
Abstract
We have previously shown that through test-tube molecular evolution, an arbitrarily chosen noncatalytic DNA sequence can be evolved into a catalytic DNA (DNAzyme) with significant RNA-cleaving activity. In this study, we aim to address the question of whether the catalytic activity of such a DNAzyme can be further optimized using in vitro selection. Several cycles of selective enrichment starting with a partially randomized DNA library have resulted in the isolation of many sequence variations that show notably improved catalytic activity. Bioinformatic analysis and activity examination of several DNAzyme-substrate constructs have led to two interesting findings about sequence mutations and the secondary structure of this DNAzyme: (1) three crucial mutations have transformed the DNAzyme into 8–17, a DNAzyme that has been discovered in multiple previous in vitro selection experiments, and (2) other mutations have allowed this special 8–17 variant to make structural fine-tuning in order to cleave an arbitrarily chosen RNA-containing substrate with a defined sequence. Our study not only showcases the combined power of directed molecular evolution and in vitro selection techniques in turning a noncatalytic nucleic acid sequence into an efficient enzyme, but it also raises the question of whether mother nature has used a similar approach to evolve natural enzymes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since its initiation on Earth, life has evolved in astonishingly complex ways. The RNA World, posited to support the transition of life from the chemical to the biotic world, advocates the self-replication of ribonucleic acid as the means by which life began—as early chemical systems capable of both information storage and reproduction. The RNA World hypothesis (Crick 1968; Orgel 1968; Gilbert 1986; Joyce 2002) would be incomplete without the emergence and evolution of enzymes.
We are interested in the mechanism by which enzymes arose from the abiotic world. In a previous study, we postulated that an enzyme can be evolved from any sequence of a functional polymer and validated this idea through a molecular evolution experiment that turned an arbitrarily chosen DNA sequence, a noncatalytic 50-nucleotide DNA fragment encoding for the Bos taurus (cattle) albumin mRNA, into a catalytic DNA (DNAzyme) with RNA-cleaving activity (Gysbers et al. 2015). This RNA-cleaving DNAzyme was named “G2501” as it was the top-ranking sequence after 25 rounds of in vitro evolution. The emergence of this catalytic sequence was made possible by low-level random errors introduced into evolving DNA molecules by Tth DNA polymerase that was used to amplify DNA during each round of evolution, and this mechanism allowed the accumulation of catalytically beneficial mutations that drove the selection of progressively more active sequence variants along the evolutionary trajectory (Gysbers et al. 2015).
Although this prior study represents the first effort of exploiting in vitro evolution to evolve a catalyst from a noncatalytic sequence, the resulting catalyst is not catalytically efficient. G2501 exhibits a first-order rate constant of ~10−4 min−1, translating into ~103-fold rate enhancement over spontaneous RNA cleavage reaction (Li and Breaker 1999). However, many previous selection experiments using random-sequence DNA pools have resulted in the isolation of RNA-cleaving DNAzymes with first-order rate constants of greater than 0.1 min−1 (Breaker and Joyce 1994; Santoro and Joyce 1997, Silverman 2005; Schlosser and Li 2009, 2010). This brings into question whether evolution from a defined sequence can lead to catalysts with a higher level of activity that might be important for the evolution of life.
The proposed secondary structure of G2501 is depicted in Fig. 1a (Gysbers et al. 2015). It is a three-way junction made of three short Watson–Crick duplexes (P1, P2, and P3; P stands for pairing element), two single-stranded (ss) sequence motifs (SS12 linking P1 and P2, and SS13 connecting P1–P3; note that rG stands for guanosine ribonucleotide, the only RNA unit in an otherwise all-DNA molecule), and a short loop (L3 at the tip of P3). This structural arrangement suggests that the functionally important nucleotides are most likely located within the 19-nucleotide P3/L3/SS13 sequence elements (pink nucleotides). Interestingly, the seven mutations acquired by G2501, which enable its noncatalytic to catalytic transformation, are all located outside of these sequence elements (Gysbers et al. 2015). Lack of mutation in the putative catalytic core suggests that G2501 has the potential to be further evolved into a more efficient catalyst. In this study, we set out to further improve the catalytic performance of G2501 using the technique of in vitro selection (Ellington and Szostak 1990; Tuerk and Gold 1990; Beaudry and Joyce 1992). Our approach involved synthesizing a DNA pool with partial randomization of the P3/L3/SS13 elements of G2501 and subjecting it to cycles of a stringent selective enrichment regime so that catalytically robust sequence variants would emerge. Here we report our findings.

a The secondary structure of G2501, an RNA-cleaving DNAzyme previously derived from a 50-nucleotide DNA fragment encoding for the Bos taurus (cattle) albumin mRNA by in vitro evolution. rG: guanosine ribonucleotide, the sole RNA unit embedded with the otherwise all-DNA sequence. Italic nucleotides in gray denote the substrate sequence. The putative catalytic core is highlighted in pink. b The secondary structure of the DNA library used for the in vitro selection experiment in this study. N: 70 % of the original nucleotide in G2501 and 10 % each of the remaining nucleotides (Color figure online)
Materials and Methods
Materials
All DNA oligonucleotides were purchased from Integrated DNA Technologies and purified by 10 % denaturing (8 M urea) polyacrylamide gel electrophoresis (dPAGE). Their concentrations were determined spectroscopically using GENESYS 10S UV–Vis Spectrophotometer (Thermo Scientific). Polynucleotide kinase (PNK) and T4 DNA ligase were purchased from Thermo Scientific. Tth DNA polymerase was purchased from Biotools. The nucleotide isotopes [γ-32P] ATP and [α-32P] dGTP were purchased from Perkin Elmer.
In Vitro Selection
A total of 500 pmol of synthetic DNA library RL1 (the last 78 nucleotides of the sequence given in Fig. 1b) was 5′-phosphorylated at 37 °C with 50 units of PNK and 4 mM ATP (200 µL, 23 °C; 40 min). The phosphorylated RL1 was incubated with 550 pmol of the RNA-containing DNA substrate RDS1 (the first 28 nucleotides of RL1, shown in gray in Fig. 1b), 550 pmol of the template oligonucleotide T1 (5′-AGTCA GTCAC TCTTG ATCGA-3′), and 10 units of T4 DNA ligase (200 µL, 23 °C, 60 min). The ligated RDS1–RL1 construct (106 nucleotides) was purified by 10 % dPAGE. For the first round of selection, 100 pmol of purified RDS1–RL1 (pool 1) were incubated in 1 × selection buffer (SB, 100 mM KCl, 300 mM NaCl, 15 mM MnCl2, 15 mM MgCl2, 55 mM HEPES, pH 7.5 at 23 °C) (500 μL, 23 °C, 4 h). The RNA cleavage reaction was terminated with the addition of 50 mM ethylenediaminetetraacetic acid (EDTA, pH 8.0). The DNAzyme-containing cleavage product (92 nucleotides) was purified by 10 % dPAGE and amplified by two consecutive PCRs (PCR1 and PCR2). PCR1 was performed with the cleavage product as the template, 0.5 µM forward primer FP1 (5′-TCCGT GAGAC CTATA CAGTG-3′), 0.5 µM reverse primer RP1 (5′-TCCGT GAGAC CTATA CAGTG-3′), 0.2 mM each dNTP (dATP, dCTP, dGTP, and dTTP), 1 × PCR buffer (7.5 mM Tris–HCl, pH 9.0, 0.2 mM MgCl2, 5 mM KCl, and 2 mM (NH4)2SO4), and 2.5 units of Tth DNA polymerase in a total reaction volume of 50 µL. An aliquot of 4 µL was removed at two cycle intervals from 6 to 12 cycles to determine the level of amplification. After full amplification, 1 µL of the PCR1 mixture was further amplified by PCR2 with FP1 and RP2 (5′-A18-iSp18-TCCGT GAGAC CTATA CAGTG-3′; iSp18 refers to 18-atom hexaethylene glycol spacer). RP2 contains a poly(dA) tail (18 adenosine deoxyribonucleotides) separated by a nonamplifiable linker, and therefore, the DNA product from PCR2 would contain a noncoding strand that is 20-nucleotide longer than the coding strand. The coding sequence from PCR2 was simply purified using 10 % dPAGE and used as the enriched pool for the second round of selection (100 pmol, pool 2). A total of 10 cycles of selection were conducted. However, the allowed RNA cleavage time was progressively reduced from 4 h (rounds 1–4), to 1 h (round 5), 30 min (round 6), 15 min (round 7), 5 min (round 8), and 1 min (rounds 9 and 10).
High-Throughput Sequencing
The PCR amplicon from round 10 was sequenced using an Illumina Miseq DNA sequencer at the Farncombe Metagenomics Facility, McMaster University. The protocol used for DNA preparation and subsequent analysis of sequence reads is the same as the one that we have described in a previous publication (Mao et al. 2015).
Activity Comparison of Pools 1, 4, 7, and 10
5′-fluorescein-labeled RDS1 was first ligated to each DNA pool (PCR amplified and dPAGE purified). Each substrate-pool construct was assessed for RNA cleavage activity in 100 µL reaction mixture containing 200 nM test construct and 1 × SB. 5 µL aliquot was removed at 5, 10, 20, 30 s, 1, 5, 10, 30 min, 1, 4, and 24 h. The cleavage reaction was immediately terminated with the addition of combined 50 mM EDTA (pH 8.0) and 2 × denaturing gel loading dye. After the last time point, all reaction mixtures were analyzed by 10 % dPAGE and imaged on Typhoon 9200 (GE Biosciences). Percent cleavage was quantified using Image Quant software (Molecular Dynamics).
Determination of RNA Cleavage Rate Constants of Trans-Acting Substrate-DNAzyme Constructs
Cleavage reactions were conducted in 100 μL of 1 × SB containing 200 nM 5′-fluorescein-labeled substrate (RDS1, RDS2, or RDS3) and 4 μM DNAzyme. 10 μL were withdrawn from the reaction mixture at the following time points: 10, 20, 30 s, 1, 5, 10, 30 min, 1, and 4 h. These DNA samples were then subjected to the same 10 % dPAGE/Typhoon/ImageQuant analysis as described above, to derive percent cleavage of the substrate. First-order rate constants were obtained by curve-fitting using Y = Y max (1 − e−kt), where Y represents the cleavage yield at the time t, Y max is the maximal cleavage yield, and k is the first-order rate constant. All the experiments were conducted in triplicate.
Results
In Vitro Selection of More Efficient RNA-Cleaving DNAzymes from a Partially Randomized DNA Pool
Using the secondary structure of G2501 depicted in Fig. 1a as a guide, we designed a DNA library, named RL1 (Fig. 1b), for the reselection experiment. Specifically, each of the 19 nucleotides located in the putative catalytic core of G2501 (pink nucleotides) was subjected to partial random mutagenesis (via chemical synthesis) as follows: 70 % of the original nucleotide and 10 % each of the remaining nucleotides.
RL1 was chemically synthesized and purified by 10 % denaturing (8 M urea) dPAGE. Approximately 6 × 1013 sequence variants (100 pmol) were taken as the initial pool (pool 1) with which in vitro selection was conducted using key steps outlined in Fig. 2a. In Step I, the RNA-containing DNA substrate (named RDS1; the first 28 nucleotides in the sequence of the DNA library shown in Fig. 1b) was joined to 5′-phosphorylated pool 1 (the rest of the sequence shown in Fig. 1b) by T4 DNA ligase-mediated DNA ligation. Following size-based purification using 10 % dPAGE, the full RDS1–RL1 construct (cis construct) was incubated in Step II with 1 × selection buffer (1 × SB; see Materials and Methods for detailed components) for 4 h. The cleaved DNAzyme strand was purified by dPAGE and amplified by polymerase chain reaction (PCR) with the use of a normal forward primer and a reverse primer containing a dA18 tail separated by a nonamplifiable linker in Step III. The use of this primer served to facilitate the purification of the DNAzyme-coding strand by dPAGE in Step IV, as the DNAzyme-coding strand was 18 nucleotides shorter. The purified coding strand, now denoted pool 2 (100 pmol), were phosphorylated at its 5′-end and subjected to the next cycle of selective amplification.

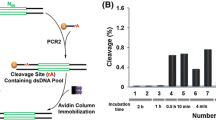
a In vitro selection strategy consisting four key steps as shown. Details are described in the main text. b Examination of catalytic activities of representative DNA pools established during the pathway of in vitro selection. Each pool was incubated in 1 × selection buffer (1 × SB) at room temperature for 1, 5, 10, 30, 60, 240, and 1440 min, followed by dPAGE analysis to determine percent cleavage. G2501 was also examined as a reference (Color figure online)
In total, 10 rounds of selection were performed using the afore-described procedure. However, the RNA cleavage reaction time (Step II) was altered from round to round as follows: 4 h in rounds 1–4; 60, 30, 15, and 5 min in rounds 5, 6, 7, and 8, respectively; and 1 min in rounds 9 and 10. This strategy was implemented in order to derive the best performing DNAzyme mutants.
Analysis of Catalytic Activities of Representative DNA Pools
We analyzed the catalytic activity of the following DNA pools along the selection path: pools 1, 4, 7, and 10. The activity of each DNA pool, measured as “% cleavage” (the percentage of cleavage of each RDS1-pool construct, detected by dPAGE) at various time points, is provided in Fig. 2b. As a reference, we also included G2501 in the same experiment. Pool 1 showed almost no detectable activity even after incubation for a long time (1440 min = 24 h). In contrast, pools 4, 7, and 10 reached 9.7, 47.3, and 54.6 % cleavage at the same time point. For comparison, only 4.1 % cleavage was observed for G2501. In fact, pool 10 exhibited 6.4 % cleavage in only 5 min. Taken together, these results indicate that the selection procedure described above led to the establishment of a catalytically efficient DNAzyme population.
Sequencing and Bioinformatic Analysis of Representative DNAzymes of Pool 10
We carried out the next-generation sequencing with pool 10 using the method we previously described (Mao et al. 2015). In total, we obtained 790,609 sequence reads, which can be clustered into 6820 unique sequences.
Careful bioinformatic analysis of these unique sequences revealed that 37.25 % of the sequence reads share the following conserved sequence motif: CCGA-N-AGT-N-TC-N-CGA (N: any nucleotide), which has 12 absolutely conserved nucleotides (bold nucleotides). Additional 19.29 % of the sequences are within one substitution mutation of this conserved sequence motif. This result indicates that a major DNAzyme family arose from the DNA pool. We named this family RF1 (Reselected DNAzyme Family 1) and chose to focus on this family for further investigation in this study.
The random-sequence elements of the top 10 sequences within RF1 are provided in Fig. 3a. Altogether, they occupied 31.29 % of pool 10. To analyze the pattern of nucleotide mutation, we have color-coded individual nucleotides of the 19-nucleotide elements as follows. Green nucleotides are conserved in both G2501 and RF1. Six nucleotides exist in this category and they are C1, C2, A4, T8, T10, and C13. Red nucleotides are mutated from G2501 but become conserved in RF1. Five nucleotides are in this grouping and they are A3G, C7G, G11C, A14G, and C15A. Based on the conservation within RF1, these eleven nucleotides must play important roles in the structure and function of RF1. A pair of nucleotides—A5 and T9—are colored blue; they are mutated from G2501 but co-varied in RF1 according to Watson–Crick rules. For example, they occur either as T5A9 or C5G9 in all the top 10 sequences within RF1. This observation suggests that nucleotide 5 and nucleotide 9 may form a Watson–Crick base pair. The remaining six nucleotides are colored gray, which appear to have mutated randomly from G2501 (as a result of partial random mutagenesis) and may not play important roles in the function of RF1.

a Nucleotide alignment of the top 10 sequences in pool 10, named RF1.E1–RF1.E10. Only the partial random region is shown. The percentage of occurrence of each sequence in the total sequence reads is provided next to the sequence. The corresponding region of G2501 is also provided at the top of alignment. Nucleotide color codes: Green, conserved nucleotides in both G2501 and RF1; red, nucleotides that are mutated from G2501 but conserved in RF1; blue, a pair of nucleotides that are mutated from G2501 but co-varied in RF1 according to Watson–Crick rules; gray: nucleotides in RF1 that mutated randomly from G2501. b The secondary structure and k obs value of trans-acting G2501. c The secondary structures and k obs values of trans-acting RF1.E1, E2, E3, E6, and E8. The fold of rate increase of each tested RF1 DNAzyme over G2501 is also provided (Color figure online)
Secondary Structure Alteration from G2501 to RF1
The putative secondary structure for G2501 with aforementioned color codes is provided in Fig. 3b. Inspection of the top 10 sequences of RF1 in the context of this secondary structure model led us to propose an altered secondary structure model depicted in Fig. 3c. Although both secondary structure models are similar three-way junctions, there are several noticeable differences. (1) P3 element is shortened from 4 to 3 base pairs. (2) SS13 is enlarged from 7 to 8 nucleotides for 9 out of the top 10 sequences; the exception is RF1.E6, which has a nucleotide deletion in SS13. (3) The size of L3 is reduced from 4 to 3 nucleotides. (4) The creation of SS23 element (made of C1 and C2). (5) The pairing partners are different in RF1 in comparison to G2501: A3 in G2501 pairs with T10; in RF1 it mutates to G3 and now pairs with C11, which is mutated from G11. A4 in G2501 pairs with T9, but in RF1 it pairs with T10. A5 is part of the AACT tetraloop in G2501; in RF1 it mutates to C or T and chooses to pair with G9 or A9 mutated from T9 in G2501. It should be noted that A5 and T8 in G2501 as well as A6 and T8 in RF1 are present at locations that can theoretically create an A-T pair. However, these pairing interactions are deemed unlikely because these arrangements would result in a very small dinucleotide (for G2501) or mononucleotide (for RF1) loop.
Comparison of Catalytic Activity of RF1 with G2501
We next investigated the catalytic activity of six representative RF1 members: RF1.E1, E2, E3, E6, and E8. Each DNAzyme was examined in the trans format as shown in Fig. 3c, under single-turnover setting (substrate/enzyme = 1/50). Percent cleavage of the substrate at various time points was determined by dPAGE analysis and the data were used to derive first-order rate constant, k obs, using the curve-fitting method described in Materials and Methods. As a reference, we also determined the k obs value for the trans-acting G2501 construct depicted in Fig. 3b using the same approach.
The k obs values of E1, E2, E3, E6, and E8 were found to be 0.254, 0.202, 0.265, 0.305, and 0.164 min−1, respectively. In contrast, G2501 only exhibited a k obs value of 4.18 × 10−4 min−1. Therefore, the reselection experiment led to isolation of DNAzyme sequences with significantly improved catalytic rate constants (392–730 fold increase in rate over G2501).
Mutational Analysis of Conserved Nucleotides in RF1
The secondary structure of RF1 (Fig. 3c) contains the following five nucleotides that were mutated from G2501 but become conserved in RF1: G3, G7, C11, G14, and A15. As we have discussed above, two of these nucleotides, G3 and C11, form a Watson–Crick base pair as part of the P3 element. The remaining three nucleotides are distributed into L3 (G7) and SS13 (G14 and A15). We speculated that these nucleotides are critically important to the catalytic function of RF1. To test for this hypothesis, we carried out mutational studies with G7 and G14 using RF1.E1 as the reference DNAzyme (Fig. 4a). The first construct, named RF1.E1M1, contains the G7C mutation (Fig. 4b), whereas the second construct, RF1.E1M2, has the G14A mutation (Fig. 4c). From the time-course study presented in Fig. 4d, it is clear that both mutations essentially abolished the catalytic activity: Noticeable cleavage was observed with RF1.E1 in as short as 10 s; in stark contrast, both mutants yielded barely detectable cleavage even after 4 h of reaction.

The secondary structure of a RF1.E1, b RF1.E1M1 with G7C mutation (purple C), and c RF1.E1M2 with G14A mutation (purple A). d Comparison of the catalytic activity of RF1.E1, RF1.E1M1, and RF1.E1M2. Each trans-acting construct was incubated at room temperature in 1 × SB for the time points indicated, followed by dPAGE analysis. %Clv: percent cleavage of the substrate RDS1 (Color figure online)
Remodeled Secondary Structure of RF1
The secondary structure of RF1 depicted in Fig. 3c has C1 and C2 as two unpaired nucleotides in SS23 that connects P2 and P3 elements. We notice that there are two unpaired guanosine nucleotides in SS12 (highlighted in light blue in Fig. 5a). This creates a possibility of two G–C base pairs as part of extended P2 element (Fig. 5b). The new arrangement would result in a T bulge (shown in purple) next to the two G–C pairs.

a The secondary structure of trans-acting RF1.E1/RDS1 construct. b Rearrangement of the secondary structure of RF1.E1/RDS1, making the two blue GG nucleotides in the substrate to form two Watson–Crick base pairs with C1 and C2 nucleotides in the DNAzyme strand. c A new DNAzyme/substrate construct, RF1.E1/RDS2. RDS2 is identical to RDS1 except for the removal of the T bulge. d Comparison of the catalytic activity of RF1.E1 in complex with RDS1 and RDS2 (Color figure online)
To test for the possible existence of these two G–C pairs, we designed a new RNA-containing DNA substrate, RDS2, in which the T bulge was eliminated. The deletion of the T residue will place GG and CC nucleotides in perfect positions for pairing. Given the fact that these two CC residues are absolutely conserved in RF1, it is expected that if the proposed two G–C pairs do not exist, the cleavage activity of RF1 towards RDS2 would be significantly reduced (as the two conserved nucleotides would otherwise have been involved in other important tertiary interactions for the function of RF1). From the kinetic comparison of the cleavage activity of RF1.E1 towards both RDS1 and RDS2 shown in Fig. 5d, it is clear that RF1.E1 exhibits the equivalent activity in cleaving both substrates. This observation is consistent with the two proposed G–C pairs.
Discussion
The RNA-cleaving DNAzyme G2501 was evolved from an arbitrarily chosen noncatalytic DNA sequence. It became a catalytic molecule after it acquired seven mutations in its 50-nucleotide sequence, two of which are deletions and five substitutions (Gysbers et al. 2015). However, the catalytic activity of G2501 was relatively poor, exhibiting a moderate k obs of 10−4 min−1, which translates into ~1000-fold rate enhancement over the background cleavage of RNA (Li and Breaker 1999). In this work, we set out to investigate whether the sequence of G2501 is encoded with a latent ability to carry out more efficient catalysis. Our findings presented above clearly indicate that, with further sequence mutations and secondary structure refining, a weakly catalytic sequence can indeed be transformed into a more robust catalyst.
The transformation from G2501 to RF1 requires mutations of only a few nucleotides. We believe three mutations, in particular, C7G, A14G, and C15C, are most critical to the enhanced DNAzyme efficiency, given their location within unpaired sequence elements (Fig. 3c). Two other mutations, A3G and G11C, also appear to be important to the improved catalytic activity because these two mutations enable different paring patterns within P3 that are responsible for reprograming the three-way junction.
Many RNA-cleaving DNAzymes have been reported (Silverman 2005; Schlosser and Li 2009). They include well-studied 10–13 (Santoro and Joyce 1997), 8–17 (Santoro and Joyce 1997), GR-5 (Breaker and Joyce 1994), and recently discovered Ce13 (Huang et al. 2014) and NaA43 (Torabi et al. 2015). Upon comparison to these reported RNA-cleaving DNAzymes, it appears that RF1 is a variant of 8–17, as we will discuss below.
The 8–17 DNAzyme has been isolated from several in vitro selection experiments conducted by different groups (Santoro and Joyce 1997; Faulhammer and Famulok 1997; Li et al. 2000; Cruz et al. 2004; Schlosser and Li 2004, 2005). Four reasons have been provided to explain the rediscovery of 8–17: its small catalytic domain (made of only ~15 nucleotides), highly variable sequence requirements, simple three-way junction secondary structure framework, and ability to cleave all 16 dinucleotide junctions of RNA (Schlosser and Li 2010). Repeated discovery of small nucleic acid enzymes such as 8–17 or the hammerhead ribozyme (Salehi-Ashtiani and Szostak 2001) has been referred as “tyranny of the small motif” (Joyce 2004).
RF1 has all the required sequence and secondary structure elements as an 8–17 DNAzyme. Specifically, RF1 has the signature three-way-junction secondary structure of 8–17 consisting of two substrate-binding arms (P1 and P2) and a small internal hairpin (P3–L3) preceding a short unpaired region (SS13). More importantly, RF1 has the four absolutely conserved nucleotides –AG and CG– at the expected locations of the secondary structure: AG in the loop of the P3–L3 hairpin (the green A6G7 in the depicted secondary structure of RF1 in Fig. 6a) and CG in the SS13 element (the green C13G14 in Fig. 6a).

The secondary structure of a RF1.E1/RDS2, b 8-17NA/RDS3, c 8-17NA/RDS2, d RF1.E1/RDS3, along with the first-order rate constant for each construct (Color figure online)
We have previously reported a specific 8–17 variant, named 8–17NA, that exhibits a high-level cleavage activity towards rG–dA chimeric dinucleotide junction (Schlosser et al. 2008), the same junction that RF1 cleaves. Therefore, RF1 and 8–17NA might share great sequence and secondary structure similarities. For this consideration, we designed an 8–17NA/RDS3 construct, as shown in Fig. 6b, and tested its cleavage activity. Note that the sequence of RD3 is identical to that of RDS2 except for the shortened SS12 element (rGA in RDS3 instead of AGCGrGA in RDS2).
In addition to the difference in the substrate sequence, RF1 and 8–17NA have subtle differences in P3-L3-SS13. 8-17NA has a smaller SS13 (5 nucleotides vs. 8 nucleotides in RF1) and a T bulge (purple) in P3. We speculate that these variations allow the two 8–17 variants to make necessary structural fine-tuning in order to accommodate the sequence differences in their substrates. The evidence supporting this idea comes from the observation that both 8–17 variants register a sharp drop in k obs towards unintended substrates. The k obs of RF1.E1 was reduced by 56-fold when the substrate was changed from RDS2 (Fig. 6a) to RDS3 (Fig. 6d), and 8–17NA was 1563-fold more active towards RDS3 (Fig. 6b) in comparison to its activity with RDS2 (Fig. 6c).
In conclusion, we have shown that a noncatalytic DNA sequence not only can be evolved into an RNA-cleaving DNAzyme through directed molecular evolution but can also be optimized into a catalytically efficient version through in vitro selection. This raises the possibility that mother nature might have used a similar approach to evolve natural enzymes. Our results presented in this study also suggest that the activity optimization has certainly benefited from the existence of structurally simple but catalytically efficient small DNAzymes such as 8–17. It remains to be determined if these conclusions can be replicated for other arbitrarily chosen DNA sequences for both RNA cleavage reaction and beyond. Finally, as we have noted earlier, the catalytic core of G2501 is made of original nucleotides in the Bos taurus albumin gene (without any mutation). Does this observation suggest that this gene is pre-adapted for an RNA-cleaving activity or is this finding purely a coincidence? Searching answers for these questions constitutes a future direction of our research program.
References
Beaudry AA, Joyce GF (1992) Directed evolution of an RNA enzyme. Science 257:635–641
Breaker RR, Joyce GF (1994) A DNA enzyme that cleaves RNA. Chem Biol 1:223–229
Crick FH (1968) The origin of the genetic code. J Mol Biol 38:367–379
Cruz RP, Withers JB, Li Y (2004) Dinucleotide junction cleavage versatility of 8–17 deoxyribozyme. Chem Biol 11:57–67
Ellington AD, Szostak JW (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346:818–822
Faulhammer D, Famulok M (1997) Characterization and divalent metal-ion dependence of in vitro selected deoxyribozymes which cleave DNA/RNA chimeric oligonucleotides. J Mol Biol 269:188–202
Gilbert W (1986) Origin of life: the RNA world. Nature 319:618
Gysbers R, Tram K, Gu J, Li Y (2015) Evolution of an enzyme from a noncatalytic nucleic acid sequence. Sci Rep 5:11405
Huang PJ, Lin J, Cao J, Vazin M, Liu J (2014) Ultrasensitive DNAzyme beacon for lanthanides and metal speciation. Anal Chem 86:1816–1821
Joyce GF (2002) The antiquity of RNA-based evolution. Nature 418:214–221
Joyce GF (2004) Directed evolution of nucleic acid enzymes. Annu Rev Biochem 73:791–836
Li Y, Breaker RR (1999) Kinetics of RNA degradation by specific base catalysis of transesterification involving the 2′-Hydroxyl Group. J Am Chem Soc 121:5364–5372
Li J, Zheng W, Kwon AH, Lu Y (2000) In vitro selection and characterization of a highly efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res 28:481–488
Mao Y, Liu M, Tram K, Gu J, Salena BJ, Jiang Y, Li Y (2015) Optimal DNA templates for rolling circle amplification revealed by in vitro selection. Chemistry 21:8069–8074
Orgel LE (1968) Evolution of the genetic apparatus. J Mol Biol 38:381–393
Salehi-Ashtiani K, Szostak JW (2001) In vitro evolution suggests multiple origins for the hammerhead ribozyme. Nature 414:82–84
Santoro SW, Joyce GF (1997) A general purpose RNA-cleaving DNA enzyme. Proc Natl Acad Sci USA 94:4262–4266
Schlosser K, Li Y (2004) Tracing sequence diversity change of RNA-cleaving deoxyribozymes under increasing selection pressure during in vitro selection. Biochemistry 43:9695–9707
Schlosser K, Li Y (2005) Diverse evolutionary trajectories characterize a community of RNA-cleaving deoxyribozymes: a case study into the population dynamics of in vitro selection. J Mol Evol 61:192–206
Schlosser K, Li Y (2009) Biologically inspired synthetic enzymes made from DNA. Chem Biol 16:311–322
Schlosser K, Li Y (2010) A versatile endoribonuclease mimic made of DNA: characteristics and applications of the 8-17 RNA-cleaving DNAzyme. Chembiochem 11:866–879
Schlosser K, Gu J, Sule L, Li Y (2008) Sequence-function relationships provide new insight into the cleavage site selectivity of the 8–17 RNA-cleaving deoxyribozyme. Nucleic Acids Res 36:1472–1481
Silverman SK (2005) In vitro selection, characterization, and application of deoxyribozymes that cleave RNA. Nucleic Acids Res 33:6151–6163
Torabi SF, Wu P, McGhee CE, Chen L, Hwang K, Zheng N, Cheng J, Lu Y (2015) In vitro selection of a sodium-specific DNAzyme and its application in intracellular sensing. Proc Natl Acad Sci USA 112:5903–5908
Tuerk C, Gold L (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–510
Acknowledgments
The funding for this work was provided by the Natural Sciences and Engineering Research Council of Canada (NSERC) via a Discovery Grant to YL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Laura Chan, Kha Tram, and Rachel Gysbers have contributed equally to this work
Rights and permissions
About this article

Cite this article
Chan, L., Tram, K., Gysbers, R. et al. Sequence Mutation and Structural Alteration Transform a Noncatalytic DNA Sequence into an Efficient RNA-Cleaving DNAzyme. J Mol Evol 81, 245–253 (2015). https://doi.org/10.1007/s00239-015-9712-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-015-9712-x