
Abstract
The formyl peptide receptors (FPRs) are a family of chemoattractant receptors with important roles in host defense and the regulation of inflammatory reactions. In humans, three FPR paralogs have been identified (FPR1, FPR2, and FPR3) and may have functionally diversified by gene duplication and adaptive evolution. However, the evolutionary mechanisms operating in the diversification of FPR family genes and the changes in selection pressures have not been characterized to date. Here, we have made a comprehensive evolutionary analysis of FPR genes from mammalian species. Phylogenetic analysis showed that an early duplication was responsible for FPR1 and FPR2/FPR3 splitting, and FPR3 originated from the latest duplication event near the origin of primates. Codon-based tests of positive selection reveal interesting patterns in FPR1 and FPR2 versus FPR3, with the first two genes showing clear evidence of positive selection at some sites while the majority of them evolve under strong negative selection. In contrast, our results suggest that the selective pressure may be relaxed in the FPR3 lineage. Of the six amino acid sites inferred to evolve under positive selection in FPR1 and FPR2, four sites were located in extracellular loops of the protein. The electrostatic potential of the extracellular surface of FPR might be affected more frequently with amino acid substitutions in positively selected sites. Thus, positive selection of FPRs among mammals may reflect a link between changes in the sequence and surface structure of the proteins and is likely to be important in the host’s defense against invading pathogens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The formyl peptide receptors (FPRs) are a family of chemoattractant receptors and are expressed mainly in mammalian phagocytic leukocytes. This family is known to be important in host defense and in the regulation of inflammatory reactions (Le et al. 2002). FPRs mediate cell chemotaxis in a pertussis toxin-sensitive manner, indicating coupling to one or more members of the inhibitory G subfamily of G proteins. Activation of FPRs induces a variety of responses, e.g., directional movement of neutrophils, lysosomal enzyme release (Schiffmann et al. 1975), degranulation, and production of superoxide anion (Le et al. 2002; Prossnitz and Ye 1997). FPR interaction with a wide range of ligands has also been associated with various diseases, including ischemia–reperfusion injury (Gavins 2010), prion disease (Zhou et al. 2009), Alzheimer’s disease (Cui et al. 2002), AIDS (Kilby et al. 1998), stomach ulcer (de Paulis et al. 2004), and some cancers (Edwards et al. 2005; Tsuruki et al. 2007).
FPRs are members of the G-protein-coupled receptor (GPCR) superfamily (Fredriksson et al. 2005). Genome-wide comparative analysis has demonstrated that GPCRs are the largest superfamily of integral membrane receptor proteins in mammals (Fredriksson and Schioth 2005). The receptors consist of a single polypeptide chain that loops through the cell membrane seven times to form a seven alpha-helical transmembrane (7TM) domain. According to the structure of the membrane-spanning 7TM domain and the functional domains within the N- and C-termini, mammalian GPCR can be assigned to one of the five basic GPCR families: glutamate, rhodopsin, adhesion, frizzled/taste, and secretin (Schioth and Fredriksson 2005). FPRs belong to the Rhodopsin family, which forms the largest GPCR subfamily in vertebrates, and receptors from this family bind chemically diverse ligands such as small molecular amines, purines, lipids, peptides, and large glycoproteins. Phylogenetic analyses demonstrate that long evolutionary processes have shaped and optimized the diversity in receptor-ligand interactions (Rompler et al. 2007).
In humans, three FPR paralogs have been identified (FPR1, FPR2, and FPR3) (Alvarez et al. 1996; Yang and Shi 2010; Ye et al. 2009). FPR1 was initially identified and shown to be a high-affinity binding site for N-formyl peptides, such as the prototypic fMLF (formyl-methionine-leucine-phenylalanine) (Ye et al. 2009). FPR2 exhibits 69 % amino acid identity with FPR1 but shows relatively low affinity for fMLF (Quehenberger et al. 1993). FPR3 exhibits 58 and 72 % amino acid identity with human FPR1 and FPR2, respectively, but does not bind N-formyl peptides (Migeotte et al. 2005). It has further been reported that FPR3 localizes in the small intracellular vesicles, suggesting distinct physiological functions (Rabiet et al. 2011). Thus, FPRs may have functionally diversified by gene duplication and adaptive evolution, but the precise nature of this evolutionary process is not known.
In order to investigate the evolutionary history and functional differentiation of the FPR gene family, we here made an evolutionary analysis of FPR genes from mammalian species. We focused on detecting positive selection and the identification of sites that were subject to natural selection. We used codon models implemented in the codeml program, contained in the PAML package (Yang 2007). The models used here assumed that selection regimes vary along the sequence but not among lineages. Additional codon models (Guindon et al. 2004) implemented in the Fitmodel software that take into account the variability of selection regimes across both sites and lineages were also considered. The results of our analysis of site- and lineage-specific selection patterns, in conjunction with 3D homology modeling analysis, revealed that the FPR1 and FPR2 genes have experienced positive selection, with significant sequence divergence among the FPR1 genes. Our study provides insights into FPR gene family evolution and information regarding potential functional diversification of amino acid residues which might be responsible for the recognition of invading pathogens.
Materials and Methods
Sequence Retrieval and Phylogenetic Reconstruction
Mammalian FPR orthologs and paralogs were initially collected from the GenBank nonredundant protein database by performing separate searches using Blastp (Altschul et al. 1997) with human FPR1, FPR2, and FPR3 amino acid sequences as query. Nucleotide and predicted amino acid sequences were retrieved for further analysis. Also, orthologs for human FPR1, FPR2, and FPR3 were downloaded from the Ensembl Compara database using the Ensembl API v72 (Stabenau et al. 2004). The resulting data sets were then screened to remove incomplete or redundant sequences as well as sequences with many ambiguous base calls. A total of 39 sequences of mammals and 8 rodent-specific sequences were selected for the final analyses. The species and accession numbers for sequences used in the analyses are listed in Supplementary Table S1.
Amino acid sequences were aligned using the MAFFT software with parameter settings optimized for data with one conserved domain and long gaps (E-INS-i) (Katoh and Toh 2008). The resulting alignment was manually checked using BioEdit (Hall 1999). A codon alignment was generated from the multiple sequence alignment of amino acid sequences (Supplementary Figure S1) and the corresponding DNA sequences by the PAL2NAL program (http://coot.embl.de/pal2nal). The phylogenetic tree was reconstructed using the neighbor-joining tree-building method (Saitou and Nei 1987) with the maximum composite likelihood model for nucleotide substitutions implemented in the program MEGA 5.05 (Tamura et al. 2011). The variability of the resulting topologies was assessed by the bootstrap method using 1,000 pseudo-replicates (Felsenstein 1985), which produced the bootstrap proportions for each interior branch in the tree. In addition, the maximum-likelihood tree for the FPRs was constructed with the PhyML v3.0 software (Guindon et al. 2010), after determining the optimal model of sequence substitution with jModeltest 2 (Darriba et al. 2012). A general time-reversible model with a proportion of invariant sites and gamma distributed among-site rate variation with four classes of rates (GTR + I + G4) was used, as selected by the Akaike information criterion. Branch support was assessed using the aLRT SH-like method.
Molecular Evolutionary Analysis
The maximum-likelihood method was used to test for traces of positive selection and to infer amino acid sites under positive selection using both the codeml program in the PAML version 4.2b (Yang 2007) and the Fitmodel program version 0.5.3 (Guindon et al. 2004). The codeml analysis was performed for FPR1, FPR2, FPR3, and the concatenation of the three FPR alignments, separately. The site models, which allow the ω ratio to vary among sites (Nielsen and Yang 1998; Yang et al. 2000), were used in the present study. Likelihood ratio tests (LRTs) using a χ 2 distribution with two degrees of freedom were conducted to test for positive selection between each pairs of models, i.e., M1a versus M2a and M7 versus M8. In the first set of models, the null model M1a allows 2 categories of codon sites in p 0 and p 1 proportions, with ω 0 < 1 and ω 1 = 1 (ω = dN/dS; dN, non-synonymous substitution rates; dS, synonymous substitution rates), whereas the alternative model M2a allows an additional category of codons (p 2) with ω 2 > 1, indicating positive selection. The second set of site models compared is M7 and M8, in which M7 specifies a null model with a beta distribution, beta (p, q), of ω values between 0 and 1, and M8 specifies an alternative model with an additional category for sites that have ω > 1, indicating positive selection. The LRT statistic equals twice the difference between the log likelihood scores of the models being compared (2ΔlnL = 2(lnL 1 – lnL 0)) and provided that the null hypothesis is correct, its distribution is approximately that of a χ 2 distribution with two degrees of freedom. For both the M1a (neutral) versus M2a (selection) and M7 (beta) versus M8 (beta + selection) comparisons, the null model has two estimated selection parameters, while the alternative model has four, resulting in a difference of two degrees of freedom. The calculations of χ 2critical values were performed using the chi2 program implemented in the PAML package. In the case of alternative models that allow for positive selection, the Bayes Empirical Bayes approach was used to calculate the posterior probability (PP) that each codon evolved under positive selection (Yang et al. 2005). The codeml program was used to calculate the PP of each amino acid site that came from the ω > 1 class. We used a cutoff of PP > 0.95 to identify sites under positive selection.
The Fitmodel program, which implements Markov-modulated Markov models of codon evolution (Guindon et al. 2004) or switching models, was used to analyze the FPR1, FPR2, FPR3, and the concatenation of FPR sequences, separately. Unlike site models, switching models allow the site-specific selection process to vary along lineages of a phylogenetic tree. Models used in this analysis included M0, M3, M3 + S1, and M3 + S2. M0 assumes that all the sites have the same ω values, whereas M3 assumes three different ω values (ω 1 < ω 2 < ω 3). If the switching rates between ω values (ω 1–ω 2, ω 1–ω 3 and ω 2–ω 3) are constrained to be equal, the switching model is designated with S1 and otherwise with S2. LRTs were performed between each pair of models M0 versus M3, M3 versus M3 + S1, and M3 + S1 versus M3 + S2, using a χ2 distribution to assess the significance of difference of log likelihoods. The posterior probabilities of the third selection class (corresponding to ω 3) were evaluated at each codon position and each branch using Fitmodel and processed using a custom script in perl.
Protein Tertiary Structure Analysis
In order to construct the three-dimensional (3D) structure models of FPRs, homology modeling was performed using the I-TASSER server, which was the best server for structural prediction in a recent critical assessment of protein structure prediction (CASP10) (Roy et al. 2010; Zhang 2008). The amino acid sequences of FPRs were submitted to the I-TASSER server for modeling based on multiple-threading alignments and iterative template fragment assembly simulations. The top threading template for FPRs was CCR5 chemokine receptor (PDB: 4MBS) which had the best Z score using all the algorithms. The server predicted 5 models of FPRs, and the best model was selected based on C score. The C score is a confidence score and ranges from −5 to 2, with higher scores representing higher confidence in the model (Roy et al. 2010). False positive and false negative rates are estimated to be below 0.1 when a C score >−1.5 is calculated (Zhang 2008). The surface electrostatic potential maps were computed using the Adaptive Poisson–Boltzmann Solver release 1.2.0 (Baker et al. 2001) by solving the nonlinear Poisson–Boltzmann equation. The visualizations of the electrostatic potential maps and the 3D structure were performed in the PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC.
Results
Phylogenetic Analysis of the FPR Family
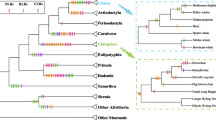
We first retrieved the available FPR sequences from the currently sequenced genomes by Blastp and the Ensembl Compara database. Querying major databases and unfinished genomes with the human FPR amino acid sequences led to the identification of many homologous proteins in vertebrate species (mammals, birds, amphibians, and fish). The wide distribution of FPR genes in various vertebrate species suggests that the genes appeared early in vertebrate evolution; however, since the sequence identity between the human and lower vertebrate proteins was relatively low, we focused on mammalian FPRs in this study. After the exclusion of partial protein sequences, the available FPR1, FPR2, and FPR3 sequences were retrieved from a total of 24 mammalian species. A phylogenetic tree with 39 mammalian FPRs was constructed by the neighbor-joining method, based on the nucleotide-coding sequences (Fig. 1). The maximum-likelihood tree was also constructed using the alignment with the Anolis carolinensis chemokine-like receptor 1 sequence as an outgroup (Supplementary Figure S2). The distance-based and maximum-likelihood tree topologies obtained are very similar with only minor differences in the FPR1 clade. The tree shows that the evolution of FPRs exhibits a divergence pattern similar to that of speciation. All mammalian FPR genes were divided into two main clusters, one that grouped all the FPR1 genes and the other with all the FPR2 and FPR3 genes. The FPR3 gene was only detected in the primate genome. This clustering strongly suggests that the two major duplications occurred during FPR evolution. The first duplication led to the emergence of two lineages which evolved into FPR1 and FPR2, and the second duplication, early in primate evolution, resulted in FPR3 (Supplementary Figure S2).

Phylogenetic relationships of 39 FPR gene sequences. The tree was constructed by the neighbor-joining method, based on the nucleotide-coding sequences. Bootstrap values are based on 1,000 replications; only those greater than 50 % are shown at the nodes. Branches with more than three codon sites under positive selection in the Fitmodel analysis are shown in red. The genes used for computing the surface electrostatic potential maps are marked with stars. Shaded boxes denote the three lineages (Color figure online)
In the rodent clade, there are several reports indicating the presence of expanded FPR family genes: FPR-rs1, FPR-rs3, FPR-rs4, FPR-rs6, and FPR-rs7 (He et al. 2013; Liberles et al. 2009; Riviere et al. 2009; Yang and Shi 2010). The extensive analyses by two independent groups (Liberles et al. 2009; Riviere et al. 2009) have demonstrated that these rodent-specific FPRs are selectively expressed in vomeronasal neurons and have an olfactory function. We constructed a maximum-likelihood tree using the alignment including rodent-specific genes (Supplementary Figure S3). The rodent-specific FPRs were clustered together within the FPR2 clade as previously reported (Liberles et al. 2009; Yang and Shi 2010), suggesting that these sequences are in fact FPR2 inparalogs. Since we focused on the FPR receptors which function in the immune system, these rodent-specific FPRs were not included in our selection analysis.
Positive Selection Drives the Diversification of FPRs
The results of the analysis employing the codeml program under site models are shown in Table 1. We performed analyses for the concatenation of FPR alignments (noted as All FPRs), as well as FPR1, FPR2, and FPR3 gene sequence alignments separately, estimating the parameters under each codon model by maximum likelihood. Model M0, which assumes a single ω for all codons in the sequence, was used to estimate a general ω value for each data set. The values of ω were very similar among the data sets used (ω = 0.352 for All FPRs genes, ω = 0.309 for FPR1 genes, and ω = 0.309 for FPR2 genes), except in the case of the FPR3 genes, where ω was estimated to have a somewhat higher value (ω = 0.738). These relatively small ω values for FPR genes suggest that the evolution of FPR genes is dominated by purifying selection operating on the majority of sites. Among the models implemented, M2a and M8 can account for positive selection by adding a class of sites where ω > 1. For the All FPRs data set, the M2a and M8 models provided significantly better fit to the data (P < 0.001) compared to the corresponding null models (M1a and M7, respectively). Model M2a assigned 2.7 % of codons to the class of positively selected sites (ω = 3.358), and model M8 assigned 5.4 % to the positive selection class (ω = 2.205), indicating that FPRs have evolved under diversifying selection in mammals. We also found strong evidence of positive selection in an analysis limited to FPR1 alone and FPR2 alone. However, we did not detect traces of positive selection in the analysis of FPR3 (Table 1). In fact, the proportion of sites evolving under a purely neutral process (ω = 1) was found to be high for FPR3 compared to FPR1 and FPR2 where most sites evolved under negative selection.
Since phylogenetic relationship of FPR1 among primate species (Fig. 1) is different from the known species tree, we tested whether the tree topology affected the phylogenetic model parameter estimates, in particular the values of ωs. A codeml site model analysis was conducted using a modified tree that matches exactly the species tree. The parameter estimates for the codon models remained essentially unchanged (results not shown). Thus, minor differences in the tree topology have little impact on our inference of positive selection under site models. Similar robustness of the site model analysis to the tree topology was also reported in other studies (Furlong and Yang 2008; Yang and Swanson 2002).
Identification of Positively Selected Sites
The Bayes Empirical Bayes approach was used to calculate the posterior probabilities and posterior means of ωs for sites under models M2a and M8. As shown in Table 1, six sites were inferred to have ω > 1 for the All FPRs data set with high posterior probabilities (PP > 0.99) under M8: 170P, 188E, 191L, 271 M, 274Y, and 281D. Site numbers and amino acids refer to the reference sequence human FPR2. Since the same amino acid sites were also inferred to have ω > 1 under model M2a with high posterior probabilities, the evidence of positive selection can be considered as strong. In the data sets limited to FPR1 and FPR2, similar amino acid sites were inferred to be positively selected, but the number of sites detected was smaller. Of the six sites found to be under positive selection in the All FPRs data set, four sites were also detected in the FPR1 data set with model M2a, and only 1 site was detected in the FPR2 data set. In the FPR1 data set, a different site specific to this family was detected (site, 84R). The posterior means of ω for sites under model M8 for FPR1, FPR2, and FPR3 are shown in Fig. 2. Overall, the majority of amino acid sites in FPR1 and FPR2 are under strong purifying selection, and FPR1 has more amino acid sites under positive selection compared to FPR2, however. The majority of sites in FPR3 showed evidence of mild negative selection. Although the LRTs failed to detect positive selection in FPR3, the posterior means of ω were elevated along broad regions exhibiting mild negative selection compared to FPR1 and FPR2 (Fig. 2). This and the higher ω value estimated under the M0 model for FPR3 strongly suggest that the selective pressure that recurrently drove positive and negative selection of mammalian FPR may be relaxed in the FPR3 lineage.

Posterior mean ω at each amino acid site across the FPR genes. Posterior means of ω were calculated as a weighted average of ω over the 11 site classes and were weighted by the posterior probabilities under the site model M8 (beta and ω). Sites with low mean ω are inferred to be under purifying selection. Sites are numbered according to the referenced amino acid sequences, a human FPR1, b human FPR2, and c human FPR3
In order to obtain some insight into the roles that positive selection might play, we mapped positively selected amino acid sites onto the 3D model of the human FPR protein. The 3D structures of FPRs were constructed by homology modeling using the I-TASSER server. The quality of the modeled protein was estimated by the C score values generated by I-TASSER software. A good predicted model is obtained when the estimated level of C score is between −5 and 2. The level of C score for all our predicted FPR models was in the range of −0.17 to −0.51, indicating that the protein structures were constructed with high accuracy. We have mapped six positively selected sites (170P, 188E, 191L, 271M, 274Y, 281D) detected in the All FPRs data set onto the 3D structure of human FPR2 (Fig. 3). Four of the six amino acids identified as having experienced positive selection were located in extracellular loops of the protein; two positively selected amino acid residues were located in the β strand, near the transmembrane/extracellular boundary. A positively selected site specific to FPR1 (84R) was also located near the transmembrane/extracellular boundary (not shown).

Location of positively selected sites in the human FPR 3D structure. Sites under positive selection identified with both M2a and M8 models in the All FPRs data set are highlighted in the predicted FPR2 structure with their side chains shown (magenta spheres) (Color figure online)
Shift in the Site-Specific Selection Process Along Lineages
To assess the variations in the site-specific selection processes along lineages, we performed maximum-likelihood analyses using the Fitmodel software under a nested set of stochastic branch-site codon substitution models (Guindon et al. 2004). As opposed to the branch-site models implemented in codeml, the stochastic branch-site models are particularly relevant in cases where there is no strong prior evidence about the lineages evolving under positive selection at individual sites (Lu and Guindon 2013). Tables 2 and 3 show the results from the analysis with substitution models M0, M3, M3 + S1, and M3 + S2. For the All FPRs, FPR1, and FPR2 data sets analyzed, the log likelihoods improved significantly as parameters were added to the nested substitution models (P < 0.001; Table 3). These results suggest that M3 + S2 (unequal switching rates among the three rate ratio classes) is the best codon substitution model in the All FPRs, FPR1, and FPR2 data sets. For the FPR3 data set, however, the LRT was not statistically significant for the M3 + S2 versus M3 + S1 model. Under the M3 + S2 model, the ω estimates for the three classes in the All FPRs data set were ω 1 = 0.03, ω 2 = 0.57, and ω 3 = 8.96 (Table 2). The switching rate between ω 2 and ω 3 (R 23 = 14.89) was significantly higher than the switching rates between ω 1 and ω 2 (R 12 = 1.05) and between ω 1 and ω 3 (R 13 = 0.00), implying that site-specific shifts between moderate purifying selection (ω 2) and positive selection (ω 3) occurred more frequently than shifts involving the most highly constrained rate ratio classes. The parameter estimates of the M3 + S2 model suggest that most sites (96 %, corresponding to p 1 + p 2) are under purifying selection with ω < 1 in the All FPRs data set, and 4 % (p 3) of sites within the FPR genes are under positive selection with a ω value considerably larger than one. This pattern of parameter estimates is similar to those inferred for the FPR1 and FPR2 data sets (Table 2).
To further characterize variation in the selective pressure on codon sites among lineages, we assessed the number of branches in the All FPRs gene tree for which each codon was placed in the ω 3 rate ratio class with high posterior probabilities. The All FPRs gene alignment included 367 codon sites, 18 of which showed evidence of positive selection at some point in the lineage of the FPR gene family (posterior probability >0.9). As shown in Table 4, among the 18 codon positions detected, the number of branches with positive selection on each codon site varied from 1 to 14, and codon sites with a higher number of branches under positive selection were also detected as positively selected sites in the codeml program using the Bayes Empirical Bayes approach (see also Table 1). Thus, the positively selected amino acids inferred by the Fitmodel program are quite compatible with those inferred by the PAML package. The branches with sites evolving under positive selection are shown in Fig. 1. Positive selection was detected at several codon sites on the branches of the FPR1 and FPR2 clade, including deep internal branches. Interestingly, branches with a higher number of sites evolving under positive selection were concentrated within the FPR1 clade.
Surface Properties of the FPR Protein Structure
Protein surface properties such as electrostatic potential play important roles in specific protein–protein and protein–ligand recognition. To gain a more precise idea of the potential functional changes during the FPR gene evolution, we studied their 3D surface electrostatic potentials. Because many branches exhibited positive selection in the FPR1 clade, we focused on five representative taxa in this clade (Fig. 1). Figure 4a shows the electrostatic surface potential for predicted FPR1 structures facing the outside of the cell for Homo sapiens, Papio anubis, Oryctolagus cuniculus, Mus musculus, and Loxodonta africana. There are significant differences in the pattern of electrostatic surface potentials among the FPR1 proteins. An amino acid substitution can achieve a different spatial organization of the electrostatic surface potential depending on their physicochemical properties (Zhang et al. 2008). When we investigated the alterations of charge and polarity for the amino acids identified as being under positive selection, five of six amino acid sites (83 %), 84R, 188K, 191I, 271L, and 280G, exhibited at least one charged state alteration across the five mammalian taxa (Fig. 4b, supplementary Table S3). In contrast, only 19 of 133 other divergent amino acid sites (14 %) exhibited at least one charged state alteration across the species (data not shown). Thus, the substitutions in positively selected sites could more potently contribute to the variations of the electrostatic potential in or near the extracellular surface of FPR1 proteins (Fig. 4b). While these predictions emphasize the evolved differences among FPR1 proteins since their divergence, the significance of these differences to the interaction between FPRs and their ligands warrants further experimental verification.

Surface electrostatic potential and amino acid substitutions of FPR1 proteins. a Distribution of surface electrostatic potential of five mammalian FPR1 proteins. The Poisson–Boltzmann electrostatic surface potential of solvent-exposed surfaces is colored from red (−2.0) to blue (+2.0) for predicted FPR1 structures for Homo sapiens, Papio anubis, Oryctolagus cuniculus, Mus musculus, and Loxodonta africana. The positions of the positively selected amino acid sites are indicated for the human protein. The extracellular surface of FPR1 is facing directly toward the viewer. b Positively selected amino acid sites and respective amino acids for five mammalian species. Charged amino acids are colored in orange (Color figure online)
Discussion
In this study, we undertook a comprehensive evolutionary analysis on the basis of the currently available data on the formyl peptide receptors (FPRs). Mammalian FPR genes have been divided into the FPR1, FPR2, and FPR3 paralogs and are considered to originate from a common ancestor and to separate after duplication events (Ye et al. 2009). Our phylogenetic analysis showed that an early duplication was responsible for FPR1 and FPR2/FPR3 splitting, and FPR3 originated from the latest duplication event near the origin of primates. Genomic organization in and around the FPR genes also supports this evolutionary scenario (Ha et al. 2011). Site-specific codeml analysis of the FPR gene family confirmed that no traces of positive selection were found for FPR3, and the evolutionary rate of that gene is higher than that of FPR1 and FPR2. In addition, Fitmodel analysis using stochastic branch-site codon models further confirmed that the parameter estimates for FPR3 are quite different from those for FPR1 and FPR2 (Table 2). These results suggest that the functional constraints on FPR3 are less stringent than that acting on the two other FPR gene families. Experimental studies on human FPR3 from Rabiet et al. (Rabiet et al. 2011) have demonstrated that FPR3 was expressed primarily in small intracellular vesicles throughout the cells, as monitored by immunofluorescence staining. This cellular distribution in the absence of agonist stimulation suggested that FPR3 was undergoing constitutive endocytosis. Furthermore, they were unable to detect G-protein activation in FPR3-expressing cells. Based on these observations, they proposed that FPR3 internalization may serve a ligand-scavenging function similar to that of the chemokine receptors D6 and CXCR7 (Graham and McKimmie 2006; Naumann et al. 2010). Thus, the FPR3 gene in primates could have a specific function, which may be distinct from that of FPR1 and FPR2. The lower selective constraints on FPR3 might have contributed to the family-specific functional evolution of this family following its diversification from FPR2.
Most genes typically evolve under negative selection (Endo et al. 1996), which is also the case for mammalian FPR genes. The selective regime on the mammalian FPR genes was predominantly negative selection, based on the estimated overall ω value being lower than 1 as determined using M0 models in PAML, reflecting the functional importance of FPRs in mammalian physiology and survival. However, site models, which analyze the sequence at the individual codons, revealed a proportion of codon sites that display evidence of positive selection (ω > 1) within the coding sequences of FPR genes. The two sets of site models (M1a vs. M2a and M7 vs. M8) revealed statistically significant results for the All FPRs data set. We performed further tests to determine whether the evidence for positive selection was restricted to different subfamilies or alternatively whether positive selection acted equally in all subfamilies. According to models M2a and M8, the FPR1 and FPR2 clades showed strong evidence for positive selection acting at a subset of sites, while neither site model indicated positive selection within FPR3. To account for the potential variation in site-specific selection patterns among branches in the FPR phylogenies, we used the program Fitmodel, which implements a maximum-likelihood phylogeny-based codon substitution model that includes parameters for switching between selection processes at individual codon sites across the phylogeny (Guindon et al. 2004; Lu and Guindon 2013). For the All FPRs, FPR1, and FPR2 data set, the M3 + S2 model was favored over the M3 and M3 + S1 models. Similar to codeml analysis using site models, the Fitmodel results indicated that purifying selection has played an important role in the evolution of FPR genes throughout most of their evolutionary history, but our results suggest branch- and site-specific shifts in selection within each FPR subfamily. Lineages with many sites under positive selection include the FPR1 and FPR2 lineages as well as many deep internal branches, implying that the FPR1 and FPR2 genes evolved most rapidly, likely resulting in functional divergence. In contrast, positive selection was not detected on any branches or at any sites within the FPR3 lineage, as in site models in PAML. Thus, the three subfamilies of FPR genes differ in evolutionary mode, with the genes being relatively conserved in FPR3 but more variable in FPR1 and FPR2, and this might have played a role in the functional divergence among them.
The domain structures of GPCRs share a conserved arrangement with three primary functional categories: extracellular loops, which mediate extracellular ligand interactions; transmembrane domains, which are important for both receptor orientation and ligand binding, and internal loops, which mediate intracellular interactions with G proteins (Wooding 2011). Previous mutagenesis and ligand-docking studies of FPR (Fujita et al. 2011; Khlebnikov et al. 2012; Mills et al. 2000) have revealed that the amino acid residues constituting the ligand-binding pocket form two cavities located inside the transmembrane domains. There were 10 amino acid residues proposed as the sites critical for ligand binding in the FPR1: F102, V105, D106, L109, R205, F206, W254, Y257, S287, and F291 (Fujita et al. 2011). Our analysis using the Bayes Empirical Bayes methods of Yang et al. (Yang et al. 2005) identified six codon positions with significantly elevated ω values (Model M2a) in the All FPRs data set, suggesting that these positions may be particularly important for the adaptive processes in FPRs. It is interesting to note that four out of the six amino acid sites that were identified as having experienced positive selection are located in the extracellular loops. Furthermore, none of the positively selected sites identified overlap with the amino acid residues proposed as the ligand-binding sites. These patterns of positive selection suggest that the amino acid residues present in the extracellular domains of mammalian FPR receptors evolve more rapidly, whereas the amino acid residues present in the transmembrane (including the ligand-binding site) and intracellular domains of these receptors are more selectively constrained.
The significance of the positively selected sites for FPR receptor function remains unclear, but their localization on the extracellular domains suggests that they might exert functional differences on ligand recognition. The present study reveals that the distribution of surface electrostatic potential is markedly different among FPR1 proteins from several species. The electrostatic potential of the surface might be affected more frequently with amino acid substitutions in the positively selected sites. It would appear that the consequence of FPR1 sequence divergence, which is at least partially driven by positive selection, has been to alter the electrostatic potential on the extracellular surface. It is unclear precisely what the functional consequences of this would be, but differences in the surface net charge of FPR receptors can be associated with changes in ligand interactions and recognition. Growing evidence indicates that FPR receptors utilize diverse ligands, most of which activate chemotactic and anti-microbial responses in neutrophils (Ye et al. 2009), and some of which differentially interact with FPR receptors in a species-dependent manner (Southgate et al. 2008). Since mammalian species are subject to a wide range of pathogenic environments, differential detection of pathogen-derived ligands by FPRs would be essential for the individual species to antagonize specific pathogens. Therefore, it is interesting to speculate that selection pressure has favored the acquisition of a distinct distribution of surface electrostatic potential for more sensitive recognition of the pathogens derived from different environments in which individual species live.
In conclusion, we reconstructed the evolutionary history of FPR genes across a diverse range of mammals and conducted evolutionary analyses to examine their functional significance. Purifying selection is the main force acting on FPRs, but signs of adaptive evolution and functional divergence were also detected. Our results indicate that FPR1 and FPR2 genes have experienced positive selection, and significant sequence divergence has occurred among the FPR1 genes. While further experimental evidence will be needed to confirm our functional predictions that FPR receptors have undergone positive selection to meet the selective pressure for antagonizing pathogens, it appears that the FPR receptors have evolved to acquire unique surface modifications at the molecular level that enable them to adapt to the extremes of different pathogenic environments.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Alvarez V, Coto E, Setien F, Gonzalez-Roces S, Lopez-Larrea C (1996) Molecular evolution of the N-formyl peptide and C5a receptors in non-human primates. Immunogenetics 44:446–452
Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98:10037–10041. doi:10.1073/pnas.181342398
Cui Y, Le Y, Yazawa H, Gong W, Wang JM (2002) Potential role of the formyl peptide receptor-like 1 (FPRL1) in inflammatory aspects of Alzheimer’s disease. J Leukoc Biol 72:628–635
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. doi:10.1038/nmeth.2109
de Paulis A et al (2004) Urokinase induces basophil chemotaxis through a urokinase receptor epitope that is an endogenous ligand for formyl peptide receptor-like 1 and -like 2. J Immunol 173:5739–5748
Edwards BS et al (2005) Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol Pharmacol 68:1301–1310. doi:10.1124/mol.105.014068
Endo T, Ikeo K, Gojobori T (1996) Large-scale search for genes on which positive selection may operate. Mol Biol Evol 13:685–690
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Fredriksson R, Schioth HB (2005) The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol Pharmacol 67:1414–1425. doi:10.1124/mol.104.009001
Fredriksson R, Lagerstrom MC, Schioth HB (2005) Expansion of the superfamily of G-protein-coupled receptors in chordates. Ann N Y Acad Sci 1040:89–94. doi:10.1196/annals.1327.011
Fujita H, Kato T, Watanabe N, Takahashi T, Kitagawa S (2011) Stimulation of human formyl peptide receptors by calpain inhibitors: homology modeling of receptors and ligand docking simulation. Arch Biochem Biophys 516:121–127. doi:10.1016/j.abb.2011.09.017
Furlong RF, Yang Z (2008) Diversifying and purifying selection in the peptide binding region of DRB in mammals. J Mol Evol 66:384–394. doi:10.1007/s00239-008-9092-6
Gavins FN (2010) Are formyl peptide receptors novel targets for therapeutic intervention in ischaemia-reperfusion injury? Trends Pharmacol Sci 31:266–276. doi:10.1016/j.tips.2010.04.001
Graham GJ, McKimmie CS (2006) Chemokine scavenging by D6: a movable feast? Trends Immunol 27:381–386. doi:10.1016/j.it.2006.06.006
Guindon S, Rodrigo AG, Dyer KA, Huelsenbeck JP (2004) Modeling the site-specific variation of selection patterns along lineages. Proc Natl Acad Sci USA 101:12957–12962. doi:10.1073/pnas.0402177101
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi:10.1093/sysbio/syq010
Ha HS, Huh JW, Gim JA, Han K, Kim HS (2011) Transcriptional variations mediated by an alternative promoter of the FPR3 gene. Mamm Genome 22:621–633. doi:10.1007/s00335-011-9341-7
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
He HQ, Liao D, Wang ZG, Wang ZL, Zhou HC, Wang MW, Ye RD (2013) Functional characterization of three mouse formyl peptide receptors. Mol Pharmacol 83:389–398. doi:10.1124/mol.112.081315
Katoh K, Toh H (2008) Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9:286–298. doi:10.1093/bib/bbn013
Khlebnikov AI, Schepetkin IA, Kirpotina LN, Brive L, Dahlgren C, Jutila MA, Quinn MT (2012) Molecular docking of 2-(benzimidazol-2-ylthio)-N-phenylacetamide-derived small-molecule agonists of human formyl peptide receptor 1. J Mol Model 18:2831–2843. doi:10.1007/s00894-011-1307-x
Kilby JM et al (1998) Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med 4:1302–1307. doi:10.1038/3293
Le Y, Murphy PM, Wang JM (2002) Formyl-peptide receptors revisited. Trends Immunol 23:541–548
Liberles SD et al (2009) Formyl peptide receptors are candidate chemosensory receptors in the vomeronasal organ. Proc Natl Acad Sci USA 106:9842–9847. doi:10.1073/pnas.0904464106
Lu A, Guindon S (2013) Performance of standard and stochastic branch-site models for detecting positive selection among coding sequences. Mol Biol Evol 31:484–495. doi:10.1093/molbev/mst198
Migeotte I et al (2005) Identification and characterization of an endogenous chemotactic ligand specific for FPRL2. J Exp Med 201:83–93. doi:10.1084/jem.20041277
Mills JS, Miettinen HM, Cummings D, Jesaitis AJ (2000) Characterization of the binding site on the formyl peptide receptor using three receptor mutants and analogs of Met-Leu-Phe and Met-Met-Trp-Leu-Leu. J Biol Chem 275:39012–39017. doi:10.1074/jbc.M003081200
Naumann U et al (2010) CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 5:e9175. doi:10.1371/journal.pone.0009175
Nielsen R, Yang Z (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Prossnitz ER, Ye RD (1997) The N-formyl peptide receptor: a model for the study of chemoattractant receptor structure and function. Pharmacol Ther 74:73–102
Quehenberger O, Prossnitz ER, Cavanagh SL, Cochrane CG, Ye RD (1993) Multiple domains of the N-formyl peptide receptor are required for high-affinity ligand binding. Construction and analysis of chimeric N-formyl peptide receptors. J Biol Chem 268:18167–18175
Rabiet MJ, Macari L, Dahlgren C, Boulay F (2011) N-formyl peptide receptor 3 (FPR3) departs from the homologous FPR2/ALX receptor with regard to the major processes governing chemoattractant receptor regulation, expression at the cell surface, and phosphorylation. J Biol Chem 286:26718–26731. doi:10.1074/jbc.M111.244590
Riviere S, Challet L, Fluegge D, Spehr M, Rodriguez I (2009) Formyl peptide receptor-like proteins are a novel family of vomeronasal chemosensors. Nature 459:574–577. doi:10.1038/nature08029
Rompler H, Staubert C, Thor D, Schulz A, Hofreiter M, Schoneberg T (2007) G protein-coupled time travel: evolutionary aspects of GPCR research. Mol Interv 7:17–25. doi:10.1124/mi.7.1.5
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. doi:10.1038/nprot.2010.5
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schiffmann E, Showell HV, Corcoran BA, Ward PA, Smith E, Becker EL (1975) The isolation and partial characterization of neutrophil chemotactic factors from Escherichia coli. J Immunol 114:1831–1837
Schioth HB, Fredriksson R (2005) The GRAFS classification system of G-protein coupled receptors in comparative perspective. Gen Comp Endocrinol 142:94–101. doi:10.1016/j.ygcen.2004.12.018
Southgate EL, He RL, Gao JL, Murphy PM, Nanamori M, Ye RD (2008) Identification of formyl peptides from Listeria monocytogenes and Staphylococcus aureus as potent chemoattractants for mouse neutrophils. J Immunol 181:1429–1437
Stabenau A, McVicker G, Melsopp C, Proctor G, Clamp M, Birney E (2004) The Ensembl core software libraries. Genome Res 14:929–933. doi:10.1101/gr.1857204
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Tsuruki T, Takahata K, Yoshikawa M (2007) Mechanism of the protective effect of intraperitoneally administered agonists for formyl peptide receptors against chemotherapy-induced alopecia. Biosci Biotechnol Biochem 71:1198–1202
Wooding S (2011) Signatures of natural selection in a primate bitter taste receptor. J Mol Evol 73:257–265. doi:10.1007/s00239-011-9481-0
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591. doi:10.1093/molbev/msm088
Yang H, Shi P (2010) Molecular and evolutionary analyses of formyl peptide receptors suggest the absence of VNO-specific FPRs in primates. J Genet Genomics 37:771–778. doi:10.1016/S1673-8527(09)60094-1
Yang Z, Swanson WJ (2002) Codon-substitution models to detect adaptive evolution that account for heterogeneous selective pressures among site classes. Mol Biol Evol 19:49–57
Yang Z, Nielsen R, Goldman N, Pedersen AM (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yang Z, Wong WS, Nielsen R (2005) Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118. doi:10.1093/molbev/msi097
Ye RD et al (2009) International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev 61:119–161. doi:10.1124/pr.109.001578
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinform 9:40. doi:10.1186/1471-2105-9-40
Zhang ZD, Weinstock G, Gerstein M (2008) Rapid evolution by positive Darwinian selection in T-cell antigen CD4 in primates. J Mol Evol 66:446–456. doi:10.1007/s00239-008-9097-1
Zhou H, Zhou X, Kouadir M, Zhang Z, Yin X, Yang L, Zhao D (2009) Induction of macrophage migration by neurotoxic prion protein fragment. J Neurosci Methods 181:1–5. doi:10.1016/j.jneumeth.2009.04.002
Conflict of interest
The authors declare that they have no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Muto, Y., Guindon, S., Umemura, T. et al. Adaptive Evolution of Formyl Peptide Receptors in Mammals. J Mol Evol 80, 130–141 (2015). https://doi.org/10.1007/s00239-015-9666-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-015-9666-z