
Abstract
There have been two distinct phases of evolution of the genetic code: an ancient phase—prior to the divergence of the three domains of life, during which the standard genetic code was established—and a modern phase, in which many alternative codes have arisen in specific groups of genomes that differ only slightly from the standard code. Here we discuss the factors that are most important in these two phases, and we argue that these are substantially different. In the modern phase, changes are driven by chance events such as tRNA gene deletions and codon disappearance events. Selection acts as a barrier to prevent changes in the code. In contrast, in the ancient phase, selection for increased diversity of amino acids in the code can be a driving force for addition of new amino acids. The pathway of code evolution is constrained by avoiding disruption of genes that are already encoded by earlier versions of the code. The current arrangement of the standard code suggests that it evolved from a four-column code in which Gly, Ala, Asp, and Val were the earliest encoded amino acids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction: Two Distinct Phases of Code Evolution
The standard genetic code (SGC) is shared by the genomes of almost all bacteria, archaea, and eukaryotes. It is therefore clear that it arose very early in evolution prior to the divergence of these domains. Although most genomes use the standard code, many variant codes have been discovered that differ by the reassignment of a small number of codons. These variant codes have arisen in specific lineages; thus, it is clear that the variants evolved from the standard code relatively recently in evolutionary history. For example, in the roughly half a billion years since the origin of Metazoans, the mitochondrial codes of vertebrates, echinoderms, arthropods, and flatworms have all become different from one another. In contrast, the divergence of prokaryotic lineages probably occurred three billion years ago or more. This means that the origin of the standard code is considerably separated in time from the period in which modern variant codes have diversified.
In this article, we will refer to the phases of code evolution before and after the establishment of the SGC as the ancient and modern phases of code evolution, respectively. Table 1 summarizes the factors that influence genetic code evolution in the two phases. The aim of this article is to show that the expected pathways by which the code evolves will be quite different in the two phases.
When studying the modern phase, we have concrete sequence data for comparative genomics and the nature of the particular changes that caused codon reassignment (usually tRNA base modifications or mutations) can be determined by experimental study of living organisms. Sengupta et al. (2007) analyzed all known cases of codon reassignments in mitochondrial genomes and updated previous surveys of both mitochondrial and other types of genomes by Knight et al. (2001) and Swire et al. (2005). The conclusions of these studies (summarized in Fig. 1 and Table 2) are that codon reassignments occur frequently in mitochondria and rarely in other types of genomes. No reassignments are known in archaea or chloroplasts, and very few are found in bacteria (despite the thousands of complete bacterial genomes now available). For the case of nuclear genomes, the ciliate group in eukaryotes account for a relatively large number of variant codes, suggesting something unusual in this group—see also Lozupone et al. (2001). However, other than this, changes are limited to a single example in each of fungi, green algae, and diplomonads. The existence of these variants is proof that the code is not completely frozen, even in modern organisms, and this makes it clear that changes to the code would also have been possible in the early stages of evolution, prior to the last universal common ancestor. However, modern variant codes differ relatively little from the standard code, which suggests that there are strong constraints that limit the possible pathways of code evolution in modern organisms.

Layout of the standard genetic code showing cases of codon reassignment in modern genomes. Alphabetical labels refer to Table 2
Studying the ancient phase is more speculative, because it occurred before the diversification of existing organisms. Therefore, comparative genomics provides no information. It seems likely that the earliest versions of the code were much simpler than the current code. Probably, there were few encoded amino acids with large blocks of codons assigned to each amino acid. Codon reassignment during the ancient phase involved the addition of a new amino acid and the subdivision of a large codon block into two smaller ones. In contrast, during the modern phase, codons are reassigned but the same set of 20 amino acids is retained. Thus one codon block increases in size while another decreases.
The centrality of mRNA, rRNA, and tRNA to the process of translation strongly suggests that the genetic code arose in an RNA world in which most important processes in the cell were controlled by RNAs. Although the RNA World hypothesis is widely accepted (Joyce 2000; Bernhardt 2012; Higgs and Lehman 2015), there are also arguments in favor of proteins coevolving with RNAs from very early on (Caetano-Anollés and Seufferheld 2013; Carter 2015; Francis 2015). If protein sequences were present before the origin of the ribosome and the genetic code, there must have been a different mechanism of specifying and synthesizing amino acid sequences, such as a direct structural interaction of proteins and RNA (Carter 2015) or the mechanism of cosynthesis of proteins and nucleic acids proposed by Francis (2011). These mechanisms seem speculative, however, and are not yet supported by experiment. Here, we are discussing the origin of the genetic code, as it operates with ribosomal protein synthesis. There must have been long RNAs present by this stage. There could also have been short peptides and/or amino acids covalently linked to RNAs (as with tRNAs). The existence of long proteins encoded by some other mechanism seems unlikely to us, although if it were the case, it would make little difference to the discussion of the evolution of the assignments between codons and amino acids, which is the main subject of this paper. We also assume that both RNA and proteins preceded the origin of DNA. In other words, RNA, not DNA, was still the genetic medium at the time of the origin of the genetic code. This is supported by the fact that the key molecules of the translation system are shared by all forms of life, whereas the key molecules of DNA replication are not the same in all domains (Lazcano et al. 1988; Burton and Lehman 2009).
It should be borne in mind that the genomes of ancient and modern organisms would be qualitatively different. The early evolution of the code would have begun in organisms with very few encoded proteins, and the development of the code would have proceeded at the same time as the number of protein coding genes increased and cells became increasingly reliant on proteins to carry out important functions. In contrast, the variant codes that arose in the modern phase evolved in organisms with DNA genomes that already coded for a large number of proteins. As the size of the encoded amino acid repertoire increased during the ancient phase, the fitness of existing proteins could increase by incorporation of the new amino acid at certain sites. The range of possible protein functions also increased. An organism that learned to synthesize proteins with a greater diversity of amino acids would be at a tremendous advantage. This is the selective driving force for code evolution in the ancient phase. However, the selective advantage of increasing the repertoire of versatile proteins by increasing the repertoire of amino acids encoded by the code is eventually offset by the cost of reassigning codon(s) from older to newer amino acids. In the modern phase, reassignment occurs only among the same set of 20 amino acids, and code evolution is not characterized by an increase in the repertoire of encoded amino acids. This suggests that selection also creates barriers to change and this may possibly explain why the amino acid repertoire of the genetic code stopped at 20. Gene sequences evolve to code for useful proteins under the current code of the organism. If the code changes, this will introduce amino acid substitutions simultaneously into many gene sequences, and many of these substitutions will be deleterious. Deleterious changes can only be eliminated by the gradual occurrence of mutations in the genes. Code changes can occur more easily in genomes with fewer encoded proteins because fewer disruptive substitutions are introduced. The genome of the ancient organisms in which the code evolved would likely have encoded only a few proteins initially, which suggests that changes to the code would have been easier in the ancient phase. This is also the main reason why, in the modern phase, changes occur in the small genomes of mitochondria much more easily than full-size bacterial and eukaryotic nuclear genomes.
These introductory considerations already point to one of the main conclusions of this paper: during the ancient phase, positive selection on new code variants can drive the evolution of the code, whereas in the modern phase, selection stabilizes the code and acts principally as a barrier that must be overcome in order to produce new code variants.
Factors Relevant to Codon Reassignment in the Modern Phase
A codon reassignment occurs when a new tRNA acquires the ability to decode a previously non-cognate codon and the original tRNA loses its ability to decode the codon in question. The gain-loss framework introduced by Sengupta and Higgs (2005) and illustrated in Fig. 2 is a useful way to view the possible mechanisms of codon reassignment. Reassignment involves two independent changes to the translation machinery. By ‘gain,’ we mean the acquisition of a new tRNA gene that interacts with the reassigned codon or the change of an existing tRNA so that it gains the ability to interact with a codon that it did not previously translate. This could occur via a mutation in the anticodon or a change in the nature of the modified bases in the anticodon. By ‘loss,’ we mean the deletion of a tRNA gene that formerly interacted with the codon or the loss of function of a tRNA (via a mutation or modified base change) such that it can no longer pair with the original codon.

a Evolutionary pathways of codon reassignment in the modern phase via the gain-loss process. b Selective cost involved in the various stages of the codon reassignment process in the modern phase
The most frequent example of codon reassignment (which occurs many times in independent lineages of mitochondria and other genomes) is the reassignment of UGA from Stop to Trp. Here, it is the release factor that formerly interacted with the Stop codon that is lost, and the gain is a mutation in the anticodon of the tRNA-Trp. For organisms using the standard code, the tRNA-Trp anticodon is CCA, which interacts with only the UGG Trp codon. The anticodon mutates to UCA, which interacts with both UGA and UGG via wobble pairing of the U in the anticodon. The reassignment of AUA from Ile to Met is similar. In the standard code, this codon is translated as Ile by a tRNA with anticodon K2CAU (Muramatsu et al. 1988), where K2C (lysidine) is a modified base that pairs only with the A at the third codon position. The standard tRNA-Met has anticodon CAU, which pairs with the single AUG Met codon. When the code changes, the anticodon either mutates to UAU or becomes modified to f5CAU, both of which are able to pair with the AUA codon and well as AUG (Tomita et al. 1999a). The structural basis of such changes are beginning to be understood (Demeshkina et al. 2012; Voorhees et al. 2013; Cantara et al. 2013). The changes corresponding to the gain and loss events are known in a large number of other cases too (Sengupta et al. 2007); thus, the biochemical details responsible for codon reassignments in the modern phase are fairly well understood.
However, in order to understand the mechanism of codon reassignment in an evolutionary sense, we need to explain how the changes in the tRNAs became fixed in the population. Figure 2a summarizes the possible routes for codon reassignment within the gain-loss framework. If the gain occurs before the loss, the codon will be translated ambiguously because there will be two different tRNAs that translate the same codon. We refer to this as the ambiguous intermediate (AI) mechanism, following Schultz and Yarus (1994, 1996). Alternatively, if the loss occurs before the gain, the codon will become unassigned because there is no tRNA that interacts well with that codon. We refer to this as the unassigned codon (UC) mechanism (Sengupta and Higgs 2005). Once both gain and loss have occurred, we have a “new code but old sequences,” as shown in Fig. 2a. The codon will be used in positions where the old amino acid was preferred. In order to for the sequences to adapt to the new code, it is necessary for the codon to be eliminated from these positions and to be introduced in positions where the new amino acid is preferred. This requires many mutations in coding sequences.
Figure 2b gives a schematic picture of the selective costs associated with these steps of codon reassignment. Moving up the diagram corresponds to a decrease in fitness. This emphasizes that there is a selective barrier to be overcome between the old and new codes. In the AI mechanism, an ambiguous codon will usually be disadvantageous because the codon will be mistranslated part of the time. Similarly, in the UC mechanism, an unassigned codon will be disadvantageous due to the absence of any cognate tRNA that can efficiently translate the codon. If there were only one tRNA that interacted with the codon, loss of this tRNA gene would be lethal and reassignments involving this loss would not occur. However, if there is another tRNA that interacts to a limited extent with the codon, then the primary tRNA for the codon can be deleted without killing the organism. However, the loss is still disadvantageous because the codon can now be translated only inefficiently by a tRNA that does not pair well with this codon. Specific examples of this were discussed by Sengupta et al. (2007).
Once we reach the “new code, old sequences” stage, there is still a selective disadvantage because the codon is used in places where the old amino acid is preferred. However, in some cases, the disadvantage may be less for an individual in which both gain and loss events have occurred than for individuals characterized by only the gain or loss event, i.e., a subsequent gain (or loss) event heavily compensates for the cost in fitness due to a prior loss (or gain) event. The gain and loss can therefore spread through the population together and the new code can get fixed in the population even without the fixation of the intermediate AI or UC stages. We termed this the compensatory change (CC) mechanism (Sengupta and Higgs 2005) because it is similar to the spread of pairs of compensatory mutations, for example, in the helical regions of RNA secondary structures—see Higgs (1998). Although the CC mechanism is distinct from AI and UC mechanisms during the time in which it is occurring, with available genomic sequence data, it is not always possible to distinguish after the fact whether a codon reassignment occurred via the AI/UC or the CC mechanism.
After both gain and loss have occurred, the genome sequence of the organism can begin to adapt to the new code by making mutations in its coding sequences where necessary, so that the codon is now used in positions where the new amino acid is preferred. At this point the new genetic code is well established. The new code has no ambiguities or unassigned codons, and it still encodes the same set of 20 amino acids as the old code. Its fitness should therefore be very similar to the fitness of the initial code. It is possible that there is a small selective difference between the new code and the initial code due to the presence of translational errors, which could either be slightly more or slightly less deleterious according to the assignment of amino acids to codons. There is a large literature discussing the selective costs of translational errors in alternative genetic codes (Freeland and Hurst 1998; Gilis et al. 2001; Freeland et al. 2003; Goodarzi et al. 2004, 2005). We will discuss this below, since this is important to the evolution of the code in the ancient phase. In our opinion, however, the small selective differences between codes that might arise from translational errors are not the drivers of code evolution in the modern phase. In the modern phase, it is the large selective barrier that arises when a gain or loss event occurs that is relevant, not the possible small selective advantage that could exist when the whole process is over.
Another possible mechanism of codon reassignment within the gain–loss framework is the codon disappearance (CD) mechanism, originally proposed by Osawa and Jukes (1989). They recognized that the selective barrier that we emphasized above could be bypassed if the codon disappears from the genome before changes occur to the tRNAs. The gain and loss events are then neutral at the time when they occur. After the codon is reassigned, it can reappear with a new meaning in different positions in the gene sequences. The CD mechanism is facilitated by mutation biases, which lead to replacement of a codon by a synonymous one. For example, the UGA stop codon tends to mutate to the UAA stop codon in mitochondrial genomes where the mutational bias favors A and U over C and G. As the number of stop codons is small in mitochondrial genomes with few genes, the number of mutations that need to occur in order for a stop codon to disappear is small. Sengupta et al. (2007) gave evidence that the CD mechanism is the most likely explanation of the reassignment of UGA from stop to Trp, and of several other reassignments involving stop codons.
On the other hand, even in a small mitochondrial genome, many sense codons occur hundreds of times. Swire et al. (2005) concluded that it was extremely unlikely that codon disappearance explains the reassignment of a sense codon from one amino acid to another. Sengupta et al. (2007) reached the same conclusion, although they did find two examples of sense codon disappearance in mitochondrial genomes. Both these cases were in yeast mitochondria, where the AU mutational bias is exceptionally strong. Apart from these two cases, the chances of codon disappearance were so remote that it seemed virtually certain that the reassignment had occurred while the codon was present in the genome. It is therefore necessary to consider the other three mechanisms.
Sengupta et al. (2007) gave several examples where there is good evidence for either the UC or the AI mechanism. A good candidate for the UC mechanism is the reassignment of AUA from Ile to Met. It is most likely that the reassignment of AUA is initiated by the deletion of the tRNA-Ile with the K2C modification (see above). A good candidate for the AI mechanism is the reassignment of AAA from Lys to Asn, which also occurs in some mitochondria. The tRNA-Asn in bacteria has a G at the wobble position, which is modified to queuosine (Q). The Q modification appears to limit the pairing of this tRNA to the Asn codons AAU and AAC codons. It is likely that if the G remains unmodified, this tRNA can also translate AAA. Changing of the modification is a gain of function because it allows pairing with a new codon. The loss in this case is the mutation of wobble position of the tRNA-Lys from U to C, so that it only pairs with AAG (Castresana et al. 1998; Tomita et al. 1999b; Yokobori et al. 2001). A further interesting example is the pair of AGR codons, which code for Arg in the standard code and are reassigned in several different ways in mitochondria. The primary factor than initiates the reassignment appears to be the deletion of the tRNA-Arg from the genome. The AGR codons are subsequently translated as Ser, Gly, or Stop codons in different groups of organisms (Sengupta et al. 2007).
In general, a majority of the sense to sense codon reassignments are between amino acids lying in the same column of the genetic code. Such reassignments require only modifications in the anticodon of one tRNA (gain) together with the loss of functionality of the original tRNA. However, there exist a few examples of sense to sense codon reassignments, such as CUN:Leu to Thr and CUU, CUA: Thr to Ala (Osawa et al. 1990; Su et al. 2011; Ling et al. 2014), where the reassigned amino acid is in a different column of the code relative to the original assignment. Such reassignments typically require tRNA duplication followed by modifications in the tRNA identity elements that lead to changes in charging specificity. This allows the duplicated tRNA to be charged with the new amino acid thereby completing the reassignment process. The multi-step nature of the process makes such cross column reassignments quite rare. Nevertheless, such changes in the tRNA are usually preceded by the disappearance of the reassigned codons. A counter-example to this rule also provides the primary evidence of ambiguous decoding of a codon has been found in Candida spp. where the CUG codon is reassigned from Leu to Ser (Massey et al. 2003; Santos et al. 2004). A double mutation in the anticodon of the tRNA-Ser from CGA to CAG enables it to decode a CUG codon, originally associated with Leucine, without any change in charging specificity of tRNA-Ser.
From our survey of the large number of reassignments that occur in mitochondria, we concluded that these are initiated by chance events—disappearance of codons, deletion or duplications of tRNAs, mutations of anticodons, or changes in base modifications. Such events are expected to be rare and hence codon reassignments are expected to be observed more frequently in genomes which have a higher mutation rate than normal. The mutation rate of animal mitochondrial genomes is almost two orders of magnitude larger than the mutation rates in plant mitochondrial genomes (Lynch et al. 2006). It is therefore interesting to note that very few code changes are observed in plant mitochondrial genomes (Knight et al. 2001; Sengupta et al. 2007) and of these, none are in land plants.
The main point of this section is that there is a selective barrier to reassigning codons in the modern phase of code evolution. Therefore, the codon reassignments we see in the modern phase are the ones for which the changes causing the gain and loss occur most frequently and the ones for which the deleterious effect in the intermediate stage is smallest. In the specific case of the CD mechanism, while there is no selective barrier, there is still probabilistic barrier, in the sense that many occurrences of the same codon have to disappear at the same time. The changes occurring in the code, regardless of the mechanism, are all unlikely events and we see the changes that are least unlikely.
Factors Relevant to Codon Reassignment in the Ancient Phase
As mentioned in the introduction and in Table 1, the factors that are most important for genetic code evolution in the ancient phase are rather different from those in the modern phase. Whereas the modern phase involves reassignment of small numbers of codons without changing the set of 20 amino acids that is encoded, the ancient phase involves the addition of new amino acids to the code until the standard code with 20 amino acids is reached. It is therefore important to consider the order of addition of amino acids to the code.
It seems highly likely that the first amino acids were those that were simplest to form by non-biological chemistry. Of the 20 amino acids in the standard code, 10 are formed in appreciable quantities in experiments on spark discharge in a mixture of atmospheric gases (Miller et al. 1976; Miller and Cleaves 2007). The amino acids that are found in carbonaceous chondrite meteorites are very similar to these (Higgs and Pudritz 2007, 2009; Cobb and Pudritz 2014). We recently surveyed many experiments related to prebiotic chemistry in which amino acid synthesis occurred and used these to construct a ranking based on relative concentrations (Higgs and Pudritz 2009). The top 10 amino acids are Gly, Ala, Asp, Glu, Val, Ser, Ile, Leu, Pro, and Thr. We refer to these as early amino acids, because we presume that these were present in the environment at the time the code evolved, and they were available to be used in the earliest proteins. We also showed that the rank order of these early amino acids is strongly correlated with the free energy of synthesis of these molecules, which was calculated previously by Amend and Shock (1998). More detailed comparisons of meteorites shows that the relative proportions of these amino acids varies significantly with the degree of aqueous alteration that has occurred in the meteorite (Cobb and Pudritz 2014). However, the main point is that the amino acids that are thermodynamically least costly to form are those that are most likely to form in prebiotic conditions.
The remaining 10 biological amino acids occur very infrequently or never in the data that we used. We refer to these as late amino acids, because we presume that they were not easy to form by prebiotic chemistry and they only arose after biosynthetic pathways evolved to synthesize them inside organisms. Trifonov (2004) also compiled a ranking of amino acids using both experimental observations and a wide range of other criteria that had been proposed by other authors. Trifonov’s ranking is Gly, Ala, Asp, Val, Pro, Ser, Glu (Leu, Thr), Arg, (Ile, Gln, Asn), His, Lys, Cys, Phe, Tyr, Met, and Trp, where parentheses indicates groups of equal rank. This is similar to that of Higgs and Pudritz (2007, 2009) for the early amino acids and also makes some predictions about the relative order in the late amino acids. Wong (2014) also emphasizes that there is a convergence between the amino acids formed in chemical experiments, those found in meteorites, and those expected from the coevolution theory of the genetic code (Wong 2005), which will be discussed below. All these results show that there is a reasonable consensus on which amino acids form most easily.
Biosynthetic pathways for amino acid synthesis are a key aspect of the coevolution theory of the genetic code (Wong 1975, 2005; Di Giulio 2008). The main point is that if an amino acid is synthesized from a precursor amino acid, then the precursor must be added to the code prior to the product. Although this is a reasonable assumption in general, it is only a logical necessity for amino acids that were not available in the environment of the early cells. The simplest amino acids, biosynthetically, are Gly, Ser, Ala, Val, Asp, and Glu. These amino acids are synthesized in modern organisms via pathways with few steps that derive from the central metabolic pathways of glycolysis and the citric acid cycle. Di Giulio (2008) has argued that these pathways existed in the organisms in which the code arose, and therefore that biosynthetic pathways also determine the first amino acids included in the code. It should be remembered; however, that the code probably arose in an RNA based organism, and we do not know whether the same metabolic pathways existed in the RNA world. Nevertheless, these 6 amino acids are also the top 6 in our ranking of early amino acids based on prebiotic chemistry. On reflection, this may not be surprising. The simplest, least thermodynamically costly amino acids that form most readily by chemical synthesis are also the ones that are simplest and least costly to form inside cells. Thus, irrespective of whether the organisms were autotrophic or heterotrophic with respect to amino acid synthesis, we may still conclude that these amino acids were early.
Another central point of the coevolution theory is that product amino acids take over codons that were previously assigned to their precursors. This means that precursor product pairs will tend to be found on neighboring codon blocks. Using statistical arguments, it has been claimed that the standard code is such that the number of precursor product pairs on neighboring codons is larger than expected (Wong 1975, 2005). Details of the statistical significance of this have subsequently been debated (Ronneberg et al. 2000; Di Gulio 2001). A related point is that certain steps of amino acid biosynthesis occur while the amino acids are attached to tRNAs (Ibba et al. 1997, 2000; Tumbula et al. 2000; Di Giulio 2002). This applies to the synthesis of Asn from Asp and Gln from Glu. In each case, a tRNA is charged with the precursor amino acid and the precursor to product reaction (changing an acid to an amine group) occurs after this. If this process arose in very early organisms, then it suggests an obvious mechanism by which the product amino acids (Asn and Gln) could take over codons that were previously assigned to their precursors (Asp and Glu). If all the amino acid synthesis pathways occurred this way when they first evolved, then it is a strong argument that product amino acids take over the codons of their precursors. Using this logic, Di Giulio and Medugno (1999) proposed a specific arrangement of codon assignments for the earliest code and deduced a series of steps by which the code could evolve to the standard code by sequential addition of amino acids. Amino acid synthesis on a tRNA may also be relevant for the pairs Met/fMet and Ser/Cys, as discussed by Di Giulio (2008). However, the other standard biological amino acids are not normally synthesized in this way in modern organisms. Several caveats regarding this idea have been given by Higgs (2009) and replied to by Di Giulio in comments on that paper.
At this point, we note that amino acid synthesis on tRNAs is relevant to the exceptional cases of the 21st (Selenocysteine) and 22nd (Pyrrolysine) naturally encoded amino acids. These were added to the code via the reassignment of the opal (UGA) and amber (UAG) codons. Both these amino acids possess their own tRNA’s and amino acyl tRNA synthetases but the recoding of UGA and UAG follows distinct molecular mechanisms. Intriguingly, Selenocysteine (Sec) synthesis occurs on a tRNA (Sheppard et al. 2008) having a UCA anticodon that is first charged with Ser and subsequently converted to Sec. Sec insertion at UGA relies on the structure of a genomic segment (the SECIS element) whose location downstream to the UGA codon varies across prokaryotes and eukaryotes. In the absence of the SECIS element, UGA is recognized as a translation termination signal. A rare case of ambiguous decoding of the UGA codon either as Cys or Sec in the ciliate Euplotes crassus has been reported (Turanov et al. 2009). Pyrrolysine (Pyl) is found in methylamine methyltransferase genes of methanogenic archaea and bacteria that utilize Pyl for methane metabolism. The tRNA-Pyl has the anticodon CUA and competes for recognition of UAG with the release factor. It has been argued (Kavran et al. 2007; Sheppard et al. 2008) that Sec and Pyl were inserted into the code before the last universal common ancestor, i.e., at the end of the ancient phase of code evolution. However, these changes did not spread through the majority of organisms.
In order to add a new amino acid to the code in the ancient phase, the charging process of the tRNA has to change. Due to wobble pairing at the third codon position, most tRNAs pair with two codons. Roughly 32 tRNAs are therefore needed to translate the full 64 codons (ignoring complications such as start and stop codons). Details on codon-anticodon pairing and the consequences that this has for codon usage frequencies and translational selection can be found in Grosjean et al. (2010) and Ran and Higgs (2010). Most bacteria possess tRNAs with at least 32 distinct anticodons, although this number can be slightly smaller in cases where a tRNA is able to pair with all four codons in a four-codon family. This exception has become the rule in mitochondria, where there is usually only one tRNA for a four-codon family and the typical number of required tRNAs is reduced to 22 (Jia and Higgs 2008). If wobble pairing in the ancient phase worked in a similar way to that of modern bacteria, then roughly 32 tRNAs would have been required for efficient translation of the full code, even if only a small number of amino acids were encoded initially. There would thus have been a larger number of distinct tRNAs charged with the same amino acid than in modern organisms. Adding a new amino acid to the code involves changing the charging of a subset of the tRNAs assigned to one amino acid. Making a biochemical change to the amino acid that is already charged, as in the Asn and Gln cases discussed above, is one way of doing this. Alternatively there must be a change to the synthetase that carries out tRNA charging. In the RNA world, modern amino acyl-tRNA synthetases could not have existed, and presumably charging was controlled by ribozymes. Another issue relevant at this point is the so-called stereochemical theory, which argues that there were direct interactions between nucleotide triplets and specific amino acids (Jukes 1973; Knight and Landweber 2000; Yarus 2000). If this were the case, it would go some way to explaining why particular amino acids became associated with tRNAs with particular anticodons.
Adding an amino acid to the code requires making an association between an amino acid and a tRNA for a particular codon block. However, for a new variant code to become established, it has to spread and become fixed in the population. This depends on the selective properties of the code. The key observation that suggests natural selection is important in the ancient phase of code evolution is that the SGC is highly unusual with respect to random codes, and apparently minimizes the effects of translational error and deleterious mutations (Freeland et al. 2003). In the following section, we turn to the question of how the arrangement of amino acids in the SGC came to be in this statistically unusual configuration.
Selective Differences and Selective Barriers Between Codes
Statistical studies have shown that the effects of deleterious mutations and translational error are lower in the standard genetic code than in almost all rearranged codes using the same set of 20 amino acids (Freeland and Hurst 1998; Gilis et al. 2001; Goodarzi et al. 2004, 2005; Novozhilov and Koonin 2009). In these studies, a matrix of costs g(a, b) is defined that represents the penalty for inserting amino acid b at a site in a protein where amino acid a is optimal. A genetic code is defined by its set of assignments between codons and amino acids—let a i be the amino acid assigned to codon i. A translational error can occur if a tRNA for codon j accidentally pairs with codon i. The cost of this error is g(a i , a j ). A code cost function Φ is defined, which measures the mean value of g(a i , a j ), averaged over all pairs of codons i and j, and weighted by the frequency with which the error is likely to occur. It is usually assumed that only single-position errors occur at an appreciable rate. We will refer to codons that differ at a single position as neighboring codons. The value of Φ depends on which amino acids are assigned to neighboring codons. Φ is calculated for the standard code, and for large numbers of randomly reshuffled codes. The fraction of random codes which have a smaller Φ than the real code is small—for example, one in a million, according to Freeland and Hurst (1998). This figure depends on the details of the cost function and the set of random codes that is considered. Nevertheless, several authors have confirmed that it is small (Gilis et al. 2001; Goodarzi et al. 2004, 2005). Hence, the result that the real code is highly non-random appears to be robust, and it strongly suggests that selection has played an important role during the evolution of the code in the ancient phase.
Above, we pointed out that there is usually a selective barrier to codon reassignment because, immediately after a codon is reassigned, it will be found in the wrong place in gene sequences, i.e., the codon will be found where the old amino acid was preferred. It will take some time until the codon is eliminated from those places and reappears in places where the new amino acid is preferred. After this process is complete, the encoded gene sequences will once again be adapted to the code. When different codes are compared using the cost functions described here, it is assumed that this adaptation has occurred, i.e., the cost function measures the selective difference between two well-adapted codes, but does not measure the selective barrier between codes. If the two codes both include the full set of 20 amino acids, the differences in the cost function between the codes are functions of the frequency of translational errors, and the cost difference would be zero if there were zero rate of translational errors. For reassignments of a single codon (or a small codon block) occurring in the modern phase, the differences in cost functions between the codes before and after the reassignment will be very small, because the differences in codon positions are small, and because the error rate is also rather very small. The barrier to selection occurring between these codes will be much larger than the selective difference between two well-adapted very similar codes. Hence, we argued above that, in the modern phase, codon reassignments are not driven by the small selective differences that might exist between codes, they are driven by random events such as changes in tRNAs and codon disappearance, and the rate at which they occur is controlled by how difficult it is to cross the selective barrier between the codes. In contrast, in the ancient phase, we need to think of possible codes that might be extremely different from current ones and will thus differ much more in their cost functions. The studies on the cost functions of different codes are much more relevant to the ancient phase of code evolution than they are to the modern phase.
In the ancient phase, we are presuming that a relatively small number of amino acids were initially encoded and that each of these would be encoded by a large number of codons. New amino acids were gradually added by subdivision of larger codon blocks into smaller ones. An alternative to this is that there were a large number unassigned or stop codons initially, and that new amino acids were added by taking over stop codons. We will proceed to discuss the idea of subdivision of codon blocks and will return to consider stop codon takeover at the end of this section. The protein sequences encoded in the genome at each step of code evolution must have been useful to the organism. An increasingly diverse and more highly functional set of proteins could be encoded as the repertoire of amino acids in the code increased. The existence of genes written in the operational code at any stage of code evolution places constraints on the way the code can subsequently evolve. This idea is termed code-message coevolution (Ardell and Sella 2001; Sella and Ardell 2002). If a block of codons is reassigned to a new amino acid, this changes the sequence of all the proteins that use this codon. The evolution of the code is therefore likely to proceed in a way that is minimally disruptive to the function of currently encoded proteins.
Higgs (2009) considered the possible pathways of amino acid addition to the code. A likely early step in code evolution would be a four-column code in which all codons with the same middle base code for the same amino acid. If the first four amino acids assigned to the four columns were Gly, Ala, Asp, and Val, as seems likely from the discussion of early and late amino acids given above, this gives the four-column code shown in Fig. 3. We can consider adding a new amino acid by reassigning a block of codons such as the CUN block shown. The modified cost function used in by Higgs (2009) accounts for this selective advantage of adding the new amino acid in addition to the cost of translational error. Codes with different numbers of amino acids differ in their cost function principally because of the different amino acids encoded. This difference would still exist, even if the error rate were zero (whereas the difference in cost of two codes with the same set of amino acids would be zero if the error rate were zero). This shows that selection can be a driving force of code evolution in the ancient phase, because there is something to be gained by adding a new amino acid, whereas in the modern phase, there is almost nothing to be gained by reassigning a codon while keeping the same set of 20 amino acids.

Reassignment of the CUN block in the 4-column code from Val to another amino acid
Figure 4 shows the steps in addition of a new amino acid to the code. If the diversity of amino acids in the current code is low, there are many different choices of additional amino acid that that could improve the diversity of the protein functions that can be encoded. By the time the new code is established, there is a significant advantage of the new code relative to the old one, in contrast to the small selective difference between initial and final codes in the modern case (Fig. 2). However, the ease with which a new amino acid can be incorporated depends on the selective barrier between codes rather than the difference between the well-adapted codes before and after addition. Consider the CUN block, assuming that is was initially Val, as in Fig. 3. In the standard code, CUN encodes Leu. If this block were reassigned to Leu, these codons will occur at positions where Val was previously the preferred amino acid from the four in the initial code. Leu is a hydrophobic amino acid with similar physico-chemical properties to Val (see discussion of amino acid property differences in Higgs 2009). Therefore changing from Val to Leu is likely to be only slightly disruptive, in which case there is only a small selective barrier for addition of Leu. There might also be sites in the encoded proteins where Val was the best available amino acid in the four amino acid code, but the incorporation of Leu instead of Val would be an improvement in protein function. If the number of these sites is large enough, then there is an advantage to changing the code, even before the positions of the codons have adjusted to the new code. This corresponds to a “negative barrier” (fitness increase) in the intermediate stage of Fig. 4. Thus we conclude that it is easy to add Leu to a codon block that was previously encoded by Val. On the other hand, suppose we add a charged amino acid like Glu. Replacing Val by Glu will be disruptive to protein function because these amino acids have very different properties. Hence there will be a large selective barrier to adding Glu in the CUN block. On the other hand, if Glu is added to a codon block previously encoded by Asp (another charged amino acid with similar properties to Glu), then the selective barrier will be low or negative.

a Evolutionary pathways of codon reassignment in the ancient phase. b Selective cost involved in the various stages of the codon reassignment process in the ancient phase. There is a significant selective advantage for replacing an old amino acid by a new (previously unencoded) one
In this way, it is possible to predict which new amino acids are most likely to be added to the code. The result is that it is easiest to add amino acids into positions that were previously occupied by earlier amino acids with similar properties. This process can continue until the diversity of amino acids encoded is quite large, and the selective advantage of adding additional ones becomes too small. One of the main features of the standard genetic code (Fig. 1) is that amino acids in the same column of the code (same middle codon base) have similar properties. For example, the hydrophobic amino acids (Phe, Leu, Ile, Val, and Met) are all in column 1, and the polar amino acids (Asp, Glu, Asn, Gln, and His) are all in column 3. Using quantitative measures of amino acid property differences, Higgs (2009) has shown that the genetic code is likely to have evolved from a four-column code with a single amino acid in each column to the standard code with groups of similar amino acids in each column. As a result of this process, the standard code ends up with similar amino acids on neighboring codons, and the cost functions that measure the effects of translational errors are minimized with respect to randomly reshuffled codes. In this theory, minimization of the cost of translational error arises as a by-product of the pathway of addition of amino acids to the code. Selection chooses the pathway of code evolution that minimizes the disruption to existing genes, and this happens to also minimize the cost of translational error.
Our argument above that there may sometimes be a selective advantage for adding an amino acid (as in Fig. 4b) is intended to apply only to the addition of an amino acid that was not previously in the code. For reassignments via the AI mechanism in the modern phase, we assumed that there was a selective barrier (as in Fig. 2b) because ambiguous codons are deleterious. We note, however, that Bender et al. (2008) have argued that the AUA: Ile to Met reassignment in animals is adaptive because it leads to accumulation of methionine in the inner membrane of animal mitochondria, which could be beneficial to cells because of the anti-oxidant and cytoprotective properties of methionine. They argue that this reassignment occurred via the AI mechanism. However, there is strong evidence (Sengupta et al. 2007) that AUA reassignment in both Metazoans and Fungi was initiated by the loss of the special tRNA-Ile used to decode AUA. Hence the reassignment mechanism responsible is UC and not AI. In our opinion, the beneficial consequence of AUA reassignment discussed by Bender et al. may have arisen subsequently instead of being the adaptive driving force behind AUA reassignment.
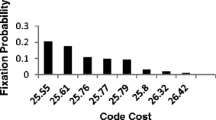
Novozhilov and Koonin (2009) also used the code cost function with certain constraints to show that a code which distributes the ten earliest amino acids among the 16 codon blocks in a manner that is consistent with the standard genetic code is also the most optimal. Most studies that have attempted to explain the optimality of the SGC have been based on infinite population models of code evolution. In such models, code cost is the sole determinant in the fixation of a code with the most optimal (lowest cost) code guaranteed to be fixed in the population. Recently, the physico-chemical hypothesis was revisited using a more realistic finite population mode of code-sequence coevolution (Bandhu et al. 2013). It was shown that selection to minimize the cost of translational and mutational errors cannot explain the structure of standard code if the pool of competing codes has similar levels of physico-chemical optimization (code cost). In a finite population, many codes having similar cost can get fixed in the population with significant probability. However, if an optimized code competes with a pool of randomly generated codes having significantly lower cost, it is easily able to out-compete those codes and get fixed with a significantly higher probability than any of the randomized code competitors. These results suggest that selection for a physico-chemically optimized code cannot be the sole explanation for the structure of the standard code. Other forces like population size, pool of competing codes, and even the rates of horizontal transfer of genetic elements (Vetsigian et al. 2006; Sengupta et al. 2014), all falling under the broad category of historical contingencies may have played an important role in the emergence of the standard code.
The discussion above assumes that new amino acids were added to the code by reassigning codons, and that large codon blocks were subdivided into smaller ones. In this picture there would be few, if any stop codons. Possibly translation terminated by a simpler mechanism that did not require specific proteins to act as release factors—for example, termination could simply occur at the end of an mRNA strand. An important alternative to this is the idea that, at the stage when few amino acids were encoded, there were large numbers of unassigned codons. These may have acted as stops, or they may simply have been non-functional. Addition of a new amino acid would then require takeover of unassigned codons rather than subdivision of an existing block (Lehman and Jukes 1988; van der Gulik and Hoff 2011). Francis (2013) has given a detailed theory that shows how the code could have built up in this way, starting from a situation in which GNC codons coded for Val, Ala, Asp, and Gly—the same four early amino acids that we considered above. In this scenario, it is expected that new amino acids with similar properties to existing ones will take over unassigned codons that are a single mutation away from the codons that are already assigned. As a result, the assignments tend to spread mostly up the columns, and the final code is expected to have similarities of amino acids in the columns. This argument leads to very similar conclusions to that of Higgs (2009), but the mechanism is slightly different. In both cases, the constraints imposed by the early code are important, and the apparent optimization of the code arises as a bi-product of the way selection acts at the time amino acids are added. Francis (2013) has also given detailed arguments as to why addition of specific amino acids might be advantageous biochemically. For example, Ile and Leu were an improvement over Val for stabilization of the hydrophobic cores of globular proteins, and Ser and Thr were an improvement in binding to anions and cations relative to Gly and Ala. These arguments are useful, and we note that they apply equally well to the case of subdivision/reassignment and the case of stop codon takeover. It should be remembered that, however, useful the new amino acid is in general and, however, similar it is to the old one, the mutation of an amino acid at one point in a given protein could be either advantageous or deleterious, depending on the specific structure and function of the protein. In the subdivision/reassignment picture, there will always be some places where reassignment is disadvantageous, and these are included in the calculation of the selective barrier (Higgs 2009). In the stop codon takeover model (Francis 2013), this is less of a problem, because mutations to the newly assigned codons can be tried out one at a time and selection will act for or against them individually. The corresponding problem with the stop codon takeover model is that there will many mutations to unassigned codons that will either cause ribosome hang-up or unexpected termination. In such a situation, it would be advantageous to quickly assign all the codons to the earliest amino acids.
With the exception of Selenocysteine and Pyrrolysine discussed above, which occur only rarely and did not become part of the SGC, the processes of addition of new amino acids to the code seems to have stopped once the standard set of 20 was reached. Nevertheless, there are many additional amino acids that are not in the standard set of 20 included in the SGC. In many cases, these have similar physico-chemical properties to the biological ones (Lu and Freeland 2006; Philip and Freeland 2011). It would appear that the 20 amino acids encoded in the standard code are sufficient to cover the range of physico-chemical properties of amino acids required in proteins in most cases, and hence there was too little further selective advantage to cause further change of the code. However, specific reasons have been given why some of the biological amino acids are preferable to some of the possible alternatives (Weber and Miller 1981; Cleaves 2010).
Conclusions
Code evolution in the ancient and modern phases follows different evolutionary pathways and is subject to distinct selective pressures. In the ancient phase characterized by organisms with small genomes and error-prone translation machinery, there is positive selection to increase the repertoire of amino acids encoded in order to synthesize functionally diverse proteins. Evolution of biosynthetic pathways as well as horizontal transfer of genetic elements, including components of the translation machinery, may have played a significant role in shaping the structure of the SGC. In contrast, the modern phase characterized by large genomes (with the exception of mitochondrial genomes), well-adapted genes, and high fidelity of translation makes it much less likely for codon reassignments to be adaptive. In this phase, the appearance of variant codes is driven by factors like codon disappearance, gain and loss of cognate tRNAs associated with the codon to be reassigned.
In the modern phase, it is often difficult to determine the precise evolutionary pathway that eventually leads to the codon reassignment event based on sequence data only. This is because information about ancestral species in which the transient intermediate stages (AI or UC) of codon reassignment are observed is unavailable in almost all cases with a few exceptions (Suzuki et al. 1997; Massey et al. 2003). However, the in vitro methods recently developed (Pape et al. 1999; Gromadski and Rodnina 2004; Gromadski et al. 2006; Cochella et al. 2007, Zaher and Green 2009a, b) to study the accuracy of the translation process can be easily adapted to test the viability of the AI and UC mechanisms. The answers are likely to depend on the codon being reassigned and the manner in which tRNAs involved in the decoding process during the transient intermediate phase interact with the ribosome. Understanding the evolutionary and structural basis of codon reassignments has profound consequences for synthetic biology. Such knowledge will make it possible to develop synthetic organisms with new functions that rely on recoding of the genetic code carried out by manipulating the association between codon(s) and amino acids. The genetic code can also be expanded by reengineering tRNAs and aaRS’ to insert synthetic amino acids. The first steps in this direction has already been taken (Lajoie et al. 2013; Rovner et al. 2015), and it is expected that this line of research will open up a brave new world of genetic code engineering.
References
Amend JP, Shock EL (1998) Energetics of amino acid synthesis in hydrothermal ecosystems. Science 281:1659–1662
Ardell DH, Sella G (2001) On the evolution of redundancy in genetic codes. J Mol Evol 53:269–281
Bandhu AV, Aggarwal N, Sengupta S (2013) Revisiting the physico-chemical hypothesis of code origin: an analysis based on code-sequence coevolution in a finite population. Orig Life Evol Biosph 43:465–489
Bender A, Hajieva P, Moosmann B (2008) Adaptive antioxidant methionine accumulation in respiratory chain complexes explains the use of a deviant genetic code in mitochondria. Proc Natl Acad Sci USA 105:16496–16501
Bernhardt HS (2012) The RNA world hypothesis: the worst theory of the early evolution of life (except for all the others). Biol Direct 7:23
Burton AS, Lehman N (2009) DNA before proteins? Recent discoveries in nucleic acid catalysis strengthen the case. Astrobiology 9:125–130
Caetano-Anollés G, Seufferheld MJ (2013) The coevolutionary roots of biochemistry and cellular organization challenge the RNA world paradigm. J Mol Microbiol Biotechnol 23:152–177
Cantara WA, Murphy FV, Demirci H, Agris PF (2013) Expanded use of sense codons is regulated by modified cytidines in tRNA. Proc Natl Acad Sci USA 110:10964–10969
Carter CW (2015) What RNA world? Why a peptide/RNA partnership merits renewed experimental attention. Life 5:294–320
Castresana J, Feldmaier-Fuchs G, Pääbo S (1998) Codon reassignment and amino acid composition in hemichordate mitochondria. Proc Natl Acad Sci USA 95:3703–3707
Cleaves HJ (2010) The origin of the biologically coded amino acids. J Theor Biol 263:490–498
Cobb AK, Pudritz RE (2014) Nature’s starships. I. Observed abundances and relative frequencies of amino acids in meteorites. Astrophys J 783:140
Cochella L, Brunelle JL, Green R (2007) Mutational analysis reveals two independent molecular requirements during transfer RNA selection on the ribosome. Nat Struct Mol Biol 14:30–36
Demeshkina N, Jenner L, Westhof E, Yusupov M, Yusupova G (2012) A new understanding of the decoding principle on the ribosome. Nature 484:256–259
Di Giulio M (2002) Genetic code origin: are the pathways of type Glu-tRNA(Gln) –> Gln-tRNA(Gln) molecular fossils or not? J Mol Evol 55:616–622
Di Giulio M (2008) An extension of the coevolution theory of the origin of the genetic code. Biol Direct 3:37
Di Giulio M, Medugno M (1999) Physicochemical optimization in the genetic code origin as the number of codified amino acids increases. J Mol Evol 49:1–10
Di Gulio M (2001) A blind empiricism against the coevolution theory of the origin of the genetic code. J Mol Evol 53:724–732
Francis BR (2011) An alternative to the RNA world hypothesis. Trends Evol Biol 3:e2
Francis BR (2013) Evolution of the genetic code by incorporation of amino acids that improved or changed protein function. J Mol Evol 77:134–158
Francis BR (2015) The hypothesis that the genetic code originated in coupled synthesis of proteins and the evolutionary predecessors of nucleic acids in primitive cells. Life 5:467–505
Freeland SJ, Hurst LD (1998) The genetic code is one in a million. J Mol Evol 47:238–248
Freeland SJ, Wu T, Keulmann N (2003) The case for an error minimizing standard genetic code. Orig Life Evol Biosph 33:457–477
Gilis D, Massar S, Cerf NJ, Rooman M (2001) Optimality of the genetic code with respect to protein stability and amino acid frequencies. Genome Biol 2:11
Goodarzi H, Nejad HA, Torabi N (2004) On the optimality of the genetic code, with the consideration of termination codons. Biosystems 77:163–173
Goodarzi H, Najafabadi HS, Hassani K, Nejad HA, Torabi N (2005) On the optimality of the genetic code, with the consideration of coevolution theory by comparison of prominent cost measure matrices. J Theor Biol 235:318–325
Gromadski KB, Rodnina MV (2004) Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol Cell 13:191–200
Gromadski KB, Daviter T, Rodnina MV (2006) A uniform response to mismatches in codon-anticodon complexes ensures ribosomal fidelity. Mol Cell 21:369–377
Grosjean H, de Crécy-Lagard V, Marck C (2010) Deciphering synonymous codons in the three domains of life: co-evolution with specific tRNA modification enzymes. FEBS Lett 584:252–264
Higgs PG (1998) Compensatory neutral mutations and the evolution of RNA. Genetica 102–103:91–101
Higgs PG (2009) A four-column theory for the origin of the genetic code: tracing the evolutionary pathways that gave rise to an optimized code. Biol Direct 4:16
Higgs PG, Lehman N (2015) The RNA world: molecular co-operation at the origin of life. Nat Rev Genet 16:7–17
Higgs PG, Pudritz RE (2007) From protoplanetary disks to prebiotic amino acids and the origin of the genetic code. In: Pudritz RE, Higgs PG, Stone J (eds) Planetary systems and the origins of life. Cambridge Series in Astrobiology, vol 3. Cambridge University Press, Cambridge
Higgs PG, Pudritz RE (2009) A thermodynamic basis for prebiotic amino acid synthesis and the nature of the first genetic code. Astrobiology 9:483–490
Ibba M, Curnow AW, Söll D (1997) Aminoacyl-tRNA synthesis: divergent routes to a common goal. Trends Biochem Sci 22:39–42
Ibba M, Becker HD, Stathopoulos C, Tumbula DL, Söll D (2000) The adaptor hypothesis revisited. Trends Biochem Sci 25:311–316
Jia W, Higgs PG (2008) Codon usage in mitochondrial genomes: distinguishing context-dependent mutation from translational selection. Mol Biol Evol 25:339–351
Joyce GF (2000) The antiquity of RNA-based evolution. Nature 418:214–221
Jukes TH (1973) Arginine as an evolutionary intruder into protein synthesis. Biochem Biophys Res Commun 53:709–714
Kavran JM, Gundllapalli S, O’Donoghue P, Englert M, Söll D, Steitz TA (2007) Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc Natl Acad Sci USA 104:11268–11273
Knight RD, Landweber LF (2000) Guilt by association: the arginine case revisited. RNA 6:499–510
Knight RD, Freeland SJ, Landweber LF (2001) Rewiring the keyboard: evolvability of the genetic code. Nat Rev Genet 2:49–58
Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, Rohland N, Schultz PG, Jacobson JM, Rinehart J, Church GM, Isaacs FJ (2013) Genomically recoded organisms expand biological functions. Science 342:357–360
Lazcano A, Guerrero R, Margulis L, Oró J (1988) The evolutionary transition from RNA to DNA in early cells. J Mol Evol 27:283–290
Lehman N, Jukes TH (1988) Genetic code development by stop codon takeover. J Theor Biol 135:203–214
Ling J, Daoud R, Lajoie MJ, Church GM, Söll D, Lang BF (2014) Natural reassignment of CUU and CUA sense codons to alanine in Ashbya mitochondria. Nucleic Acids Res 42:499–508
Lozupone CA, Knight RD, Landweber LF (2001) The molecular basis of nuclear genetic code change in ciliates. Curr Biol 11:65–74
Lu Y, Freeland S (2006) On the evolution of the standard amino-acid alphabet. Genome Biol 7:102
Lynch M, Koskella B, Schaack S (2006) Mutation pressure and the evolution of organelle genomic architecture. Science 311:1727–1730
Massey SE, Moura G, Beltrão P, Almeida R, Garey JR, Tuite MF, Santos MA (2003) Comparative evolutionary genomics unveils the molecular mechanism of reassignment of the CTG codon in Candida spp. Genome Res 13:544–557
McCutcheon JP, McDonald BR, Moran NA (2009) Origin of an alternative genetic code in the extremely small and GC-rich genome of a bacterial symbiont. PLoS Genet 5:e1000565
Miller SL, Cleaves HJ (2007) Prebiotic chemistry on the primitive earth. Orig Life 1:3–56
Miller SL, Urey HC, Oró J (1976) Origin of organic compounds on the primitive earth and in meteorites. J Mol Evol 9:59–72
Muramatsu T, Yokoyama S, Horie N, Matsuda A, Yamaizumi Z, Kuchino Y, Nishimura S, Miyazawa T (1988) A novel lysine-substituted nucleoside in the first position of the anticodon of minor isoleucine tRNA from Escherichia coli. J Biol Chem 263:9261–9267
Novozhilov AS, Koonin EV (2009) Exceptional error minimization in putative primordial genetic codes. Biol Direct 4:44
Osawa S, Jukes TH (1989) Codon reassignment (codon capture) in evolution. J Mol Evol 28:271–278
Osawa S, Collins D, Ohama T, Jukes TH, Watanabe K (1990) Evolution of the mitochondrial genetic code III. Reassignment of CUN codons from leucine to threonine during evolution of yeast mitochondria. J Mol Evol 30:322–328
Pape T, Wintermeyer W, Rodnina M (1999) Induced fit in initial selection and proofreading of aminoacyl-tRNA on the ribosome. EMBO J 18:3800–3807
Philip GK, Freeland SJ (2011) Did evolution select a nonrandom “alphabet” of amino acids? Astrobiology 11:235–240
Ran W, Higgs PG (2010) The influence of anticodon-codon interactions and modified bases on codon usage bias in bacteria. Mol Biol Evol 27:2129–2140
Ronneberg TA, Landweber LF, Freeland SJ (2000) Testing a biosynthetic theory of the genetic code: fact or artifact? Proc Natl Acad Sci USA 97:13690–13695
Rovner AJ, Haimovich AD, Katz SR, Li Z, Grome MW, Gassaway BM, Amiram M, Patel JR, Gallagher RR, Rinehart J, Isaacs FJ (2015) Recoded organisms engineered to depend on synthetic amino acids. Nature 518:89–93
Santos MA, Moura G, Massey SE, Tuite MF (2004) Driving change: the evolution of alternative genetic codes. Trends Genet 20:95–102
Schultz DW, Yarus M (1994) Transfer RNA mutation and the malleability of the genetic code. J Mol Biol 235:1377–1380
Schultz DW, Yarus M (1996) On malleability in the genetic code. J Mol Evol 42:597–601
Sella G, Ardell DH (2002) The impact of message mutation on the fitness of a genetic code. J Mol Evol 54:638–651
Sengupta S, Higgs PG (2005) A unified model of codon reassignment in alternative genetic codes. Genetics 170:831–840
Sengupta S, Yang X, Higgs PG (2007) The mechanisms of codon reassignments in mitochondrial genetic codes. J Mol Evol 64:662–688
Sengupta S, Aggarwal N, Bandhu AV (2014) Two perspectives on the origin of the standard genetic code. Orig Life Evol Biosph 44:287–291
Sheppard K, Yuan J, Hohn MJ, Jester B, Devine KM, Söll D (2008) From one amino acid to another: tRNA-dependent amino acid biosynthesis. Nucleic Acids Res 36:1813–1825
Su D, Lieberman A, Lang BF, Simonovic M, Söll D, Ling J (2011) An unusual tRNAThr derived from tRNAHis reassigns in yeast mitochondria the CUN codons to threonine. Nucleic Acids Res 39:4866–4874
Suzuki T, Ueda T, Watanabe K (1997) The “polysemous” codon—a codon with multiple amino acid assignment caused by dual specificity of tRNA identity. EMBO J 16:1122–1134
Swire J, Judson OP, Burt A (2005) Mitochondrial genetic codes evolve to match amino acid requirements of proteins. J Mol Evol 60:128–139
Tomita K, Ueda T, Ishiwa S, Crain PF, McCloskey JA, Watanabe K (1999a) Codon reading patterns in Drosophila melanogaster mitochondria based on their tRNA sequences: a unique wobble rule in animal mitochondria. Nucleic Acids Res 27:4291–4297
Tomita K, Ueda T, Watanabe K (1999b) The presence of pseudouridine in the anticodon alters the genetic code: a possible mechanism for assignment of the AAA lysine codon as asparagine in echinoderm mitochondria. Nucleic Acids Res 27:1683–1689
Trifonov EN (2004) The triplet code from first principles. J Biomol Struct Dyn 22:1–11
Tumbula DL, Becker HD, Chang WZ, Söll D (2000) Domain-specific recruitment of amide amino acids for protein synthesis. Nature 407:106–110
Turanov AA, Lobanov AV, Fomenko DE, Morrison HG, Sogin ML, Klobutcher LA, Hatfield DL, Gladyshev VN (2009) Genetic code supports targeted insertion of two amino acids by one codon. Science 323:259–261
van der Gulik PT, Hoff WD (2011) Unassigned codons, nonsense suppression, and anticodon modifications in the evolution of the genetic code. J Mol Evol 73:59–69
Vetsigian K, Woese C, Goldenfeld N (2006) Collective evolution and the genetic code. Proc Natl Acad Sci USA 103:10696–10701
Voorhees RM, Mandal D, Neubauer C, Köhrer C, RajBhandary UL, Ramakrishnan V (2013) The structural basis for specific decoding of AUA by isoleucine tRNA on the ribosome. Nat Struct Mol Biol 20:641–643
Weber AL, Miller SL (1981) Reasons for the occurrence of the twenty coded protein amino acids. J Mol Evol 17:273–284
Wong JT (1975) A co-evolution theory of the genetic code. Proc Natl Acad Sci USA 72:1909–1912
Wong JT (2005) Coevolution theory of the genetic code at age thirty. BioEssays 27:416–425
Wong JT (2014) Emergence of life: from functional RNA selection to natural selection and beyond. Front Biosci 19:1117–1150
Yarus M (2000) RNA-ligand chemistry: a testable source for the genetic code. RNA 6:475–484
Yokobori S, Suzuki T, Watanabe K (2001) Genetic code variations in mitochondria: tRNA as a major determinant of genetic code plasticity. J Mol Evol 53:314–326
Zaher HS, Green R (2009a) Quality control by the ribosome following peptide bond formation. Nature 457:161–166
Zaher HS, Green R (2009b) Fidelity at the molecular level: lessons from protein synthesis. Cell 136:746–762
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sengupta, S., Higgs, P.G. Pathways of Genetic Code Evolution in Ancient and Modern Organisms. J Mol Evol 80, 229–243 (2015). https://doi.org/10.1007/s00239-015-9686-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-015-9686-8