McFadden G, Esteves PJ (2013) Positive evolutionary selection on the RIG-I-like receptor genes in mammals. PLoS ONE 8:e81864
Doronina L, Churakov G, Kuritzin A, Shi J, Baertsch R, Clawson H, Schmitz J (2017) Speciation network in Laurasiatheria: retrophylogenomic signals. Genome Res 276:997–1003
Escalera-Zamudio M, Zepeda-Mendoza ML, Loza-Rubio E, Rojas-Anaya E, Méndez-Ojeda ML, Arias CF, Greenwood AD (2015) The evolution of bat nucleic acid-sensing toll-like receptors. Mol Ecol 2423:5899–5909
Fornůsková A, Vinkler M, Pagès M, Galan M, Jousselin E, Cerqueira F, Morand S, Charbonnel N, Bryja J, Cosson JF (2013) Contrasted evolutionary histories of two toll-like receptors (Tlr4 and Tlr7) in wild rodents (MURINAE). BMC Evol Biol 13:194
Geijtenbeek TBH, Gringhuis SI (2009) Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol 97:465–479
Graham LM, Brown GD (2009) The dectin-2 family of C-type lectins in immunity and homeostasis. Cytokine 481:148–155
Grueber CE, Wallis GP, Jamieson IG (2014) Episodic positive selection in the evolution of avian toll-like receptor innate immunity genes. PLoS ONE 93:e89632
Ishengoma E, Agaba M (2017) Evolution of toll-like receptors in the context of terrestrial ungulates and cetaceans diversification. BMC Evol Biol 171:54
Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 3275963:291–295
Jann OC, Werling D, Chang JS, Haig D, Glass EJ (2008) Molecular evolution of bovine toll-like receptor 2 suggests substitutions of functional relevance. BMC Evol Biol 8:288
Jiménez-Dalmaroni MJ, Gerswhin ME, Adamopoulos IE (2016) The critical role of toll-like receptors-from microbial recognition to autoimmunity: a comprehensive review. Autoimmun Rev 151:1–8
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36:W5–W9
Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S (2005) Cell type-specific involvement of RIG-I in antiviral response. Immunity 231:19–28
Lafferty KD, Allesina S, Arim M, Briggs CJ, De Leo G, Dobson AP, Martinez ND (2008) Parasites in food webs: the ultimate missing links. Ecol Lett 11:533–546
Lang AS, Rise ML, Culley AI, Steward GF (2009) RNA viruses in the sea FEMS. Microbiol Rev 332:295–323
Lewis SH, Obbard DJ (2014) Recent insights into the evolution of innate viral sensing in animals. Curr Opin Microbiol 20:170–175
Luis AD, Hayman DT, O’Shea TJ, Cryan PM, Gilbert AT, Pulliam JR, Mills JN, Timonin ME, Willis CK, Cunningham AA, Fooks AR, Charles E, Rupprecht CE, Wood JL, Webb CT (2013) A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc R Soc B 280:20122753
Luis AD, O’Shea TJ, Hayman DT, Wood JL, Cunningham AA, Gilbert AT, Mills JN, Webb CT (2015) Network analysis of host-virus communities in bats and rodents reveals determinants of cross-species transmission. Ecol Lett 1811:1153–1162
McCallum H, Harvell D, Dobson A (2003) Rates of spread of marine pathogens. Ecol Lett 612:1062–1067
Medzhitov R (2001) Toll-like receptors and innate immunity. Nat Rev Immunol 12:135–145
Mucha R, Bhide MR, Chakurkar EB, Novak M, Mikula I (2009) Toll-like receptors TLR1, TLR2 and TLR4 gene mutations and natural resistance to Mycobacterium avium subsp. paratuberculosis infection in cattle. Vet Immunol Immunop 1284:381–388
Nakajima T, Ohtani H, Satta Y, Uno Y, Akari H, Ishida T, Kimura A (2008) Natural selection in the TLR-related genes in the course of primate evolution. Immunogenetics 60:727–735
Pond SLK, Frost SDW (2005) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533
Quéméré E, Galan M, Cosson JF, Klein F, Aulagnier S, Gilot-Fromont E, Joël Merlet J, Bonhomme M, Hewison AJM, Charbonnel N (2015) Immunogenetic heterogeneity in a widespread ungulate: the European roe deer (Capreolus capreolus). Mol Ecol 2415:3873–3887
Ranwez V, Delsuc F, Ranwez S, Belkhir K, Tilak MK, Douzery EJ (2007) OrthoMaM: a database of orthologous genomic markers for placental mammal phylogenetics. BMC Evol Biol 7:241
Schad J, Voigt CC (2016) Adaptive evolution of virus-sensing toll-like receptor 8 in bats. Immunogenetics 6810:783–795
Shen T, Xu S, Wang X, Yu W, Zhou K, Yang G (2012) Adaptive evolution and functional constraint at TLR4 during the secondary aquatic adaptation and diversification of cetaceans. BMC Evol Boil 121:39
Sironi M, Cagliani R, Forni D, Clerici M (2015) Evolutionary insights into host-pathogen interactions from mammalian sequence data. Nat Rev Gene 164:224–236
Slade RW, McCallum HI (1992) Overdominant vs. frequency-dependent selection at MHC loci. Genetics 132:861–862
Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Nat Acad Sci 10016:9440–9445
Swanson WJ, Nielsen R, Yang Q (2003) Pervasive adaptive evolution in mammalian fertilization proteins. Mol Biol Evol 20:18–20
Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Suzanne M, Louis S, Sosathya S, Jason G, Lena A, Richard FA, Bruce B (2004) Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA 10110:3516–3521
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 1406:805–820
Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S (2002) Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol 1691:10–14
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tschirren B, Råberg L, Westerdahl H (2011) Signatures of selection acting on the innate immunity gene Toll-like receptor 2 (TLR2) during the evolutionary history of rodents. J Evol Biol 246:1232–1240
Voogdt CG, Bouwman LI, Kik MJ, Wagenaar JA, Van Putten JP (2016) Reptile Toll-like receptor 5 unveils adaptive evolution of bacterial flagellin recognition. Sci Rep 6 19046
Werdelin L (2007) The origin and evolution of mammals. Acta Zool 88:179–180
Wlasiuk G, Nachman MW (2010) Adaptation and constraint at toll-like receptors in primates. Mol Biol Evol 27:2172–2186
Woolley S, Johnson J, Smith MJ, Crandall KA, McClellan DA (2003) TreeSAAP: selection on amino acid properties using phylogenetic trees. Bioinformatics 195:671–672
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang Z, Wong WS, Nielsen R (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Yilmaz A, Shen S, Adelson DL, Xavier S, Zhu JJ (2005) Identification and sequence analysis of chicken toll-like receptors. Immunogenetics 56:743–753
Yoneyama M, Fujita T (2009) RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev 2271:54–65
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40
Zhang J, Nielsen R, Yang Z (2005) Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol 22:2472–2479
Zhang G, Cowled C, Shi Z, Huang Z, Bishop-Lilly KA, Fang X et al (2013) Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science 339:456–460
Acknowledgements
We thank Mr. Xinrong Xu for help with collecting samples for many years. We also thank members of the Jiangsu Key Laboratory for Biodiversity and Biotechnology, Nanjing Normal University, for their contributions to this paper.
Funding
This work was funded by the Key Project of the National Natural Science Foundation of China (NSFC) (Grant No. 31630071 to G.Y. and Grant No. 31570379; 31772448 to S.X.), the National Science Fund for Distinguished Young Scholars (Grant No. 31325025 to G.Y.), the National Key Program of Research and Development, Ministry of Science and Technology (Grant No. 2016YFC0503200 to G.Y. and S.X.), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) to G.Y. and S.X., and the Natural Science Foundation of Jiangsu Province of China (Grant No. BK20141449) to S.X., and the Cultivation Plan for Excellent Doctorial Dissertations of Nanjing Normal University (NNU) to R.T.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict interests.
Electronic Supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tian, R., Chen, M., Chai, S. et al. Divergent Selection of Pattern Recognition Receptors in Mammals with Different Ecological Characteristics. J Mol Evol 86, 138–149 (2018). https://doi.org/10.1007/s00239-018-9832-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-018-9832-1