
Abstract
Most of the largest vertebrate genomes are found in salamanders, a clade of amphibians that includes 686 species. Salamander genomes range in size from 14 to 120 Gb, reflecting the accumulation of large numbers of transposable element (TE) sequences from all three TE classes. Although DNA loss rates are slow in salamanders relative to other vertebrates, high levels of TE insertion are also likely required to explain such high TE loads. Across the Tree of Life, novel TE insertions are suppressed by several pathways involving small RNA molecules. In most known animals, TE activity in the germline is primarily regulated by the Piwi-interacting RNA (piRNA) pathway. In this study, we test the hypothesis that salamanders’ unusually high TE loads reflect the loss of the ancestral piRNA-mediated TE-silencing machinery. We characterized the small RNA pool in the female and male adult gonads, testing for the presence of small RNA molecules that bear the characteristics of TE-targeting piRNAs. We also analyzed the amino acid sequences of piRNA pathway proteins from salamanders and other vertebrates, testing whether the overall patterns of sequence divergence are consistent with conserved pathway function across the vertebrate clade. Our results do not support the hypothesis of piRNA pathway loss; instead, they suggest that the piRNA pathway is expressed in salamanders. Given these results, we propose hypotheses to explain how the extraordinary TE loads in salamander genomes could have accumulated, despite the expression of TE-silencing machinery.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Most of the largest vertebrate genomes are found in salamanders, a clade of amphibians that includes 686 recognized species (AmphibiaWeb 2015). Salamander genomes range in size from 14 to 120 Gb. These sizes are an order of magnitude larger than bird, mammal, reptile, and frog genomes, as well as all fish genomes with the exception of the six species of lungfish (Gregory 2016). The phenotypic correlates of genomic gigantism in salamanders include reduced neural, visual, and skeletal complexity, unusual blood cell morphology, slow developmental rate, and low metabolic rate (Jockusch 1997; Mueller et al. 2008; Roth et al. 1997; Szarski 1983).
Paleohistological analyses of fossil cell size in extinct tetrapods confirm that the gigantic genomes of living salamanders are a derived trait (Organ et al. 2011). All species of extant crown salamanders examined to date, as well as one of the earliest known stem salamanders, have large genomes, suggesting that genome sizes in the clade have been gigantic for at least 150 to 200 million years (Gregory 2016; Laurin et al. 2015; Marjanović and Laurin 2007; Mueller 2006). Salamander genomes are diploid; their enormous sizes do not reflect increases in ploidy (Sessions 2008; Sessions and Kezer 1991). Instead, they reflect the accumulation of unusually large numbers of transposable element (TE) sequences (Sun and Mueller 2014; Sun et al. 2012b).
Within the salamander clade, genome size has both increased and decreased across lineages, with increases outnumbering decreases by a factor of at least three (Sessions 2008; Sessions and Larson 1987). Additionally, the TE landscape varies across salamander lineages (Sun and Mueller 2014). These patterns demonstrate that the substantial repetitive portion of salamander genomes is dynamic; these genomes have been large for hundreds of millions of years, but not because the same repetitive sequences have persisted for hundreds of millions of years. Rather, each salamander lineage has experienced accumulation and removal of TE sequences, as is the case for all eukaryotes. In salamanders, however, the long-term balance between these two processes has been struck at a different point than in other vertebrates, producing unusually high—albeit variable—TE loads across the clade.
What explains the accumulation and persistence of such high TE levels in salamanders? DNA loss rate has been proposed to be a major determinant of genome size (Petrov 2002). Under this model, the mutational spectrum drives genome size towards an equilibrium value set, in large part, by the deletion rate. DNA loss rates from salamander genomes through small deletions (i.e., <30 bp) are slower than from vertebrate genomes of typical size, suggesting that slow rates of DNA loss through small deletions contribute to genomic gigantism in salamanders (Sun et al. 2012a; Sun and Mueller 2014). However, slow rates of DNA loss through small indels are likely insufficient, in and of themselves, to explain salamanders’ enormous genomes; this result is consistent with analyses of DNA loss rates and genome sizes across other taxa (Gregory 2004). LTR retrotransposon deletion through ectopic recombination also appears to be less common in salamanders than in other vertebrates (Frahry et al. 2015). Thus, slow rates of DNA loss through large deletions also contribute to genomic gigantism in salamanders, although the total contribution cannot be quantified with existing data. However, given that as much as 75 % of salamander genomes consist of relatively recently active repeat elements (i.e., repeats that can be identified by sequence similarity-based methods) (Sun and Mueller 2014; Sun et al. 2012b), it is likely that slow rates of DNA loss, through both small and large deletions, cannot fully explain the TE accumulation underlying salamanders’ gigantic genomes.
The strength of genetic drift, reflecting the effective population size (N e), has also been hypothesized to be a major determinant of genome size (Lynch 2007; Lynch and Conery 2003). Under this model, the deleterious fitness consequences of non-coding DNA (including TEs) are weak enough that, in lineages with low N e, the power of genetic drift overwhelms selection’s ability to purge non-coding sequences from the population. To date, there is no evidence that crown salamanders have experienced stronger genetic drift throughout their evolutionary history than related amphibians with more typically sized vertebrate genomes (Mohlhenrich and Mueller 2016). This result suggests that strong persistent genetic drift cannot explain the TE accumulation underlying salamanders’ gigantic genomes.
Taken together, the results from the analyses of deletion and drift suggest that high levels of TE insertion are required to explain salamanders’ high TE loads. Despite the fact that neither deletion nor drift alone appears sufficient to explain persistent genomic gigantism in salamanders, these two processes interact in an important way. Deletions involving TEs are a major determinant of their mutational hazard and, by extension, their negative impacts on fitness. Thus, TE sequences that mediate/sustain fewer deletions are less deleterious than those that mediate/sustain more deletions. For example, TE insertions more likely to mediate ectopic recombination events—which can produce large deletions as well as duplications—are stronger targets of purifying selection than TEs less likely to mediate such events (Barrón et al. 2014). Additionally, TEs that sustain more small deletions have a higher probability of mutating to a harmful gain-of-function allele. Because of their low rates of both large and small deletions, TEs in salamander genomes likely have a smaller negative impact on fitness—based solely on mutational hazard—than TEs in other vertebrate genomes. This low mutational hazard is consistent with high levels of TE insertion being tolerated in salamander genomes.
What might facilitate high levels of TE insertion in salamander genomes? Across the Tree of Life, novel TE insertions are suppressed by several pathways involving small RNA molecules (Malone and Hannon 2009; Siomi et al. 2011). Differences in these pathways are apparent among different model organisms, both within and among major eukaryotic clades (Dumesic and Madhani 2014). In most animals, TE activity in the germline (i.e., the activity that directly impacts genome evolution) is primarily regulated by the Piwi-interacting RNA (piRNA) pathway (Grimson et al. 2008; Siomi et al. 2011). Relative to other endogenous small RNA classes (e.g., microRNAs and endogenous small interfering RNAs), piRNAs are the largest and most diverse class of non-coding RNAs, and they are produced by a distinct biogenesis pathway (Iwasaki et al. 2015). piRNAs are bound by proteins in the Piwi clade of the Argonaute family and guide their suppression of TE activity (Clark and Lau 2014; Dumesic and Madhani 2014). piRNAs also regulate germline gene expression (Castel and Martienssen 2013). Although much about piRNA biology remains incompletely understood (Clark and Lau 2014), models of transcriptional and post-transcriptional TE suppression are becoming established (Yamanaka et al. 2014).
In this study, we test the hypothesis that salamanders’ unusually high TE loads reflect the loss of the ancestral piRNA-mediated TE-silencing machinery. We deeply sequenced small RNA molecules in the female and male adult gonads in order to identify sequences that bear the characteristics of TE-targeting piRNAs —a length of 27–31 nt, a bias towards 5′ U, base complementarily to transposable element sequences, and, in some cases, an antisense binding partner with a 10-nucleotide overlap. We also examined the amino acid sequences of 12 piRNA pathway proteins from salamanders and other vertebrates, testing whether the overall patterns of sequence divergence are consistent with conserved pathway function across the vertebrate clade. Our results do not support the hypothesis of piRNA pathway loss; instead, they suggest that the piRNA pathway is expressed in salamanders. Given these results, we propose hypotheses to explain how the extraordinary TE loads in salamander genomes could have accumulated, despite the expression of TE-silencing machinery.
Materials and Methods
Sample Information
We obtained two adult Desmognathus fuscus (one female, one male) from Wilkes County, North Carolina on May 13–15, 2012. GPS coordinates for the samples are 36.116072, −81.128333 for the female and 36.07151, −81.176845 for the male. Desmognathine salamanders have the smallest genomes among salamanders (i.e., ~15 Gb); thus, they are a reasonable system to begin exploring small RNA-mediated TE suppression in salamanders because future research incorporating genomic information (e.g., on piRNA clusters) will be as tractable as possible. Animals were euthanized by immersion in chloretone or benzocaine, decapitated, and dissected immediately in accordance with the Colorado State University Institutional Animal Care and Use Committee (IACUC) protocol #11-2775A and the Brandeis University IACUC protocol #13008. Ovaries and testes were dissected from female and male, respectively, flash frozen, and stored at −80 °C. Based on visual inspection, the majority of the ovarian tissue was comprised of late-stage eggs; thus, the bulk of RNA from the female sample likely reflects maternal deposition into eggs. However, our dataset will also include a small fraction of piRNAs produced in the somatic gonadal tissue of females. RNA was extracted using TRIzol® according to manufacturer’s protocols. RNA quality was assessed on a Bioanalyzer (Agilent).
Library Construction and Sequencing
Small RNA libraries and standard RNA-Seq libraries were constructed for both samples. All samples were Turbo-DNased (Ambion) prior to library construction. For small RNA libraries, library construction was performed with the IntegenX PrepX RNA library preparation kit with the PrepX small RNA 8 protocol. For standard RNA-Seq libraries, samples were treated with Invitrogen’s RiboMinus Eukaryote Kit for RNA-Seq. Double-stranded cDNA was synthesized using both poly-A enrichment (i.e., oligo-dT priming) and non-poly-A enrichment (i.e., random priming) because of our interest in capturing diverse transcripts to aid in identifying piRNA targets (e.g., transposons) and precursor loci (e.g., piRNA clusters, which may or may not be polyadenylated). The resulting cDNA was used to make shotgun libraries with the IntegenX PrepX DNA library preparation kit; the Chip-Seq library prep method was used, rather than the standard shotgun method, to avoid biasing the library based on insert length. RNA-Seq libraries and small RNA libraries were sequenced on the Illumina MiSeq (2 × 300 PE) platform. Small RNA libraries were then sequenced to greater depth of coverage on an Illumina HiSeq 2000 (50 SR). RNA quality assessment, library construction, and sequencing were all performed by the Genomics Resources Core of the Institute for Bioinformatics and Evolutionary Studies (IBEST) at the University of Idaho.
Transcriptome Assembly and Annotation
For each individual (i.e., female, male), low-quality reads were eliminated with Trimmomatic (Bolger et al. 2014), and the remaining shotgun reads were assembled with Trinity using default parameters (Grabherr et al. 2011). For the resulting female and male assemblies, we annotated contigs containing TEs using BLASTx against TE-encoded proteins (http://www.repeatmasker.org/RepeatProteinMask.html#database), with an e-value cutoff of 1e−5. We annotated contigs containing non-TE-derived protein-coding genes using BLASTx against the Swiss-Prot protein database (http://www.uniprot.org/uniprot/), with an e-value cutoff of 1e−5.
Small RNA Pool Characterization
Following quality filtering and adapter trimming, we sorted all small RNAs by size and focused our analyses on those between 18 and 36 nt in length. We plotted the length distribution of these sequences in the female and male samples and looked for a peak at the expected size of piRNAs (27–31 nt). We also looked for a peak at the expected size of siRNAs (22–23 nt), as this class of small RNAs has also been shown to target TEs in Xenopus tropicalis (Armisen et al. 2009). We used Bowtie 1.1.2, allowing 0 mismatches, to map the 22–23 nt RNAs from each sample against miRBase to identify microRNAs (Langmead et al. 2009). We used the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) to determine the number of unique small RNA sequences.
Characterization of Putative piRNAs
We tested whether the sequences comprising the peaks between 27 and 31 nt (i.e., the putative piRNAs) in the female and male samples show the characteristic piRNA bias towards having a uracil in the 5′ position by calculating the proportion of sequences with a 5′ U using in-house Perl scripts.
Identification of Small RNA Targets
Small RNAs guide effector proteins to their target sequences by base complementarity. Accordingly, we mapped the four relevant subsets of the total small RNA dataset (i.e., the female and male putative piRNAs, and the female and male putative siRNAs) to several other datasets to identify their targets using Bowtie (version: bowtie-0.12.9; command: bowtie-a -v 1 -best -strata). First, we mapped each small RNA dataset to its respective reference transcriptome. Next, we mapped each small RNA dataset to the reference transcriptome from the other sex. Finally, we mapped each small RNA dataset to the TE-containing contigs we assembled from our previously published 454 genomic shotgun sequence data from the related species Desmognathus ochrophaeus. Our goal in mapping to the latter two datasets was to identify piRNA targets and precursor transcripts that were not present in the reference transcriptome dataset derived from the same individual as the small RNA dataset. This absence could reflect either methodology (i.e., the transcript was not sequenced and/or assembled, despite the locus being expressed) or true biological reality (i.e., the transcript was not present in the tissue because the locus was transcriptionally silenced). We allowed up to one mismatch when mapping small RNAs to the two reference transcriptomes. When mapping to the D. ochrophaeus genomic shotgun contigs, however, we allowed up to two mismatches to reflect the evolutionary distance between the two species. We suggest that these annotations should be interpreted with less confidence. For each small RNA dataset, we calculated the proportion of small RNAs mapping to the three classes of transposable elements—LTR retrotransposons, non-LTR retrotransposons, and DNA transposons. Within each class, we ranked transposable element superfamilies by the density of mapped small RNAs. We also calculated the proportion of small RNAs mapping to protein-coding genes.
Measurement of Ping-Pong Cycle Activity in Putative piRNAs
In certain gonadal cell developmental stages and compartments, piRNAs can be generated by a ping-pong cycle that yields TE-matching sense and antisense piRNAs. The piRNAs generated by this mechanism exhibit a signature: two piRNAs of opposite polarity that display a 10-nucleotide overlap in their genomic position (Aravin et al. 2007; Brennecke et al. 2007; Gunawardane et al. 2007; Wang et al. 2014; Zhang et al. 2011). In male mice, ping-pong amplification is most prevalent during embryogenesis (Aravin et al. 2007, 2008), but a ping-pong signature is present in adult male and female gonads of other mammals, zebrafish, and Xenopus (Armisen et al. 2009; Houwing et al. 2007; Robine et al. 2009; Roovers et al. 2015). Accordingly, we calculated the overall ping-pong fraction in the female and male putative piRNA datasets. We used RepeatMasker (version 3.2.9; http://www.repeatmasker.org), with the TE sequences annotated from the RNA-Seq datasets as a custom repeat library, to identify TE-mapping piRNAs and determine their sense/antisense orientation relative to the TE transcripts. We used the intersectBed function in BEDTools (Quinlan and Hall 2010) to identify sense/antisense piRNA pairs with a 10-nucleotide overlap. We then identified the individual TE families targeted by ping-pong pairs in both samples.
Analysis of piRNA Pathway Proteins
To complement our analysis of the piRNA pool in salamanders, we also analyzed the amino acid sequences of proteins known to be involved in the piRNA pathway. We chose 12 proteins with diverse roles in piRNA-mediated TE silencing whose patterns of molecular evolution were previously analyzed in diverse vertebrates—ASZ1/GASZ, DDX4, FKBP6, HEN1, KIF17, MOV10L1, PIWIL1, PIWIL2, PLD6, PRMT5, TDRD1, and TDRKH (Yi et al. 2014). We used reciprocal best hit BLAST searches to identify the contigs containing orthologs of each gene from transcriptome datasets for three salamander species: D. fuscus (present study), Ensatina eschscholtzii (Mohlhenrich and Mueller 2016), and Cryptobranchus alleganiensis (Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.kv57r) using the human protein sequences as queries. We obtained the amino acid sequences of orthologs from other non-salamander vertebrates (Anolis carolinensis, Bos taurus, Danio rerio, Gallus gallus, Gasterosteus aculeatus, Homo sapiens, Monodelphis domestica, Monopterus albus, Mus musculus, Oreochromis niloticus, Oryzias latipes, Petromyzon marinus, Python bivittatus, Taeniopygia guttata, Takifugu rubripes, Tetraodon nigroviridis, Thamnophis sirtalis, Xenopus (Silurana) tropicalis, and Xiphophorus maculatus) as well as the salamander Ambystoma mexicanum from GenBank and EMBL. For each protein-coding gene, we performed multiple sequence alignments of amino acid sequences using PSI-coffee, an aligner in the T-coffee package that aligns distantly related sequences based on homology extension. We trimmed alignments to positions receiving a T-coffee alignment score of “good” or better and estimated phylogenetic trees from each alignment in MrBayes 3.2, using a mixed model of amino acid substitution (Notredame et al. 2000; Ronquist et al. 2012). We ran each analysis for 10,000,000 generations, sampling every 1000, with three heated chains. We discarded 25 % of the sampled trees as burn-in and verified convergence by comparison of the average deviation of split frequencies between two independent runs. Our goal in estimating these phylogenies was to determine whether overall patterns of sequence divergence are consistent with conserved pathway function across the vertebrate clade. To this end, we asked whether salamander branch lengths differ from those in other taxa. For unrooted trees (i.e., proteins for which we were unable to obtain an ortholog from P. marinus to use as outgroup), we restricted this comparison to the tetrapod clade. To complement the phylogenetic analyses, we also tested for the presence of relevant functional domains in each salamander protein. We used the amino acid sequences of the twelve proteins from salamanders, as well as from Mus musculus and Xenopus (Silurana) tropicalis, to search against the NCBI conserved domain database using RPS-BLAST (http://www.ncbi.nlm.nih.gov/cdd).
Results
Small RNA-Seq and RNA-Seq Datasets
Summary statistics for the small RNA-Seq and RNA-Seq datasets are presented in Table 1. The small RNA-Seq dataset includes 9,364,709 reads and 13,903,326 reads for the female and male samples, respectively, in the 18–36 nt size range. We annotated contigs in the assembled RNA-Seq datasets by 19,100 protein-coding genes and 13,611 protein-coding genes in the female and male samples, respectively. We annotated 11,376 and 15,727 contigs as transposable elements in the female and male samples, respectively. All small RNA-Seq and RNA-Seq datasets are deposited in the NCBI short read archive (SRA) under BioSample accessions SAMN05785596-99.
Small RNA Pool Characterization
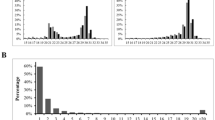
The length distributions for small RNAs between 18 and 36 nt for both female and male samples are shown in Fig. 1. There is a large peak at 27–31 nt in both samples, corresponding to the expected size of piRNAs; for brevity, we refer to these putative piRNAs as piRNAs hereafter. There are 3,859,744 total piRNA sequences (1,119,179 unique) in the female and 6,624,981 total piRNA sequences (1,006,994 unique) in the male. In the female, 67 % of piRNAs have a 5′ U. In the male, 72 % of piRNAs have a 5′ U. These data suggest that piRNAs are (1) transcribed in the adult male gonad (germline and/or somatic tissue) and (2) transcribed in the female and likely deposited into the egg. There is a smaller peak at 22–23 nt in both samples, corresponding to the expected size of siRNAs and miRNAs. In the female, 43,517 of the 22–23 nt sequences are miRNAs. In the male, 49,795 of the 22–23 nt sequences are miRNAs. For brevity, we refer to the remaining 22–23 nt RNAs as siRNAs hereafter. In the female, 41 % of the small RNA reads between 18 and 36 nt in length are piRNAs and 13 % are siRNAs. In the male, 48 % of the small RNA reads between 18 and 36 nt in length are piRNAs and 11 % are siRNAs (Table 1). Consistent with other vertebrates, piRNAs are the most abundant class of small RNAs in both female and male D. fuscus gonads.

The length distribution of small RNAs between 18 and 36 nt in a male and female D. fuscus. The peaks at 27–31 nt likely correspond to piRNAs, whereas the peaks at 22–23 nt likely correspond to siRNAs and miRNAs
Small RNA Targets or Precursors
The success rates of mapping the gonadal small RNAs to the two D. fuscus transcriptomes and the heterospecific genomic TE contigs are summarized in Table 2. The proportions of small RNAs mapping to protein-coding transcripts and transposable element classes are summarized in Table 3. We were able to map 25.1 and 31.2 % of female and male piRNAs, respectively, and 27.0 and 22.0 % of female and male siRNAs, respectively. Overall, a higher percentage of siRNAs than piRNAs mapped to protein-coding genes, and a higher percentage of piRNAs than siRNAs mapped to transposable elements. TE superfamilies with mapped small RNAs in each dataset are ranked by density of reads in Electronic Supplementary Material 1. For all small RNA datasets, Gypsy and L2 are the most frequently mapped superfamilies; these are the most abundant superfamilies in the genomes of salamanders of the family Plethodontidae (which includes D. fuscus) and among the top three most abundant superfamilies in other salamanders (Sun and Mueller 2014; Sun et al. 2012b).
Detection of the Ping-Pong Signature in Putative piRNAs
Results from the analysis of piRNA ping-pong cycle signatures are summarized in Table 4. We detected ping-pong pairs mapping to TEs in both the female and male samples. In the female, we identified 796 unique piRNAs that were a part of ping-pong pairs. These piRNAs comprised 0.07 % of the total unique piRNAs and 0.12 % of the total piRNA pool; they mapped to 87 different families representing all three TE classes. In the male, we identified 902 unique piRNAs that were a part of ping-pong pairs. These piRNAs comprised 0.09 % of the total unique piRNAs and 0.43 % of the total piRNA pool; they mapped to 60 families representing all three TE classes. Taken together, these results suggest that the ping-pong cycle occurs at low levels in the adult salamander gonads, contributing to the silencing of all three classes of transposable elements.
Analysis of piRNA Pathway Proteins
Phylogenies estimated for the twelve selected piRNA pathway proteins are shown in Fig. 2 (PIWIL1) and Electronic Supplementary Material 2 (all other proteins). All alignments are available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.kv57r. All proteins were identified in at least one salamander dataset, and all have intact ORFs. For 11 of these proteins, the branch lengths estimated for salamanders fall within the range of lengths estimated for other vertebrates; for FKBP6, all salamander branch lengths are shorter than other lineages. Although this is a conservative test, none of these patterns of sequence evolution suggests unusual rates of amino acid replacement in salamanders. Functional domains identified in each salamander protein are summarized in Table 5. For all proteins examined, all functional domains were identified with significant e-values.

Phylogeny estimated for PIWIL1 amino acid sequences. Numbers at nodes are Bayesian posterior probabilities. PIWIL1 salamander branch lengths are among the shortest on the tree; however, for the majority of piRNA pathway proteins, branch lengths for salamanders fall within those estimated for the other vertebrates, consistent with conserved pathway function across the vertebrate clade
Discussion
Our data suggest that the piRNA pathway is expressed in the salamander germline, and that the piRNA pool includes molecules complementary to TE transcripts of all three classes. Although putative piRNAs mapping to TEs have been identified in the model salamander Ambystoma mexicanum in the blastema, a dedifferentiated structure formed at the onset of limb regeneration (Zhu et al. 2012), to our knowledge, no previous studies have reported putative TE-mapping piRNAs in salamander gonads. Given our results, how is it that such high TE levels accumulate in salamander genomes? Below, we briefly summarize piRNA-mediated TE suppression and pose several hypotheses for the evolution of high TE loads, despite the expression of TE-silencing machinery.
piRNA-Mediated TE Suppression
TEs and the suppression machinery of their hosts are engaged in an arms race, resulting in a dynamic relationship that oscillates between higher and lower activity levels of individual TE families (Blumenstiel 2011). When a novel TE appears in a naïve host genome, its initial activity level can be high. Novel TEs can result from horizontal transfer or from sequence divergence during vertical transmission; in salamanders, the most abundant TE superfamily (Gypsy/Ty3) shows evidence of the latter (Sun and Mueller 2014). After this initial appearance, the host’s piRNA-mediated silencing pathway may adapt to target the novel TE through the following steps: First, the novel TE may transpose into an existing piRNA cluster locus. piRNA cluster loci are genomic regions transcribed into long RNA molecules that are processed into mature piRNAs; this processing pathway is called primary piRNA biogenesis (Brennecke et al. 2007; Girard et al. 2006; Lau et al. 2006; Malone et al. 2009; Robine et al. 2009; Vagin et al. 2006). Some piRNA cluster loci contain fragments of active and previously active TEs; a subset of the piRNAs produced from such loci is therefore complementary to TE sequences. Second, the PIWI/piRNA complex can enter the nucleus of gonadal cells and guide transcriptional silencing of complementary genomic TE loci through epigenetic modification (Huang et al. 2013; Le Thomas et al. 2013; Rozhkov et al. 2013; Sienski et al. 2012; Sytnikova et al. 2014). piRNAs bound by AUB or AGO3 proteins can also act in the cytoplasm to guide destruction of TE transcripts (Brennecke et al. 2007; Gunawardane et al. 2007; Li et al. 2009). The piRNAs formed by primary biogenesis can feed into the production of additional piRNAs through the ping-pong cycle (i.e., secondary amplification) (Brennecke et al. 2007; Gunawardane et al. 2007; Han et al. 2015; Senti et al. 2015; Wang et al. 2015). These so-called secondary piRNAs also guide TE suppression through associations with Piwi proteins. In addition, secondary piRNAs initiate phased production of diverse primary piRNAs from cleaved TE transcripts (Han et al. 2015; Mohn et al. 2015). Once piRNAs are activated against a TE family through these processes, transposition of the TE is suppressed, and its activity level in the host decreases.
How Might piRNA-Mediated TE Suppression Differ in Salamanders?
In salamanders, although LTR retrotransposons are overrepresented relative to the composition of other vertebrate genomes, all three classes of TEs are abundant (Keinath et al. 2015; Sun and Mueller 2014; Sun et al. 2012b). This suggests that salamanders’ gigantic genomes do not reflect the “escape” of a few TE families from detection/targeting by the host silencing machinery. Rather, it suggests that salamanders differ from other vertebrates in global (i.e., genome wide) TE suppression. Our analysis of gonadal gene and small RNA expression suggests the presence of the Piwi pathway in salamanders, allowing us to infer that complete loss of the Piwi pathway is an unlikely mechanism for global TE expansion in their gigantic genomes.
The global level of piRNA-mediated TE suppression reflects the extent to which all individual TE families in a genome are suppressed by the pathway. More specifically, it reflects (1) the proportion of novel TEs that becomes targeted, (2) the speed with which novel TEs become targeted, and/or (3) the extent to which transposition of targeted TEs is suppressed. Based on our results, we hypothesize that salamanders have evolved less comprehensive TE suppression through changes in one or more of these variables; relative to other vertebrates, we would predict that (1) proportionally fewer TEs ultimately become targeted in salamanders, (2) TEs that do become targeted remain untargeted for a longer period in salamanders, and/or (3) residual transposition levels of targeted TEs are higher in salamanders. Such differences, in turn, could result from many evolved changes to the piRNA pathway machinery. For example, (1) and (2) could result from piRNA clusters in salamanders being smaller in size, fewer in number, or different in some other way that similarly reduces their efficacy as TE “traps.” Our third prediction of higher residual transposition levels in salamanders could result from the guide RNA/effector protein complexes being less likely to interact with and suppress their target TE loci/transcripts in salamanders. Future efforts aimed at (1) recovering genomic piRNA cluster sequences, despite the inherent challenges of assembling large repetitive genomes; and (2) assessing function of all piRNA pathway proteins, through both sequence-based and functional analyses, will allow refinement and testing of these hypotheses. However, consistent with less comprehensive TE suppression, the proportion of piRNAs relative to siRNA/miRNAs (Fig. 1) is lower in D. fuscus than in other vertebrate gonads (Chirn et al. 2015). In addition, the levels of TE-directed piRNAs in salamanders (14 to 18 %; Table 3) are low compared with the levels in other species that have much less substantial genomic TE loads; for example, in Xenopus and some mammals, ~20 to 25 % of the piRNAs map to TEs, whereas TEs comprise only ~30 to 45 % of the genome (Girard et al. 2006; Lau et al. 2006, 2009). In Drosophila melanogaster, ~70 % of the piRNAs map to TEs, whereas TEs comprise only ~12 % of the genome (Adams et al. 2000; Brennecke et al. 2007; dos Santos et al. 2015). Additionally, within the salamander genome, the most abundant class of TEs—LTR retrotransposons—is not disproportionately targeted in the piRNA pool. These patterns suggest that expansion of TE load in salamanders has not been accompanied by an increase in TE-targeting piRNA levels; however, we interpret these numbers with extreme caution because our piRNA mapping was necessarily completed without a reference genome.
With the evolution of less comprehensive TE suppression through any means, overall TE transposition rate increases, resulting in the accumulation of more new TE insertions in populations. For neutral/effectively neutral TE insertions, this translates into an increased rate of fixation of new TE loci. However, the fitness disadvantages of a TE insertion reflect, in part, whether or not it is transpositionally active; silenced loci are less disadvantageous, all else being equal. Because of this, less comprehensive TE suppression decreases the proportion of TE insertions that are effectively neutral, allowing a smaller proportion of novel TE insertions to drift to fixation (Lu and Clark 2010). Despite this lower proportion of fixation, less comprehensive TE suppression is predicted to increase the number of new TE insertions accumulating over evolutionary time (Lu and Clark 2010). Based on our results, we suggest that less comprehensive TE suppression by an expressed piRNA pathway has contributed to genomic gigantism in salamanders.
References
Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers Y-HC, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR, Gabor GL, Miklos Abril JF, Agbayani A, An H-J, Andrews-Pfannkoch C, Baldwin D, Ballew RM, Basu A, Baxendale J, Bayraktaroglu L, Beasley EM, Beeson KY, Benos PV, Berman BP, Bhandari D, Bolshakov S, Borkova D, Botchan MR, Bouck J, Brokstein P, Brottier P, Burtis KC, Busam DA, Butler H, Cadieu E, Center A, Chandra I, Cherry JM, Cawley S, Dahlke C, Davenport LB, Davies P, Pablos Bd, Delcher A, Deng Z, Mays AD, Dew I, Dietz SM, Dodson K, Doup LE, Downes M, Dugan-Rocha S, Dunkov BC, Dunn P, Durbin KJ, Evangelista CC, Ferraz C, Ferriera S, Fleischmann W, Fosler C, Gabrielian AE, Garg NS, Gelbart WM, Glasser K, Glodek A, Gong F, Gorrell JH, Gu Z, Guan P, Harris M, Harris NL, Harvey D, Heiman TJ, Hernandez JR, Houck J, Hostin D, Houston KA, Howland TJ, Wei M-H, Ibegwam C, Jalali M, Kalush F, Karpen GH, Ke Z, Kennison JA, Ketchum KA, Kimmel BE, Kodira CD, Kraft C, Kravitz S, Kulp D, Lai Z, Lasko P, Lei Y, Levitsky AA, Li J, Li Z, Liang Y, Lin X, Liu X, Mattei B, McIntosh TC, McLeod MP, McPherson D, Merkulov G, Milshina NV, Mobarry C, Morris J, Moshrefi A, Mount SM, Moy M, Murphy B, Murphy L, Muzny DM, Nelson DL, Nelson DR, Nelson KA, Nixon K, Nusskern DR, Pacleb JM, Palazzolo M, Pittman GS, Pan S, Pollard J, Puri V, Reese MG, Reinert K, Remington K, Saunders RDC, Scheeler F, Shen H, Shue BC, Sidén-Kiamos I, Simpson M, Skupski MP, Smith T, Spier E, Spradling AC, Stapleton M, Strong R, Sun E, Svirskas R, Tector C, Turner R, Venter E, Wang AH, Wang X, Wang Z-Y, Wassarman DA, Weinstock GM, Weissenbach J, Williams SM, Woodage T, Worley KC, Wu D, Yang S, Yao QA, Ye J, Yeh R-F, Zaveri JS, Zhan M, Zhang G, Zhao Q, Zheng L, Zheng XH, Zhong FN, Zhong W, Zhou X, Zhu S, Zhu X, Smith HO, Gibbs RA, Myers EW, Rubin GM, Venter JC (2000) The genome sequence of Drosophila melanogaster. Science 287:2185–2195
AmphibiaWeb (2015) Information on amphibian biology and conservation. Berkeley. http://amphibiaweb.org/
Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ (2007) Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 316:744–747
Aravin AA, Sachidanandam R, Bourc’his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ (2008) A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31:785–799
Armisen J, Gilchrist MJ, Wilczynska A, Standart N, Miska EA (2009) Abundant and dynamically expressed miRNAs, piRNAs, and other small RNAs in the vertebrate Xenopus tropicalis. Genome Res 19:1766–1775
Barrón MG, Fiston-Lavier A-S, Petrov DA, González J (2014) Population genomics of transposable elements in Drosophila. Ann Rev Genet 48:561–581
Blumenstiel JP (2011) Evolutionary dynamics of transposable elements in a small RNA world. Trend Genet 27:23–31
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ (2007) Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128:1089–1103
Castel SE, Martienssen RA (2013) RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Gen 14:100–112
Clark JP, Lau NC (2014) Piwi proteins and piRNAs step onto the systems biology stage. In: Yeo GW (ed) Systems biology of RNA binding proteins. Springer, New York, pp 159–197
dos Santos G, Schroeder AJ, Goodman JL, Strelets VB, Crosby MA, Thurmond J, Emmert DB, Gelbart WM, Flybase Consortium (2015) FlyBase: introduction of the Drosophila melanogaster Release 6 reference genome assembly and large-scale migration of genome annotations. Nucleic Acids Res 43:D690–D697
Dumesic PA, Madhani HD (2014) Recognizing the enemy within: licensing RNA-guided genome defense. Trend Biochem Sci 39:25–34
Frahry MB, Sun C, Chong R, Mueller RL (2015) Low levels of LTR retrotransposon deletion by ectopic recombination in the gigantic genomes of salamanders. J Mol Evol 80:120–129
Girard A, Sachidanandam R, Hannon GJ, Carmell MA (2006) A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442:199–202
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Gregory TR (2004) Insertion-deletion biases and the evolution of genome size. Gene 324:15–34
Gregory TR (2016) Animal Genome Size Database. http://www.genomesize.com.
Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS, Bartel DP (2008) Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature 455:1193–1197
Gunawardane LS, Saito K, Nishika KM, Kawamura Y, Nagami T, Siomi H, Siomi MC (2007) A slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science 315:1587–1590
G-w Chirn, Rahman R, Sytnikova YA, Matts JA, Zeng M, Gerlach D, Yu M, Berger B, Naramura M, Kile BT, Lau NC (2015) Conserved piRNA expression from a distinct set of piRNA cluster loci in eutherian mammals. PLoS Genet 11:e1005652
Han BW, Wang W, Li C, Weng Z, Zamore PD (2015) piRNA-guided transposon cleavage initiates Zucchini-dependent, phased piRNA production. Science 348:817–821
Houwing S, Kamminga LM, Berezikov E, Cronembold D, Girard A, van den Elst H, Filippov DV, Blaser H, Raz E, Moens CB, Plasterk RHA, Hannon GJ, Draper BW, Ketting RF (2007) A role for Piwi and piRNAs in germ gell maintenance and transposon silencing in zebrafish. Cell 129:69–82
Huang Xiao A, Yin H, Sweeney S, Raha D, Snyder M, Lin H (2013) A major epigenetic programming mechanism guided by piRNAs. Dev Cell 24:502–516
Iwasaki YW, Siomi MC, Siomi H (2015) PIWI-interacting RNA: its biogenesis and functions. Annu Rev Biochem 84:405–433
Jockusch EL (1997) An evolutionary correlate of genome size change in plethodontid salamanders. Proc R Soc Lond B 264:597
Keinath MC, Timoshevskiy VA, Timoshevskaya NY, Tsonis PA, Voss SR, Smith JJ (2015) Initial characterization of the large genome of the salamander Ambystoma mexicanum using shotgun and laser capture chromosome sequencing. Sci Rep 5:16413
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25
Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T, Bartel DP, Kingston RE (2006) Characterization of the piRNA complex from rat testes. Science 313:363–367
Lau NC, Ohsumi T, Borowsky M, Kingston RE, Blower MD (2009) Systematic and single cell analysis of Xenopus Piwi-interacting RNAs and Xiwi. EMBO J 28:2945–2958
Laurin M, Canoville A, Struble M, Organ C, de Buffrénil V (2016) Early genome size increase in urodeles. CR Palevol 15(1–2):74–82
Le Thomas A, Rogers AK, Webster A, Marinov GK, Liao SE, Perkins EM, Hur JK, Aravin AA, Tóth KF (2013) Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev 27:390–399
Li C, Vagin VV, Lee S, Xu J, Ma S, Xi H, Seitz H, Horwich MD, Syrzycka M, Honda BM, Kittler ELW, Zapp ML, Klattenhoff C, Schulz N, Theurkauf WE, Weng Z, Zamore PD (2009) Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell 137:509–521
Lu J, Clark AG (2010) Population dynamics of PIWI-interacting RNAs (piRNAs) and their targets in Drosophila. Genome Res 20:212–227
Lynch M (2007) The origins of genome architecture. Sinauer Associates Inc, Sunderland
Lynch M, Conery JS (2003) The origins of genome complexity. Science 302:1401–1404
Malone CD, Hannon GJ (2009) Small RNAs as guardians of the genome. Cell 136:656–668
Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ (2009) Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell 137:522–535
Marjanović D, Laurin M (2007) Fossils, molecules, divergence times, and the origin of lissamphibians. Syst Biol 56:369–388
Mohlhenrich E, Mueller RL (2016) Genetic drift, mutational hazard, and the evolution of genomic gigantism in salamanders. Evolution. doi:10.1111/evo.13084
Mohn F, Handler D, Brennecke J (2015) piRNA-guided slicing specifies transcripts for Zucchini-dependent, phased piRNA biogenesis. Science 348:812–817
Mueller RL (2006) Evolutionary rates, divergence dates, and the performance of mitochondrial genes in Bayesian phylogenetic analysis. Syst Biol 55:289
Mueller RL, Gregory TR, Gregory SM, Hsieh A, Boore JL (2008) Genome size, cell size, and the evolution of enucleated erythrocytes in attenuate salamanders. Zoology 111:218–230
Notredame C, Higgins D, Heringa J (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217
Organ CL, Canoville A, Reisz RR, Laurin M (2011) Paleogenomic data suggest mammal-like genome size in the ancestral amniote and derived large genome size in amphibians. J Evol Biol 24:372–380
Petrov DA (2002) Mutational equilibrium model of genome size evolution. Theor Popul Biol 61:533–546
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842
Robine N, Lau NC, Balla S, Jin Z, Okamura K, Kuramochi-Miyagawa S, Blower MD, Lai EC (2009) A broadly conserved pathway generates 3′ UTR-directed primary piRNAs. Curr Biol 19:2066–2076
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Roovers EF, Rosenkranz D, Mahdipour M, Han C-T, He N, de Sousa Lopes SMC, van der Westerlaken LA, Zischler H, Butter F, Roelen BA (2015) Piwi proteins and piRNAs in mammalian oocytes and early embryos. Cell Rep 10:2069–2082
Roth G, Nishikawa KC, Wake DB (1997) Genome size, secondary simplification, and the evolution of the brain in salamanders. Brain Behav Evol 50:50–59
Rozhkov NV, Hammell M, Hannon GJ (2013) Multiple roles for Piwi in silencing Drosophila transposons. Genes Dev 27:400–412
Senti K-A, Jurczak D, Sachidanandam R, Brennecke J (2015) piRNA-guided slicing of transposon transcripts enforces their transcriptional silencing via specifying the nuclear piRNA repertoire. Genes Dev 29:1747–1762
Sessions SK (2008) Evolutionary cytogenetics in salamanders. Chromosome Res 16:183–201
Sessions SK, Kezer J (1991) Evolutionary cytogenetics of bolitoglossine salamanders (family Plethodontidae). In: Green DM, Sessions SK (eds) Amphibian cytogenetics and evolution. Academic Press, San Diego, pp 89–130
Sessions SK, Larson A (1987) Developmental correlates of genome size in plethodontid salamanders and their implications for genome evolution. Evolution. doi:10.2307/2409090
Sienski G, Dönertas D, Brennecke J (2012) Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell 151:964–980
Siomi MC, Sato K, Pezic D, Aravin AA (2011) PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol 12:246–258
Sun C, Mueller RL (2014) Hellbender genome sequences shed light on genome expansion at the base of crown salamanders. Gen Biol Evol 6:1818–1829
Sun C, Arriaza JRL, Mueller RL (2012a) Slow DNA loss in the gigantic genomes of salamanders. Gen Biol Evol 4:1340–1348
Sun C, Shepard DB, Chong RA, Arriaza JL, Hall K, Castoe TA, Feschotte C, Pollock DD, Mueller RL (2012b) LTR retrotransposons contribute to genomic gigantism in plethodontid salamanders. Gen Biol Evol 4:168–183
Sytnikova YA, Rahman R, G-w Chirn, Clark JP, Lau NC (2014) Transposable element dynamics and PIWI regulation impacts lncRNA and gene expression diversity in Drosophila ovarian cell cultures. Genome Res 24:1977–1990
Szarski H (1983) Cell size and the concept of wasteful and frugal evolutionary strategies. J Theor Biol 105:201–209
Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD (2006) A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313:320–324
Wang W, Yoshikawa M, Han Bo W, Izumi N, Tomari Y, Weng Z, Zamore Phillip D (2014) The initial uridine of primary piRNAs does not create the tenth adenine that is the hallmark of secondary piRNAs. Mol Cell 56:708–716
Wang W, Han Bo W, Tipping C, Ge Daniel T, Zhang Z, Weng Z, Zamore Phillip D (2015) Slicing and binding by Ago3 or Aub trigger Piwi-bound piRNA production by distinct mechanisms. Mol Cell 59:819–830
Yamanaka S, Siomi M, Siomi H (2014) piRNA clusters and open chromatin structure. Mob DNA 5:22
Yi M, Chen F, Luo M, Cheng Y, Zhao H, Cheng H, Zhou R (2014) Rapid evolution of piRNA pathway in the teleost fish: implication for an adaption to transposon diversity. Gen Biol Evol 6:1393–1407
Zhang Z, Xu J, Koppetsch Birgit S, Wang J, Tipping C, Ma S, Weng Z, Theurkauf William E, Zamore Phillip D (2011) Heterotypic piRNA ping-pong requires qin, a protein with both E3 ligase and Tudor domains. Mol Cell 44:572–584
Zhu W, Kuo D, Nathanson J, Satoh A, Pao GM, Yeo GW, Bryant SV, Voss SR, Gardiner DM, Hunter T (2012) Retrotransposon long interspersed nucleotide element-1 (LINE-1) is activated during salamander limb regeneration. Dev Growth Differ 54:673–685
Acknowledgments
This research was supported by NSF–DBI 1103746 to MM-V and NSF-DEB 1021489 to RLM. NCL was supported by the Searle Scholars Foundation and the NIH (R00HD057298). J. Krakovil and D. Weisrock (U Kentucky) provided the tissues and access to unpublished Cryptobranchus alleganiensis transcriptome data; funding for collecting trips was provided by Highlands Biological Station and the University of Kentucky Department of Biology to J. Krakovil. D. New at IBEST provided critical technical expertise. Suggestions from anonymous reviewers improved the quality of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. J. Madison-Villar and Cheng Sun have contributed equally.
An erratum to this article is available at http://dx.doi.org/10.1007/s00239-016-9769-1.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 1 (PDF 483 kb)
Transposable element families targeted by piRNAs and siRNAs in the female and male samples. Results were obtained from mapping each small RNA dataset to its respective reference transcriptome. TE families are ranked by density of mapped small RNA reads
Supplementary material 2 (PDF 1550 kb)
Phylogenies estimated for eleven piRNA pathway protein sequences obtained from salamanders and other vertebrates
Rights and permissions
About this article

Cite this article
Madison-Villar, M.J., Sun, C., Lau, N.C. et al. Small RNAs from a Big Genome: The piRNA Pathway and Transposable Elements in the Salamander Species Desmognathus fuscus . J Mol Evol 83, 126–136 (2016). https://doi.org/10.1007/s00239-016-9759-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-016-9759-3