
Abstract
Non-coding small RNAs (sRNAs) are critical to post-transcriptional gene regulation in bacteria. However, unlike for protein-coding genes, the evolutionary forces that shape sRNAs are not understood. We investigated sRNAs in enteric bacteria and discovered that recently emerged sRNAs evolve at significantly faster rates than older sRNAs. Concomitantly, younger sRNAs are expressed at significantly lower levels than older sRNAs. This process could potentially facilitate the integration of newly emerged sRNAs into bacterial regulatory networks. Furthermore, it has previously been difficult to trace the evolutionary histories of sRNAs because rapid evolution obscures their original sources. We overcame this challenge by identifying a recently evolved sRNA in Escherichia coli, which allowed us to determine that novel sRNAs could emerge from vestigial bacteriophage genes, the first known source for sRNA origination.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Non-coding RNAs (ncRNAs) regulate gene expression in all domains of life. In Eukaryotes, post-transcriptional control of gene expression by ncRNA such as microRNA (miRNA) and small interfering RNA is now recognized as a fundamental layer of gene regulation (Wilson and Doudna 2013). While less studied, Archaea also contain a large repertoire of ncRNAs (Bernick et al. 2012). In Bacteria, a major class of ncRNA is small RNAs (sRNAs), which are around 50–200 nucleotides in length, and regulate gene expression by binding to messenger RNAs (mRNAs) (Gottesman and Storz 2011). Broadly, sRNAs are classified into two major types: (1) trans-acting sRNAs that are encoded in intergenic regions and regulate the expression of distantly located genes via imperfect complementarity, and (2) cis-acting sRNAs (also called antisense RNAs) that are transcribed from the opposite strand of adjacent target genes and regulate gene expression through perfectly complementary regions (Thomason and Storz 2010; Georg and Hess 2011). Some of the other major classes of bacterial ncRNA are riboswitches and RNA thermometers that are located in the untranslated regions of certain mRNAs (Breaker 2011; Kortmann and Narberhaus 2012), and intraRNAs that originate from within protein-coding genes (Miyakoshi et al. 2015).
Several advantages of sRNAs over proteins have led to their emergence as important gene regulators in bacteria. For instance, relatively lower energy is required to synthesize sRNAs; they act rapidly and their co-degradation along with target mRNAs allows precise control of regulatory circuits (Storz et al. 2011; Updegrove et al. 2015). Each sRNA typically controls multiple genes belonging to interconnected processes, including metabolic pathways, quorum sensing, biofilm formation, and virulence (Michaux et al. 2014), and because there are hundreds of sRNAs in each bacterium, their contribution to bacterial adaptation and phenotypic diversity could be tantamount to that of protein-coding genes (Gottesman and Storz 2011; Raghavan et al. 2011; Kröger et al. 2013). Hence, differences in sRNA sequences and contents between closely related bacteria could have a substantial impact on bacterial physiology and pathogenicity. However, we have minimal knowledge about the evolutionary forces that shape bacterial sRNAs. An analysis of the conservation of sRNAs in Escherichia coli showed that variation in sRNA contents between strains is mainly due to deletions (Skippington and Ragan 2012), and a broader examination of sRNA conservation across bacteria revealed that most E. coli sRNAs originated after enteric bacteria split from other Gammaproteobacteria (Peer and Margalit 2014). In addition to this cycle of birth and loss, sRNA genes evolve at faster rates than protein-coding genes, making it difficult to identify sRNA homologs in distantly related bacteria (Gardner et al. 2005; Hoeppner et al. 2012). One group of bacteria that was shown to be at optimum evolutionary distance for effective evolutionary analysis of sRNAs is the family Enterobacteriaceae (Lindgreen et al. 2014), which includes E. coli and Salmonella enterica, two model organisms in which sRNAs have been investigated thoroughly (e.g., Raghavan et al. 2011; Kröger et al. 2013).
In this study, we estimated the evolutionary ages of >200 sRNAs present in E. coli and S. enterica, and show that younger sRNAs are expressed at significantly lower levels than older sRNAs, and that younger sRNAs have significantly higher rates of evolution than older sRNAs. The low expression of new sRNAs could mitigate the negative effects of nascent sRNA–mRNA interactions, whereas their rapid evolution could generate beneficial interactions that facilitate their integration into bacterial regulatory networks. We also show that most sRNAs are evolving under purifying selection, and discovered that a young sRNA in E. coli originated from a vestigial bacteriophage protein-coding gene, thereby revealing the first known source for sRNA origination in bacteria.
Results
Newly Evolved sRNAs have Low Expression and Rapid Rate of Evolution
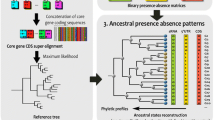
We identified the homologs of 81 E. coli sRNAs (Raghavan et al. 2011, 2015) and 127 S. enterica Typhimurium sRNAs (Kröger et al. 2013) in Citrobacter freundii, Klebsiella pneumoniae, Serratia marcescens, and Yersinia enterocolitica using BLASTn. This approach has been shown to be effective at identifying sRNA homologs within Enterobacteriaceae (Skippington and Ragan 2012; Peer and Margalit 2014). We utilized maximum parsimony to estimate the age of each sRNA along a 16S rDNA phylogenetic tree that encompasses the six enteric bacteria (Fig. 1), as done previously to study miRNA evolution (Lyu et al. 2014). We assigned the sRNAs into three age groups: old (those that originated in the common ancestor of all six bacteria), middle-aged (those that originated in the common ancestor of E. coli, S. enterica, C. freundii, and K. pneumoniae), and young (those that originated in the E. coli, S. enterica branch) (Fig. 1). Based on this classification, E. coli contained 21 young, 27 middle-aged, and 33 old sRNAs, whereas S. enterica contained 53 young, 48 middle-aged, and 26 old sRNAs (Supplemental dataset 1).

sRNA age groups. sRNAs were binned based on their presence in six enteric bacteria using maximum parsimony
We analyzed the expression of sRNAs using RNA-seq data for exponential phase growth of E. coli in Lysogeny Broth (Raghavan et al. 2011), and S. enterica Typhimurium in Lennox Broth (Kröger et al. 2013), and discovered that sRNA expression correlated with sRNA age: younger sRNAs have significantly lower expression than older sRNAs (Fig. 2, Supplemental dataset 1). In order to rule out the possibility that the observed relationship between sRNA age and expression is an artifact of the growth conditions, we expanded our analysis of S. enterica sRNAs to all 22 “infection-relevant” growth conditions described by Kroger et al. (2013). Significantly reduced expression of young sRNAs in comparison to older sRNAs was observed under all growth conditions (Table S1), showing that the relationship between sRNA age and expression is not dependent on the growth condition (p = 0.818, Permutational ANOVA). Furthermore, to confirm that our conclusions are independent of the BLAST algorithm’s ability to locate sRNA homologs in other enteric species, we performed a similar analysis using 49 E. coli sRNAs described in the Rfam database, which uses a different approach (covariance model) to identify homologous sRNAs (Nawrocki et al. 2015). As shown previously (Peer and Margalit 2014), we got comparable results using either BLASTn or Rfam (Supplemental dataset 1).

Younger sRNAs have lower expression. a Mean expression (±SE) of 81 sRNAs in E. coli grown in Lysogeny Broth, and b 127 sRNAs in S. enterica grown in Lennox Broth are shown. sRNA expression was measured using RNA-seq (Raghavan et al. 2011; Kröger et al. 2013). sRNAs were binned as described in Fig. 1 and p-values were calculated using Kruskal–Wallis test and Dunn’s test for multiple comparisons
We also measured the rate of evolution of each sRNA by calculating the nucleotide diversity index π, the average number of nt differences per site, using homologs in 85 E. coli and 112 S. enterica strains (Supplemental dataset 1) (Nei 1987; Jovelin and Cutter 2014). We discovered that the rate of sRNA evolution inversely correlated with age i.e., younger sRNAs evolved at significantly higher rates than older sRNAs (Fig. 3; Supplemental dataset 1). Although we examined sRNAs only in enteric bacteria, the observed relationship among sRNA age, expression, and rate of evolution is likely to be a widespread phenomenon because previous studies in humans, nematodes, and Drosophila have shown that younger miRNAs have lower expression and faster rate of evolution than older miRNAs (Chen and Rajewsky 2007; Jovelin and Cutter 2014; Lyu et al. 2014).

Younger sRNAs have higher rates of evolution. a Average (±SE) π values for 81 sRNAs in E. coli, and b 127 sRNAs in S. enterica are shown. sRNAs were binned as described in Fig. 1 and p-values were calculated using Kruskal–Wallis test and Dunn’s test for multiple comparisons
A Young sRNA in E. coli Originated from a Degraded Bacteriophage Gene
One of the young sRNAs that had low expression and rapid rate of evolution was EcsR2 (Fig. 4), an sRNA found exclusively in E. coli (Raghavan et al. 2015). In order to understand the origin of EcsR2, we traced the evolutionary history of the yagU–ykgJ intergenic region (IGR) that contains this sRNA. The arrangement in which yagU neighbors ykgJ is found only in E. coli, whereas an alternate, and likely ancestral, gene order (yciC–ykgJ–ompW) is present in most other enteric bacteria (Fig. 5). The ancestral gene arrangement is also present in E. albertii, which is one of E. coli’s closest relatives, indicating that ykgJ moved to its current location in E. coli after the two bacteria split from a common ancestor. Additionally, the ykgJ ORF (open reading frame) is smaller in E. coli than in E. albertii, and ~ 90 bp remnant of the gene’s 3′ end is still recognizable in the yciC-ompW IGR in E. coli, confirming that ykgJ was translocated recently to its current location in E. coli to create the unique yagU–ykgJ IGR (Fig. 5).

EcsR2 has low expression and rapid evolution. a Expression levels, and b nucleotide divergence index (π) of EcsR2 compared to two old sRNAs (RyhB and SsrS)

EcsR2 evolved in a novel IGR in E. coli. a The yagU–ykgJ IGR is present only in E. coli. b Relocation of ykgJ next to yagU in E. coli resulted in the formation of a novel IGR that contains EcsR2. Gene locations based on E. albertii (NZ_CP007025.1) and E. coli (NC_000913.2)
Due to their rapid rate of evolution, it is usually difficult to trace the ancestry of sRNAs (Gottesman and Storz 2011). However, because EcsR2 emerged in an IGR that was formed recently, we were able to identify through sequence alignment that the sRNA evolved from a vestigial bacteriophage gene (Fig. 6). To identify genes that are potentially regulated by EcsR2, we used RNA-seq to detect changes in mRNA levels in cells transiently expressing EcsR2 (cells with empty vector was used as control). This approach has been used previously to identify sRNA targets because pulse expression of sRNA limits indirect regulatory effects (e.g., Zhang et al. 1998; Wang et al. 2015). The RNA-seq analysis identified 26 genes that were significantly downregulated in the EcsR2-expressing strain (Table S2). Further, we combined in vivo RNA crosslinking (Lustig et al. 2010; Liu et al. 2015) with RNA-seq to identify mRNAs that could directly interact with EcsR2 (Fig. S1). This approach (Crosslink-seq) identified nine mRNAs that were potentially bound to EcsR2 (Table S3).

Origination of EcsR2 from a degraded prophage gene. a A pseudogenized version of a prophage gene (tfaR) evolved into EcsR2 by gaining a promoter-like sequence and an intrinsic terminator. Putative −10 and −35 sequences are in green, and the transcription start site of EcsR2 is in bold. b Alignment of EcsR2 gene and its homologous region in tfaR gene. The putative mRNA-binding region and the intrinsic terminator of EcsR2 are shown in red and purple, respectively. The tfaR stop codon is in bold. The 5′ end of EcsR2 (nucleotides 1–107) has ~75% identity, whereas the 3′ end (nucleotides 108–166) has ~50% identity to tfaR (Color figure online)
One gene that was identified through both RNA-seq and Crosslink-seq as a potential direct target of EcsR2 was ansB (downregulated ~16 fold in RNA-seq, and enriched >3 fold in Crosslink-seq). In silico modeling predicted that EcsR2 could bind to AnsB mRNA via nucleotides within an unstructured region: positions +52 to +83 (Fig. S2); coincidentally, using a sliding-window analysis that mapped the rate of polymorphism across EcsR2, we identified the same region to be evolving at a much lower rate than the rest of the sRNA (Fig. 7). Previous studies have shown that mRNA-binding sites are the most conserved regions within sRNAs (Peer and Margalit 2011; Richter and Backofen 2012), indicating that the +50 to +80 region is the potential AnsB-binding site. Additionally, although this region is highly conserved among E. coli strains (Fig. 7), it seems to have evolved considerably from its progenitor tfaR gene (Fig. 6, S3), potentially due to its functional importance. To verify EcsR2′s ability to regulate ansB expression, we pulse induced the expression of full-length EcsR2 and a version of EcsR2 in which the putative binding site was deleted. We quantified ansB expression using qRT-PCR and observed significant reduction in ansB expression only with the full-length version of EcsR2 (Fig. 8), suggesting that the putative mRNA-binding region is required for gene regulation.

EcsR2 sequence conservation and predicted structure. a Nucleotide differences calculated for the yagU–ykgJ IGR using a sliding-window analysis is represented by the black line, and the flanking green lines indicate the 95% confidence interval. The locations of the 3′ ends of yagU and ykgJ genes are shown in yellow. The most conserved region (blue) within the IGR correlates with yagU’s intrinsic terminator. The location of EcsR2 within the IGR is highlighted in purple. The blue and red arrows on y-axis indicate the average nucleotide polymorphism for EcsR2 and IGR, respectively. bThe predicted secondary structure of EcsR2 was generated using mfold web server (Color figure online)

EcsR2 downregulates ansB expression. Fold difference in ansB expression in the presence of full-length EcsR2 or EcsR2 without nucleotides +51 to +80, in comparison to a strain lacking EcsR2 (normalized to 1, dashed line). Data represent the means of three experiments ± standard deviations. Asterisk indicates p < 0.01 (unpaired t test)
Conserved sRNAs are Under Purifying Selection
To assess the impact of natural selection on sRNAs, we analyzed a subset of sRNAs (n = 38) that are conserved in E. coli and S. enterica (74 and 102 strains, respectively) (Supplemental dataset 1). As expected, younger sRNAs have higher rates of polymorphism and divergence than older sRNAs (Fig. 9). Interestingly, both young and old sRNAs are evolving at significantly lower rates than genome-wide four-fold degenerate sites (proxy for neutral evolution), showing that purifying selection is acting to preserve sRNAs in both bacteria, probably due to their contribution to bacterial fitness, as shown previously for miRNAs in humans, Drosophila, and Caenorhabditis (Quach et al. 2009; Jovelin and Cutter 2014; Lyu et al. 2014).

sRNAs are under purifying selection. Selective constrains are stronger on sRNAs (n = 38) than on neutral sites (4-fold degenerate sites; n = 100). sRNAs present in the common ancestor of all six species were considered old and the rest as young. p-values calculated using Kruskal–Wallis test and Dunn’s test for multiple comparisons
Discussion
Although sRNAs are critical to gene regulation, we lack a clear understanding of how they originate and evolve in bacteria. In this study, we show that young sRNAs are expressed at low levels and evolve at faster rates than older sRNAs, thereby uncovering a novel process that potentially facilitates the establishment of new sRNAs in bacterial genomes. We also discovered that an sRNA (EcsR2) emerged from a degraded bacteriophage protein-coding gene, thus revealing the first known source for sRNA origination in bacteria. Similar to the origination of EcsR2, new ncRNA genes in eukaryotes have arisen from the remnants of protein-coding genes by gaining regulatory motifs (Kaessmann 2010; Ruiz-Orera et al. 2015). Additionally, in eukaryotes, the evolution of a spurious transcript into a functional ncRNA is associated with changes in the RNA’s secondary structure (Heinen et al. 2009). In concordance with this observation, in EcsR2, the putative mRNA-binding region appears to have become more unstructured, whereas the intrinsic terminator likely became more structured (Figs. 6, S3). Analogous to EcsR2, another E. coli-specific sRNA IsrA (McaS) (Jørgensen et al. 2013) is also evolving at a rapid rate (π = 0.050), validating the observation that young sRNAs evolve swiftly in bacteria. Most of the other sRNAs with similarly high rates of evolution are antisense RNAs that are part of toxin–antitoxin systems (Fozo et al. 2008). Interestingly, SgrS, an sRNA present in several Gammaproteobacteria (Horler and Vanderpool 2009), displayed an elevated rate of evolution (π = 0.060). However, a closer examination revealed that the sRNA has diverged considerably in 30 out of the 85 E. coli strains used in our analysis. If we consider only the other 55 genomes, the nucleotide diversity value for SgrS falls within the expected range for older sRNAs (π = 0.0029). The reasons for the disparity in SgrS evolutionary rates in the two E. coli cohorts are unknown.
Similar to protein-coding genes, most sRNA genes are evolving under purifying selection, suggestive of their importance to bacterial fitness; however, young sRNAs are evolving much more rapidly than evolutionarily older sRNAs, and young sRNAs are expressed at significantly lower levels than established sRNAs. One of the probable causes for the low expression could be that their promoters are not yet fully functional. In bacteria, promoter-like sequences arise spontaneously through point mutations, especially in IGRs (Mendoza-Vargas et al. 2009), and inefficient transcription from these promoters is the main source of pervasive transcripts i.e., RNAs originating from all across the genome (Dornenburg et al. 2010; Raghavan et al. 2012; Thomason et al. 2015). The functions, if any, of these genome-wide transcripts are not yet understood, but they could serve as the raw material for the emergence of new functional RNAs (Gottesman and Storz 2011; Wade and Grainger 2014; Lybecker et al. 2014). Pervasive transcription has been observed in all domains of life, and recently it was shown that new functional RNAs could evolve from such transcripts in humans (Ruiz-Orera et al. 2015). Our data also point towards such a scenario where the emergence of a promoter-like sequence resulted in the production of a transcript that evolved into EcsR2 by gaining regulatory motifs.
To be functional, an sRNA only requires a small seed sequence with partial complementarity to an mRNA; therefore, several such target mRNAs should occur in a bacterial genome just through chance. Although a few nascent sRNA–mRNA interactions might have positive outcomes, most are likely deleterious, which could be mitigated by the low expression of incipient sRNAs, while new beneficial interactions could arise through rapid sRNA evolution. Similar to what we show in young enterobacterial sRNAs, low expression and rapid evolution have been observed in young miRNAs (Chen and Rajewsky 2007; Jovelin and Cutter 2014; Lyu et al. 2014), suggesting that this a universal process that facilitates the emergence of new non-coding regulatory RNAs in all domains of life.
Materials and Methods
Bacterial Strains and Plasmids
Escherichia coli K-12 MG1655 was used in all experiments. For EcsR2 expression vector construction, EcsR2 gene was amplified using the following primers: 5′ATGCTAGCGCAGATAGTCAGTGAGTATATC3′, 5′GACGTCGCAGATAGTC-AGTGAGTATATC3′, and cloned into the plasmid pBAD (Guzman et al. 1995) by digesting both the PCR product and pBAD with NheI and AatII restriction enzymes (restriction sites on primers are underlined). EcsR2-deletion strain was constructed using λ Red-mediated recombination (Datsenko and Wanner 2000).
RNA-Seq and Crosslink-Seq
Highest level of EcsR2 expression was observed during exponential phase growth (Fig. S4). Hence, for the RNA-seq analysis, E. coli transformed with either empty pBAD (control) or pBAD with cloned EcsR2 (test) that were grown in Lysogeny Broth (LB) aerobically to OD600 of ~0.5. Cultures were supplemented with arabinose (0.2%) for 10 min to induce the expression of EcsR2, 0.2 volume stop solution (5% water-saturated phenol, 95% ethanol) was added, and the cells were harvested by centrifugation. Total RNA was extracted using TRI reagent, treated with DNase, and ribosomal RNAs were removed using MICROBExpress kit (Life Technologies). RNA-seq (Illumina HiSeq 2000, 100 cycles, single-end) was performed using two control and test samples at the Genomic Sequencing and Analysis Facility at the University of Texas at Austin. The trimmed reads were mapped to the E. coli genome (NC_000913.2) using CLC Genomics Workbench to identify genes that were differentially expressed. The RNA-seq reads are available on NCBI SRA (accession: SRP044074).
Crosslink-seq was adapted from Lustig et al. 2010 and Liu et al. 2015. EcsR2-deletion strain containing either empty pBAD (control) or pBAD with cloned EcsR2 (test) were grown in LB aerobically to OD600 of ~0.5 and cultures were supplemented with arabinose (0.2%) for 10 min to induce the expression of EcsR2. Cells were washed twice with PBS, resuspended in 8 mL of fresh PBS, and 0.2 mg/mL 4′- Aminomethyltrioxsalen (Cayman Chemicals) was added. The cells were incubated on ice for 10 min, and were irradiated with UV light at 365 nm for 1 h on ice. The cells were washed once with PBS and total RNA was isolated using TRI reagent. RNA treated with DNase was mixed in hybridization buffer (20 nM HEPES pH8, 5 mM MgCl2, 300 mM KCl, 0.01% NP-40, 1 mM DTT) and heated at 80 °C for 2 min followed by immediate cooling on ice. 10 nmol biotinylated oligonucleotides that were antisense to a portion of EcsR2 were added to the samples and incubated at room temperature overnight. 150 μL of NeutrAvidin agarose resin (Thermo Fisher) was washed twice in WB100 buffer (20 mM HEPES pH 8, 10 mM MgCl2, 100 mM KCl, 0.01% NP-40, 1 mM DTT) followed by blocking the beads for 2 h (blocking buffer: WB100, 50 μL BSA (10 mg/mL), 40 μL tRNA (10 mg/mL), 10 μL glycogen (20 mg/mL)). The blocked beads were once again washed with blocking buffer and added to the hybridized RNAs bound to the biotinylated oligos. Samples were incubated for 4 h at 4 °C and then washed five times with WB400 buffer (20 mM HEPES pH 8, 10 mM MgCl2, 400 mM KCl, 0.01% NP40, 1 mM DTT). The hybridized RNAs bound to the beads were isolated using TRI reagent. The affinity-selected, crosslinked mRNAs were released from EcsR2 using UV light at 254 nm on ice for 15 min. The RNA samples were deep-sequenced at Oregon Health and Science University Massively Parallel Sequencing Shared Resource (Illumina HiSeq 2500, 100 cycles, single-end), and the trimmed reads were mapped to E. coli genome (NC_000913.2) using CLC Genomics Workbench to determine the genes that were enriched in test samples (expressing EcsR2) in comparison to controls (no EcsR2). Gene expression was calculated from two independent experiments, and the RNA-seq reads are available on NCBI SRA (accession: SRP074317).
For qRT-PCR confirmation, EcsR2-deletion strain containing empty pBAD, or pBAD with cloned full-length EcsR2, or pBAD with EcsR2 in which the +51 to +80 region was deleted using inverse PCR were used. Bacteria were grown in LB aerobically to OD600 of ~0.5. Cultures were supplemented with arabinose (0.2%) for 10 min to induce the expression of EcsR2, and 0.2 volume stop solution was immediately added, and the cells were harvested by centrifugation. Total RNA was extracted using TRI reagent, treated with DNase, and qRT-PCR was performed as previously described (Raghavan et al. 2011).
Expression and Evolution of sRNAs
We used previously published RNA-seq data to determine the expression of sRNAs (Raghavan et al. 2011; Kröger et al. 2013). To identify the homologs of 92 sRNAs described in E. coli K-12 MG1655 (NC_000913.2) (Raghavan et al. 2011, 2015), we used BLASTn (E value < 10−5 and target length ≥60% of query length) to search 146 fully sequenced E. coli genomes available on NCBI. We ultimately chose 81 sRNAs that were conserved in 85 E. coli genomes in order to maximize the number of genomes and sRNAs (Supplemental dataset 1). Similarly, we searched 151 S. enterica genomes to identify the homologs of 170 sRNAs described in S. enterica Typhimurium SL1344 (NC_016810.1) (Kröger et al. 2013), and chose 127 sRNAs that are conserved in 112 S. enterica genomes for further analyses (Supplemental dataset 1). Sequences were aligned using Clustal Omega (Sievers et al. 2011), and nucleotide differences were quantified using nucleotide diversity index π (Nei 1987; Jovelin and Cutter 2014) with DnaSP 5.10 (Librado and Rozas 2009). Briefly, π was calculated by summing, over all distinct pairs of sequences in the sample, the proportion of different nucleotides between a pair of sequences multiplied by the respective frequencies of those sequences. To calculate the average nucleotide differences throughout the yagU–ykjG IGR, we used a sliding window of 35 bp and a step size of 15 bp using DnaSP. RNA secondary structure and minimum free energy were predicted using Vienna RNA webserver (Gruber et al. 2008) and Mfold webserver (Zuker 2003), and EcsR2–AnsB interaction was modeled using IntaRNA (Wright et al. 2014).
To determine whether the sRNAs in E. coli and S. enterica were present in other enteric bacteria, sRNA gene sequences were searched (BLASTn, E < 10−5 and target length ≥60% of query length) against the following representative genomes (as denoted by NCBI Genome database): Citrobacter freundii (NZ_CP007557.1), Klebsiella pneumoniae (NC_016845.1), Serratia marcescens (NZ_HG326223.1), and Yersinia enterocolitica (NC_008800.1). PMCMR R package was used to perform both Kruskal–Wallis test (non-parametric 1-way ANOVA) to assess differences in expression and nucleotide diversity between sRNA age classes, and the post hoc pairwise comparison Dunn’s test. For analyzing S. enterica expression data from 22 growth conditions (Kröger et al. 2013), Permutational ANOVA (non-parametric 2-way ANOVA) was conducted using perm.anova, and post hoc pairwise comparisons were conducted using pairwise.perm.t.test with FDR correction in the RVAideMemoire R package. To analyze sRNA evolution in more detail, 38 sRNAs that were conserved in at least 50% of currently available complete genomes of E. coli (74 strains) and S. enterica (102 strains) were chosen (Supplemental dataset 1). To detect purifying selection, within-species polymorphism and between-species divergence were calculated using DnaSP; four-fold degenerate sites (in hundred randomly selected genes; Supplemental dataset 1) were used as control because they are considered to evolve neutrally (Ochman and Wilson 1987).
References
Bernick DL, Dennis PP, Lui LM, Lowe TM (2012) Diversity of antisense and other non-coding RNAs in archaea revealed by comparative small RNA sequencing in four Pyrobaculum species. Front Microbiol 3:1–18. doi:10.3389/fmicb.2012.00231
Breaker RR (2011) Prospects for riboswitch discovery and analysis. Mol Cell 43:867–879. doi:10.1016/j.molcel.2011.08.024
Chen K, Rajewsky N (2007) The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 8:93–103. doi:10.1038/nrg1990
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci 97:6640–6645. doi:10.1073/pnas.120163297
Dornenburg J, DeVita A, Palumbo M, Wade J (2010) Widespread antisense transcription in Escherichia coli. MBio 1:e00024. doi:10.1128/mBio.00024-10.Updated
Fozo EM, Hemm MR, Storz G (2008) Small toxic proteins and the antisense RNAs that repress them. Microbiol Mol Biol Rev 72:579–589. doi:10.1128/MMBR.00025-08
Gardner PP, Wilm A, Washietl S (2005) A benchmark of multiple sequence alignment programs upon structural RNAs. Nucleic Acids Res 33:2433–2439. doi:10.1093/nar/gki541
Georg J, Hess WR (2011) cis-antisense RNA, another level of gene regulation in bacteria. Microbiol Mol Biol Rev 75:286–300. doi:10.1128/MMBR.00032-10
Gottesman S, Storz G (2011) Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol 3:a003798. doi:10.1101/cshperspect.a003798
Gruber AR, Lorenz R, Bernhart SH et al (2008) The Vienna RNA websuite. Nucleic Acids Res 36:W70–W74. doi:10.1093/nar/gkn188
Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promoter. J Bacteriol 177:4121–4130. doi:10.1128/jb.177.14.4121-4130.1995
Heinen TJAJ, Staubach F, Häming D, Tautz D (2009) Emergence of a new gene from an intergenic region. Curr Biol 19:1527–1531. doi:10.1016/j.cub.2009.07.049
Hoeppner MP, Gardner PP, Poole AM (2012) Comparative analysis of RNA families reveals distinct repertoires for each domain of life. PLoS Comput Biol 8:e1002752. doi:10.1371/journal.pcbi.1002752
Horler RSP, Vanderpool CK (2009) Homologs of the small RNA SGRS are broadly distributed in enteric bacteria but have diverged in size and sequence. Nucleic Acids Res 37:5465–5476. doi:10.1093/nar/gkp501
Jørgensen MG, Thomason MK, Havelund J et al (2013) Dual function of the McaS small RNA in controlling biofilm formation. Genes Dev 27:1132–1145. doi:10.1101/gad.214734.113
Jovelin R, Cutter AD (2014) Microevolution of nematode miRNAs reveals diverse modes of selection. Genome Biol Evol 6:3049–3063. doi:10.1093/gbe/evu239
Kaessmann H (2010) Origins, evolution, and phenotypic impact of new genes. Genome Res 20:1313–1326. doi:10.1101/gr.101386.109
Kortmann J, Narberhaus F (2012) Bacterial RNA thermometers: molecular zippers and switches. Nat Rev Microbiol 10:255–265. doi:10.1038/nrmicro2730
Kröger C, Colgan A, Srikumar S et al (2013) An infection-relevant transcriptomic compendium for Salmonella enterica serovar Typhimurium. Cell Host Microbe 14:683–695. doi:10.1016/j.chom.2013.11.010
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi:10.1093/bioinformatics/btp187
Lindgreen S, Umu SU, Lai ASW et al (2014) Robust identification of noncoding RNA from transcriptomes requires phylogenetically-informed sampling. PLoS Comput Biol 10:e1003907. doi:10.1371/journal.pcbi.1003907
Liu N, Niu G, Xie Z et al (2015) The Streptococcus mutans irvA gene encodes a trans-acting riboregulatory mRNA. Mol Cell 57:179–190. doi:10.1016/j.molcel.2014.11.003
Lustig Y, Wachtel C, Safro M et al (2010) “RNA walk” a novel approach to study RNA-RNA interactions between a small RNA and its target. Nucleic Acids Res 38:e5. doi:10.1093/nar/gkp872
Lybecker M, Bilusic I, Raghavan R (2014) Pervasive transcription: detecting functional RNAs in bacteria. Transcription 5:e944039. doi:10.4161/21541272.2014.944039
Lyu Y, Shen Y, Li H et al (2014) New microRNAs in Drosophila-birth, death and cycles of adaptive evolution. PLoS Genet 10:e1004096. doi:10.1371/journal.pgen.1004096
Mendoza-Vargas A, Olvera L, Olvera M et al (2009) Genome-wide identification of transcription start sites, promoters and transcription factor binding sites in E. coli. PLoS ONE 4:e7526. doi:10.1371/journal.pone.0007526
Michaux C, Verneuil N, Hartke A, Giard J-C (2014) Physiological roles of small RNA molecules. Microbiology 160:1007–1019. doi:10.1099/mic.0.076208-0
Miyakoshi M, Chao Y, Vogel J (2015) Regulatory small RNAs from the 3′ regions of bacterial mRNAs. Curr Opin Microbiol 24:132–139. doi:10.1016/j.mib.2015.01.013
Nawrocki EP, Burge SW, Bateman A et al (2015) Rfam 12.0: updates to the RNA families database. Nucleic Acids Res 43:D130–D137. doi:10.1093/nar/gku1063
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Ochman H, Wilson AC (1987) Evolution in bacteria: evidence for a universal substitution rate in cellular genomes. J Mol Evol 26:74–86. doi:10.1007/BF02111283
Peer A, Margalit H (2011) Accessibility and evolutionary conservation mark bacterial small-RNA target-binding regions. J Bacteriol 193:1690–1701. doi:10.1128/JB.01419-10
Peer A, Margalit H (2014) Evolutionary patterns of Escherichia coli small RNAs and their regulatory interactions. RNA 20:994–1003. doi:10.1261/rna.043133.113
Quach H, Barreiro LB, Laval G et al (2009) Signatures of purifying and local positive selection in human miRNAs. Am J Hum Genet 84:316–327. doi:10.1016/j.ajhg.2009.01.022
Raghavan R, Groisman EA, Ochman H (2011) Genome-wide detection of novel regulatory RNAs in E. coli. Nucleic Acids Res 10:1487–1497. doi:10.1101/gr.119370.110.21
Raghavan R, Sloan DB, Ochman H (2012) Antisense transcription is pervasive but rarely conserved in enteric bacteria. MBio 3:e00156. doi:10.1128/mBio.00156-12.Editor
Raghavan R, Kacharia Fenil R, Millar Jess A et al (2015) Genome rearrangements can make and break small RNA genes. Genome Biol Evol 7:557–566. doi:10.1093/gbe/evv009
Richter AS, Backofen R (2012) Accessibility and conservation: general features of bacterial small RNA-mRNA interactions? RNA Biol 9:954–965. doi:10.4161/rna.20294
Ruiz-Orera J, Hernandez-Rodriguez J, Chiva C et al (2015) Origins of de novo genes in human and chimpanzee. PLoS Genet 11:1–24. doi:10.1371/journal.pgen.1005721
Sievers F, Wilm A, Dineen D et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi:10.1038/msb.2011.75
Skippington E, Ragan MA (2012) Evolutionary dynamics of small RNAs in 27 Escherichia coli and Shigella genomes. Genome Biol Evol 4:330–345. doi:10.1093/gbe/evs001
Storz G, Vogel J, Wassarman KM (2011) Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43:880–891. doi:10.1016/j.pestbp.2011.02.012.Investigations
Thomason M, Storz G (2010) Bacterial antisense RNAs: how many are there and what are they doing? Annu Rev Genet 44:167–188. doi:10.1146/annurev-genet-102209-163523.Bacterial
Thomason MK, Bischler T, Eisenbart SK et al (2015) Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol 197:18–28. doi:10.1128/JB.02096-14
Updegrove TB, Shabalina SA, Storz G (2015) How do base-pairing small RNAs evolve? FEMS Microbiol Rev 39:379–391. doi:10.1093/femsre/fuv014
Wade JT, Grainger DC (2014) Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat Rev Microbiol 12:647–653. doi:10.1038/nrmicro3316
Wang J, Rennie W, Liu C et al (2015) Identification of bacterial sRNA regulatory targets using ribosome profiling. Nucleic Acids Res 43:10308–10320. doi:10.1093/nar/gkv1158
Wilson RC, Doudna J (2013) Molecular mechanisms of RNA interference. Annu Rev Biophys 42:217–239. doi:10.1146/annurev-biophys-083012-130404
Wright PR, Georg J, Mann M et al (2014) CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res 42:W119–W123. doi:10.1093/nar/gku359
Zhang A, Altuvia S, Tiwari A et al (1998) The OxyS regulatory RNA represses rpoS translation and binds the Hfq (HF-I) protein. EMBO J 17:6061–6068. doi:10.1093/emboj/17.20.6061
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi:10.1093/nar/gkg595
Acknowledgements
We thank Justin Merritt, Nan Liu, Abraham Moses, and Jim Archuleta for technical help. This work was supported in part by Portland State University. J.A.M. was supported by the Forbes-Lea Research Fund and by a Sigma Xi Grants-in-Aid of Research award (G201510151633590).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kacharia, F.R., Millar, J.A. & Raghavan, R. Emergence of New sRNAs in Enteric Bacteria is Associated with Low Expression and Rapid Evolution. J Mol Evol 84, 204–213 (2017). https://doi.org/10.1007/s00239-017-9793-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-017-9793-9