
Abstract
The leadzyme refers to a small ribozyme that cleaves a RNA substrate in the presence of Pb2+. In an optimized form, the enzyme strand contains only two unpaired nucleotides. Most RNA-cleaving DNAzymes are much longer. Two classical Pb2+-dependent DNAzymes, 8–17 and GR5, both contain around 15 nucleotides in the enzyme loop. This is also the size of most RNA-cleaving DNAzymes that use other metal ions for their activity. Such large enzyme loops make spectroscopic characterization difficult and so far no high-resolution structural information is available for active DNAzymes. The goal of this work is to search for DNAzymes with smaller enzyme loops. A simple replacement of the ribonucleotides in the leadzyme by deoxyribonucleotides failed to produce an active enzyme. A Pb2+-dependent in vitro selection combined with deep sequencing was then performed. After sequence alignment and DNA folding, a new DNAzyme named PbE22 was identified, which contains only 5 nucleotides in the enzyme catalytic loop. The biochemical characteristics of PbE22 were compared with those of the leadzyme and the two classical Pb2+-dependent DNAzymes. The rate of PbE22 rises with increase in Pb2+ concentration, being 1.7 h−1 in the presence of 100 μM Pb2+ and reaching 3.5 h−1 at 500 µM Pb2+. The log of PbE22 rate rises linearly in a pH-dependent fashion (20 µM Pb2+) with a slope of 0.74. In addition, many other abundant sequences in the final library were studied. These sequences are quite varied in length and nucleotide composition, but some contain a few conserved nucleotides consistent with the GR5 structure. Interestingly, some sequences are active with Pb2+ but none of them were active with even 50 mM Mg2+, which is reminiscent of the difference between the GR5 and 8–17 DNAzymes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Most RNA-cleaving ribozymes and DNAzymes require divalent metal ions for catalysis (Lu 2002; Sigel and Pyle 2007; Ward et al. 2014). Among the different metal ions used for RNA cleavage, Pb2+ has greatly fueled the growth of this field. The interaction between Pb2+ and nucleic acids was observed more than 50 years back. In 1959, the hydrolysis of RNA by lead hydroxide was demonstrated (Dimroth et al. 1959). A detailed investigation was published in 1968 showing that the rate of RNA depolymerization as well as its pH optimum, both varied with the Pb2+ concentration (Farkas 1968). It first became known that Pb2+ can bring about site-specific cleavage of tRNA in 1973 (Winterme and Zachau 1973). The mechanism of cleavage was proposed based on biochemical data and the crystal structure of the yeast tRNAPhe soaked in lead acetate (Brown et al. 1985; Werner et al. 1976).
Multiple variations of this well-studied yeast tRNA were used to design RNA selection libraries to isolate RNAs that undergo autolytic cleavage in the presence of Pb2+ (Pan and Uhlenbeck 1992a). Among the sequences derived from this selection, one was truncated and optimized into a minimal motif known as the ‘leadzyme.’ The leadzyme is a very small but interesting ribozyme with only two unpaired nucleotides 5′rGrA3′ in the enzyme loop and four unpaired nucleotides in the substrate strand (Fig. 1b). This enzyme is highly specific for lead and its reaction produces a 5′-OH end along with a 2′3′ cyclic phosphate product (Pan et al. 1994; Pan and Uhlenbeck 1992b). Deeper insights have been obtained about this enzyme through biochemical studies (Chartrand et al. 1997; Ohmichi et al. 1998), NMR (Hoogstraten et al. 1998, 2000; Legault et al. 1998), X-ray crystallography (Wedekind and McKay 1999, 2003), and other biophysical characterizations (Kadakkuzha et al. 2009).

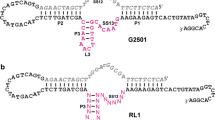
The secondary structures of the a PbE22, b leadzyme, c 17E, and d GR5 DNAzymes. The enzyme strands are in blue and substrate in green. The cleavage junction is indicated by the arrowheads (Color figure online)
Parallel to the ribozyme field, Pb2+ has been a very important metal ion in DNAzyme research as well. The first DNAzyme, GR5 (Fig. 1d), was obtained as a result of a Pb2+-dependent selection (Breaker and Joyce 1994), and it is highly specific and active with Pb2+ (Lan et al. 2010). The most extensively studied 8–17 DNAzyme, initially discovered from a Mg2+-dependent selection (Santoro and Joyce 1997), is also highly active in the presence of Pb2+. A commonly used variant of the 8–17 DNAzyme is named 17E (Fig. 1c) (Cruz et al. 2004; Li et al. 2000). It has been used as a model for biosensor development (Brown et al. 2003; Li and Lu 2000; Liu et al. 2009; Schlosser and Li 2009, 2010; Zhang et al. 2011).
While a few biochemical studies have been carried out on these DNAzymes, only little structure-related information is known (Brown et al. 2003; Huang and Liu 2014; Kim et al. 2007; Nowakowski et al. 1999). These two Pb2+-dependent DNAzymes (8–17 and GR5) have a similar size, containing 14 or 15 nucleotides in the enzyme loop (Fig. 1c, d). This relatively large size makes it difficult to carry out X-ray crystallography or NMR studies. We reason that a shorter DNAzyme similar to the leadzyme might help in detailed spectroscopic and structural analysis. Since even two nucleotides can perform the catalytic function in the leadzyme, an interesting question is whether it is possible to achieve similar catalysis in a short DNAzyme. In this work, we employed both rational design and in vitro selection to search for very short DNAzymes.
Materials and Methods
Chemicals
The in vitro selection-related DNA samples were purchased from Integrated DNA Technologies (IDT, Coralville, IA). For characterization, the enzyme strands were from Eurofins (Huntsville, AL). Pb(OAc)2 and other metal salts were from Sigma–Aldrich at the highest possible purity. Sodium acetate, 2-(N-morpholino)ethanesulfonic acid (MES), 3-(N-morpholino)propanesulfonic acid (MOPS), ethylenediaminetetraacetic acid (EDTA) disodium salt dihydrate, sodium chloride, and ammonium acetate were from Mandel Scientific Inc. (Guelph, Ontario, Canada). Sso Fast EvaGreen supermix was from Bio-Radfor real-time polymerase chain reaction (PCR). T4-DNA ligase, deoxynucleotide (dNTP) mix, Taq DNA polymerase with ThermoPol buffer and low molecular weight DNA ladder were from New England Biolabs.
In Vitro Selection
The method of in vitro selection is similar to the one we reported previously (Huang et al. 2014b). In brief, for each cleavage step, the DNA library was incubated with freshly prepared Pb2+ solutions. For all the selection rounds, the metal incubation time was maintained at 60 min and was reduced to 5 min only in the last round. For all rounds, 60 µM Pb2+ was used. After incubation, the solution was mixed with 8 M urea and purified by 10 % dPAGE (denaturing polyacrylamide gel electrophoresis). The position corresponding to the cleaved product was excised from the gel, the DNA was extracted by crushing and soaking the gel and was further desalted with a Sep-Pak C18 column (Waters). After drying in an Eppendorf Vacufuge at 45 °C overnight, the dried DNA was re-suspended in 60 µL of 5 mM HEPES buffer (pH 7.5). The round 6 library was sent out for deep sequencing. The PCR protocols for DNA amplification were also identical to the previously published and will not be repeated here.
Gel-Based Activity Assays
Gel-based activity assays were performed with a final concentration of 1 µM of the FAM-labeled substrate strand and 2 µM of the enzyme. The DNAzyme complexes were prepared by annealing them in buffer (50 mM MES, pH 6.0, 25 mM NaCl) and a final concentration of 100 µM Pb2+ was added. For the Mg2+ assay, 50 mM Mg2+ ions were added in pH 7.5 HEPES buffer to initiate the cleavage reaction. The products were separated on a denaturing polyacrylamide gel and analyzed using a Bio-Rad Chemi-Doc MP imaging system. For pH-dependent activity assay, the sodium acetate, 2-(N-morpholino)ethanesulfonic acid (MES) and 3-(Nmorpholino) propanesulfonic acid (MOPS) buffers (50 mM buffer with 25 mM NaCl) were used.
Deep Sequencing
The round 6 selected library was subjected to PCR, and the full-length library generated from this step was used to prepare the sample for deep sequencing. The PCR product was subjected to another PCR reaction so that the Illumina sequencing adaptors can be added. The forward primer P701: (5′-CAAGCAGAAGACGGCATACGAGATTCGCCTTAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTGCAGAATTCTAATACGAGTCAC) and reverse primer P501: (5′-AATGATACGGCGACCACCGAGATCTACACTAGATCGCACACTCTTTCCCTACACGACGCTCTTCCGATCTGTGCCAAGCTTACCG), each containing a unique index sequence were used. The PCR product was purified with 2 % agarose gel and extracted using a gel extraction kit from IBI Scientific. The extracted DNA was eluted in 25 µL Milli-Q water and quantified using a NanoDrop Spectrophotometer.
Results and Discussion
Rational Designed DNAzymes
From the study of the 8–17 and GR5 DNAzymes, it is known that both contain a few highly conserved nucleotides which are important for catalysis (Breaker and Joyce 1994; Brown et al. 2003; Peracchi et al. 2005; Schlosser et al. 2008b; Schlosser and Li 2010). In particular, the AG and CG dinucleotides in their enzyme loops have been identified to be critical. The sequence of the leadzyme also contains the unpaired AG in the enzyme loop. Based on these, we designed a few putative DNAzyme sequences which can bind to the substrate (Fig. 2a). However, when these sequences were assayed in the presence of 100 µM Pb2+ at pH 7.0 for 2 h, very low amount of cleavage was observed (Fig. 2b). The fastest Pb7 has a rate of only ~0.05 h−1, which is close to the background RNA cleavage rate by Pb2+ (vide infra) and is significantly slower than that of the leadzyme. Therefore, a simple combination of such nucleotides is insufficient for catalysis.

a The secondary structures of the eight rationally designed DNAzymes. b The cleavage yield of these enzymes in the presence of 100 μM Pb2+ at pH 7.0 for 2 h
In Vitro Selection
Since rational design failed to produce sufficiently active DNAzymes, we next resorted to in vitro selection (Fig. 3a). Since it is difficult to predict the optimal minimal length of the enzyme loop and also the number of unpaired nucleotides in the substrate strand, instead of using a very short randomized region, we employed a library containing 35 random nucleotides. We reason that a larger size can offer more flexibility and sequence diversity. If shorter DNAzymes exist, they may still be reflected in the final library by hiding redundant sequences as overhangs or hairpins.

a Schematic of the in vitro selection procedure. The library contains 35 random nucleotides (N35) and a single RNA linkage (rA) serving as the cleavage site. Sequences cleaved by Pb2+ are amplified by two PCR steps to seed the next round of selection. b The secondary structure of the library for in vitro selection. c Selection progress at each round. d The secondary structure of the original cis-cleaving enzyme; it has been engineered to the trans-cleaving PbE22 enzyme (shown in Fig. 1a). e Two other examples of short DNAzyme candidates from the selection, but they are inactive
The library design is shown in Fig. 3b. A single RNA linkage (rA) is embedded in this DNA library to serve as the cleavage site. Since RNA is about 1-million-fold less stable compared to DNA (Li and Breaker 1999), cleavage is most likely to take place at the RNA position. This library was incubated with Pb2+ and sequences that can be cleaved were separated from the rest using gel electrophoresis and amplified by two rounds of PCR to seed for the next round of selection (Fig. 3a). A reaction condition of 60 µM Pb2+ for 1 h was pursued up to the 5th round. The incubation time was reduced to 5 min in round 6, which was then subjected to sequencing. The cleavage yield at each round is shown in Fig. 3c. We did not push for very fast enzymes in this selection since we aim to obtain shorter DNAzymes, which may not cleave very efficiently. In other words, our goal was to maximize sequence diversity in the resulting library.
Instead of using conventional cloning and Sanger sequencing, we chose to use deep sequencing for this project to search for very short DNAzymes, which may not be highly active and may not represent the major population in the library. As a result, we needed to exhaustively search for all possible sequences. From the sequencing results of our final library, a total of 32,144 sequences were obtained.
Sequence Analysis
It is interesting to note that while the first DNAzyme selection was carried out using Pb2+, Pb2+ has not been used as a metal cofactor in any subsequent selections. Therefore, this work represents a second example of such an effort. The selection condition is also quite different: while GR5 was selected using 1 mM PbOAc in a high salt buffer (0.5 M NaCl, 0.5 M KCl, 50 mM MgCl2 at pH 7) (Breaker and Joyce 1994), our selection used only 60 µM Pb2+ in a low salt buffer (50 mM MES pH 6.0, 25 mM NaCl). Instead of only 20 sequences reported in the previous paper, we have obtained over thirty thousand sequences.
The sequenced library was aligned into different families based on their sequence similarity, and we observed quite high sequence diversity. Even the most abundant family of DNAzyme represents only 6.79 % of the final sequences. The sequences from the first ninety families (76.4 % of the total sequences) were individually folded using Mfold (Zuker 2003). Out of these, 32 of the resulting trans-cleaving enzymes display a reasonable fold, and their trans-cleaving sequences are shown in Fig. 4a. These sequences represent 46.3 % of the ninety families analyzed and 35.4 % of the total sequences. Some of them have the bases 5′–AGCG–CG–3′ conserved exactly as they are in GR5, while a few have nucleotide insertions, mutations, or deletions from these conserved ones. Each of these 32 sequences were tested with 10 µM Pb2+ and 50 mM Mg2+, respectively. Their cleavage fraction after 1 h is plotted in Fig. 4b. Interestingly, like GR5, cleavage was observed only with Pb2+; while unlike the 8–17 DNAzyme, none of them was active with Mg2+.

a Sequences of 32 tested DNAzymes from the selected library and their abundance in the library. The nucleotides that can be aligned with the conserved nucleotides in GR5 are marked in red. b The cleavage fraction after 1 h of reaction for the 32 trans-cleaving enzymes, tested with 10 µM Pb2+ (red) and 50 mM Mg2+ (black). The 8–17 and GR5 DNAzymes are also included for comparison (Color figure online)
PbE22
Since the goal of this work is to identify very short DNAzymes, after the general understanding of the activity of all representing sequences, we focused our attention to very short enzyme loops. Out of the many sequences, we found only one short enzyme (family C22 in Fig. 4) that exhibited decent activity, achieving ~50 % cleavage in the above assay. This enzyme motif has appeared 285 times out of the 32,144 sequences. The truncation of this enzyme from its cis-cleaving form is shown in Fig. 3d, and the trans-cleaving construct is shown in Fig. 1a, which was re-named to be PbE22. PbE22 has 5 nucleotides 5′GAAGC3′ in the catalytic loop of the enzyme and 6 unpaired nucleotides in the substrate strand 5′rAGGAAGA3′ including the cleavage dinucleotide junction. It is interesting to note that most of these unpaired nucleotides are purines. Aside from PbE22, two other sequences can also fold into a short enzyme loop structure (Fig. 3e). However, they are inactive when tested with Pb2+. Therefore, PbE22 was used for the subsequent studies.
Biochemical Characterization of PbE22
To characterize this new DNAzyme, we next performed preliminary biochemical studies. First, the cleavage kinetics of PbE22 was measured in the presence of 100 µM Pb2+ (Fig. 5a, black dots). The time-dependent cleavage yield can be fitted to first-order reaction kinetics with a rate constant of 1.7 h−1. Under the same condition, the free substrate was cleaved at a rate of ~0.0082 h−1. Therefore, the rate enhancement brought by PbE22 in the presence of 100 µM Pb2+ at pH 7.0 is ~210-fold. For comparison, the GR5 DNAzyme (Fig. 1d) has a reported rate enhancement of ~105 (Breaker and Joyce 1994), while the leadzyme has a rate enhancement of 1100 (Pan and Uhlenbeck 1992b). Therefore, PbE22 has the lowest catalytic efficiency, and GR5 has the highest.

Biochemical characterization of PbE22. a Kinetics of PbE22 and GR5 cleavage at pH 7. b Cleavage rate constant of PbE22 as a function of [Pb2+] at pH 7.0. Inset = gel showing cleavage at various [Pb2+] concentrations after 30 min reaction at pH 6.5. Log scale plot of the rate as a function of pH for c PbE22 with 20 µM Pb2+ and d GR5 with 1 µM Pb2+
The inset of Fig. 5b is a gel image showing the cleavage yield at 30 min with increasing Pb2+ concentrations. Indeed more Pb2+ induced more cleavage, confirming this is a Pb2+-dependent DNAzyme. To quantitatively understand the effect of Pb2+ concentration, we next measured the enzyme kinetics at various Pb2+ concentrations at pH 7.0 (Fig. 5b). An apparent dissociation constant (K d) of 77 µM Pb2+ was obtained. For comparison, the leadzyme has a rate of 0.4–0.5 min−1 at 25 µM Pb2+ and pH 7.0. This is faster than PbE22 but less as compared to GR5 (Pan et al. 1994). This comparison suggests that a bigger catalytic loop might be required for optimum activity of the enzyme.
pH-Dependent Activity
To further characterize this enzyme, we studied the DNAzyme rate with increasing pH at 20 µM Pb2+ concentration. Again we compared it to that of the GR5 using 1 µM Pb2+. The log of rate increased linearly with increasing pH in the low pH region with a slope of 0.74 (Fig. 5c). Beyond pH 7, the increase in rate slowed down. The slope of GR5 was calculated to be 0.82 and it maintained a good linearity up to pH 7.6 (Fig. 5d). Beyond pH 7.6, we could not measure the rate since it was too fast for manual pipetting. The reason for the narrower linear range for PbE22 might be related to the use of higher Pb2+ concentration (20 µM), as Pb2+ tends to precipitate more easily at such a high concentration. Similar to PbE22, the leadzyme also exhibits a linear increase in log (rate) with increase in pH up to 7.0 at 25 µM Pb2+.11 Therefore, this indicates all these enzymes have a similar mechanism in terms of a single deprotonation at the rate-limiting step of this reaction, and this is often directly or indirectly linked to the deprotonation of the 2′-OH at the cleavage site.
Metal Specificity Test
GR5 has excellent selectivity for Pb2+, and an impressive fact is that GR5 is inactive even with 50 mM Mg2+. For comparison, the 8–17 DNAzyme is quite active with such a high concentration of Mg2+ (~1.6 min−1) (Brown et al. 2003; Santoro and Joyce 1997; Wang et al. 2010). We also carried out metal specificity tests on PbE22. We first studied the cleavage of the substrate in the presence of an array of divalent and trivalent metal ions (10 µM each, Fig. 6a) and found that like GR5, PbE22 too has a high selectivity for Pb2+. Furthermore, even with 500 µM of other metals still no observable activity was found (Fig. 6b). Therefore, PbE22 is also highly specific for Pb2+.

Metal specificity test of the PbE22 DNAzyme in the presence of a 10 and b 500 µM of all the metals compared to 10 µM of Pb2+ at pH 6.2
It is worth mentioning here that we tested up to 0.5 mM Pb2+ and 50 mM Mg2+ for PbE22. Based on a rough estimation, at the same metal concentration, the rate of Pb2+ is ~33,800-fold faster than that of Mg2+ for cleaving the 17E DNAzyme (Brown et al. 2003). This difference is even larger for the GR5 DNAzyme. Since our PbE22 is much slower, it would require over 1 M of Mg2+ to conclude that PbE22 is inactive with Mg2+ if we use 17E as the standard. We tested the PbE22 in 4 M Mg2+ and found that it has no cleavage as well (data not shown). Therefore, PbE22 is highly specific for Pb2+.
Further Discussion
The PbE22 DNAzyme gives an example for the better understanding of the effect of the size of the catalytic loop on the activity of DNAzyme. This study indicates that although site-specific and metal-specific catalysis is possible with short loops, the presence of extra nucleotides is probably needed for optimal activity. It suggests the significance of bigger catalytic loops for better folding or scaffolding for utilizing the metal cofactor. For example, in the 8–17 DNAzyme, in addition to the four nucleotides identified to be critical for the cleavage reaction, additional nucleotides are found to have other roles to assist DNAzyme folding (Schlosser et al. 2008b; Wang et al. 2010). In this PbE22 DNAzyme, the number of nucleotides on the enzyme strand is very limited. At the same time, DNA lacks the structural versatility present in RNA due to the lack of the 2′-OH group, which may explain the faster cleavage by the leadzyme despite its even smaller size.
Currently, most DNAzyme selections use 30–50 random nucleotides in the selection library. When the catalytic core is examined, however, the required sequences are much shorter. Usually the core contains ~15 nucleotides for RNA-cleaving DNAzymes. To deal with the remaining nucleotides, one of the common strategies adopted by the enzymes is to form a hairpin. This hairpin may or may not play an important role in catalysis. The DNAzyme folding may turn out to be very different if no hairpin is added or deleted and this may affect DNAzyme activity. For example, in the UO2 2+ specific DNAzyme, removal of the hairpin completely suppresses its activity (Brown et al. 2009; Liu et al. 2007). On the other hand, for the lanthanide-specific DNAzyme Ce13d, removal of the hairpin has relatively less effect (Huang et al. 2014a). Another strategy to deal with the redundant nucleotides is by forming alternative substrate binding regions (e.g., a portion of the library is used for the substrate binding purpose).
Given the ability of longer libraries to hide extra bases, we decided to use the N35 library in this work. In addition, it may allow more flexibility in substrate binding than directly using very short libraries. For example, some nucleotides nearby the cleavage site may need to be non-paired, and this is particularly true for the short enzymes (the leadzyme and PbE22 are the examples). A large library may better support such possibility by forming alternative base pairing with the substrate. Finally, since we cannot predict the minimal length to use, we counted on the sequence diversity and flexibility of the long library to obtain short DNAzymes in a single selection experiment. The successful isolation of the PbE22 DNAzyme has indicated the success and reliability of this strategy. There have been studies using a shorter random region of N20 for carrying out DNAzyme selections (Schlosser et al. 2008a). One of the future directions is to test even smaller randomized regions and this may directly eliminate GR5 like sequences.
Traditionally, at the end of in vitro selection, the selected library is cloned into plasmid vectors and transformed into the bacterial cells which are further grown and disrupted for isolating the amplified plasmids. These purified plasmids are then subjected to sequencing. Although the method is widespread in usage, it poses certain limitations upon the number of sequences and diversity of sequences that can be obtained. As a result, only a small fraction of the selected library and mostly the most abundant sequences can be obtained. This leads to loss of valuable information regarding the selection and can prove to be a great hindrance in the discovery of new enzymes. However, the technology of deep sequencing is bringing about a revolution in this area. Through this, thousands of sequences can be obtained from the selected library which can further be subjected to clustering based on their sequence similarities. A comprehensive understanding of the diversity and abundance of each sequence family can be obtained and significant sequence variations within each type of family can also be dug out and analyzed. This gives a huge pool of information. For example, in the above-mentioned selection, PbE22 could be fished out of 32,144 sequences in spite of its abundance being only 0.89 % among the entire pool. Thus, this demonstrates the power of the technology of deep sequencing. Several other selection studies have already explored this deep sequencing technique to reveal diverse sequence information (Ameta et al. 2014; Majerfeld et al. 2010; Mao et al. 2015; Pitt and Ferré-D’Amaré 2010).
The PbE22 DNAzyme is not a very efficient DNAzyme, but it may provide a scaffold for studying Pb2+ binding to DNAzyme given its much smaller size. Subsequent work will be focused on testing its structure and spectroscopic properties.
Conclusions
In summary, we had a goal of obtaining a very short RNA-cleaving DNAzyme. To achieve this goal, both rational design and in vitro selection were performed. We isolated a very short RNA-cleaving DNAzyme that uses Pb2+ as a cofactor named PbE22. It has a rate enhancement of ~200-fold and this is lower compared to that for the leadzyme or the GR5 DNAzyme. These enzymes share a similar cleavage mechanism as deduced from pH-dependent studies. PbE22 has excellent selectivity for Pb2+. In the rest of the library too, all the active sequences are only active with Pb2+ but not with Mg2+. This study has provided insights into the significance of the size of the DNAzymes, Pb2+-dependent activity, and metal specificity. We expect that this DNAzyme can be used for spectroscopic and structural analysis. These experiments have been difficult to carry out with the current DNAzymes that bear relatively large catalytic loops.
References
Ameta S, Winz M-L, Previti C, Jäschke A (2014) Next-generation sequencing reveals how RNA catalysts evolve from random space. Nucleic Acids Res 42:1303–1310
Breaker RR, Joyce GF (1994) A DNA enzyme that cleaves RNA. Chem Biol 1:223–229
Brown RS, Dewan JC, Klug A (1985) Crystallographic and biochemical investigation of the lead(II)-catalyzed hydrolysis of yeast phenylalanine tRNA. Biochemistry 24:4785–4801
Brown AK, Li J, Pavot CMB, Lu Y (2003) A lead-dependent DNAzyme with a two-step mechanism. Biochemistry 42:7152–7161
Brown AK, Liu JW, He Y, Lu Y (2009) Biochemical characterization of a uranyl ion-specific DNAzyme. ChemBioChem 10:486–492
Chartrand P, Usman N, Cedergren R (1997) Effect of structural modifications on the activity of the leadzyme. Biochemistry 36:3145–3150
Cruz RPG, Withers JB, Li Y (2004) Dinucleotide junction cleavage versatility of 8–17 deoxyribozyme. Chem Biol 11:57–67
Dimroth K, Witzel H, Hulsen W, Mirbach H (1959) Uber die hydrolyse von ribonucleinsauren in gegenwart von metallhydroxyden. Annalen Der Chemie-Justus Liebig 620:94–108
Farkas WR (1968) Depolymerization of ribonucleic acid by plumbous ion. Biochimica et biophysica acta (BBA)—nucleic acids and protein. Synthesis 155:401–409
Hoogstraten CG, Legault P, Pardi A (1998) NMR solution structure of the lead-dependent ribozyme: evidence for dynamics in RNA catalysis. J Mol Biol 284:337–350
Hoogstraten CG, Wank JR, Pardi A (2000) Active site dynamics in the lead-dependent ribozyme. Biochemistry 39:9951–9958
Huang P-JJ, Liu J (2014) Two Pb2+-specific dnazymes with opposite trends in split-site-dependent activity. Chem Commun 50:4442–4444
Huang P-JJ, Lin J, Cao J, Vazin M, Liu J (2014a) Ultrasensitive DNAzyme beacon for lanthanides and metal speciation. Anal Chem 86:1816–1821
Huang P-JJ, Vazin M, Liu J (2014b) In vitro selection of a new lanthanide-dependent DNAzyme for ratiometric sensing lanthanides. Anal Chem 86:9993–9999
Kadakkuzha BM, Zhao L, Xia T (2009) Conformational distribution and ultrafast base dynamics of leadzyme. Biochemistry 48:3807–3809
Kim HK, Rasnik I, Liu JW, Ha TJ, Lu Y (2007) Dissecting metal ion-dependent folding and catalysis of a single DNAzyme. Nat Chem Biol 3:762–768
Lan T, Furuya K, Lu Y (2010) A highly selective lead sensor based on a classic lead DNAzyme. Chem Commun 46:3896–3898
Legault P, Hoogstraten CG, Metlitzky E, Pardi A (1998) Order, dynamics and metal-binding in the lead-dependent ribozyme. J Mol Biol 284:325–335
Li Y, Breaker RR (1999) Kinetics of RNA degradation by specific base catalysis of transesterification involving the 2′-hydroxyl group. J Am Chem Soc 121:5364–5372
Li J, Lu Y (2000) A highly sensitive and selective catalytic DNA biosensor for lead ions. J Am Chem Soc 122:10466–10467
Li J, Zheng W, Kwon AH, Lu Y (2000) In vitro selection and characterization of a highly efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res 28:481–488
Liu J, Brown AK, Meng X, Cropek DM, Istok JD, Watson DB, Lu Y (2007) A catalytic beacon sensor for uranium with parts-per-trillion sensitivity and millionfold selectivity. Proc Natl Acad Sci USA 104:2056–2061
Liu J, Cao Z, Lu Y (2009) Functional nucleic acid sensors. Chem Rev 109:1948–1998
Lu Y (2002) New transition metal-dependent DNAzymes as efficient endonucleases and as selective metal biosensors. Chem Eur J 8:4588–4596
Majerfeld I, Chocholousova J, Malaiya V, Widmann J, McDonald D, Reeder J, Iyer M, Illangasekare M, Yarus M, Knight R (2010) Nucleotides that are essential but not conserved; a sufficient l-tryptophan site in RNA. RNA 16:1915–1924
Mao Y, Liu M, Tram K, Gu J, Salena BJ, Jiang Y, Li Y (2015) Optimal DNA templates for rolling circle amplification revealed by in vitro selection. Chem Eur J 21:8069–8074
Nowakowski J, Shim PJ, Prasad GS, Stout CD, Joyce GF (1999) Crystal structure of an 82-nucleotide RNA–DNA complex formed by the 10-23 DNA enzyme. Nat Struct Biol 6:151–156
Ohmichi T, Okumoto Y, Sugimoto N (1998) Effect of substrate RNA sequence on the cleavage reaction by a short ribozyme. Nucleic Acids Res 26:5655–5661
Pan T, Uhlenbeck OC (1992a) In vitro selection of rnas that undergo autolytic cleavage with Pb(II). Biochemistry 31:3887–3895
Pan T, Uhlenbeck OC (1992b) A small metalloribozyme with a two-step mechanism. Nature 358:560–563
Pan T, Dichtl B, Uhlenbeck OC (1994) Properties of an in vitro selected Pb2+ cleavage motif. Biochemistry 33:9561–9565
Peracchi A, Bonaccio M, Clerici M (2005) A mutational analysis of the 8-17 deoxyribozyme core. J Mol Biol 352:783–794
Pitt JN, Ferré-D’Amaré AR (2010) Rapid construction of empirical RNA fitness landscapes. Science 330:376–379
Santoro SW, Joyce GF (1997) A general purpose RNA-cleaving DNA enzyme. Proc Natl Acad Sci USA 94:4262–4266
Schlosser K, Li YF (2009) Biologically inspired synthetic enzymes made from DNA. Chem Biol 16:311–322
Schlosser K, Li Y (2010) A versatile endoribonuclease mimic made of DNA: characteristics and applications of the 8-17 RNA-cleaving DNAzyme. ChemBioChem 11:866–879
Schlosser K, Gu J, Lam JCF, Li YF (2008a) In vitro selection of small RNA-cleaving deoxyribozymes that cleave pyrimidine-pyrimidine junctions. Nucleic Acids Res 36:4768–4777
Schlosser K, Gu J, Sule L, Li YF (2008b) Sequence-function relationships provide new insight into the cleavage site selectivity of the 8-17 RNA-cleaving deoxyribozyme. Nucleic Acids Res 36:1472–1481
Sigel RKO, Pyle AM (2007) Alternative roles for metal ions in enzyme catalysis and the implications for ribozyme chemistry. Chem Rev 107:97
Wang B, Cao L, Chiuman W, Li Y, Xi Z (2010) Probing the function of nucleotides in the catalytic cores of the 8-17 and 10-23 dnazymes by abasic nucleotide and C3 spacer substitutions. Biochemistry 49:7553–7562
Ward WL, Plakos K, DeRose VJ (2014) Nucleic acid catalysis: metals, nucleobases, and other cofactors. Chem Rev 114:4318–4342
Wedekind JE, McKay DB (1999) Crystal structure of a lead-dependent ribozyme revealing metal binding sites revelant to catalysis. Nat Struct Biol 6:261–268
Wedekind JE, McKay DB (2003) Crystal structure of the leadzyme at 1.8 Å resolution: metal ion binding and the implications for catalytic mechanism and allo site ion regulation. Biochemistry 42:9554–9563
Werner C, Krebs B, Keith G, Dirheimer G (1976) Specific cleavages of pure tRNAs by plumbous ions. Biochim Biophys Acta 432:161–175
Winterme W, Zachau HG (1973) Mg2+-catalyzed specific splitting of transfer-RNA. Biochim Biophys Acta 299:82–90
Zhang X-B, Kong R-M, Lu Y (2011) Metal ion sensors based on DNAzymes and related DNA molecules. Annu Rev Anal Chem 4:105–128
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415
Acknowledgments
Funding for this work was from the University of Waterloo, Ontario Ministry of Research & Innovation, and the Natural Sciences and Engineering Research Council (NSERC) of Canada (Discovery Grant and Strategic Project Grant).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Saran, R., Chen, Q. & Liu, J. Searching for a DNAzyme Version of the Leadzyme. J Mol Evol 81, 235–244 (2015). https://doi.org/10.1007/s00239-015-9702-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-015-9702-z