
Abstract
The evolution of cancer suppression is essential for the maintenance of multicellularity. The lack of correlation between body size and cancer risk across species, known as Peto’s paradox, suggests that genetic variation in cancer resistance is sufficient to compensate for increases of cell numbers in bigger animals. To assess evolutionary dynamics of cancer-related genes, we analyzed Ka, Ks,and Ka/Ks values in 120 oncogenes and tumor suppressor genes (TSG) among seven hominoid species, including two extinct species, Neanderthal and Denisovan. Ka/Ks of tumor suppressor genes tended to be higher relative to that of oncogenes, consistent with relaxed purifying selection acting on the former. Ka/Ks values were positively correlated with TSG scores, but negatively correlated with oncogene scores, suggesting opposing selection pressures operating on the two groups of cancer-related genes. Additionally, we found 108 species-divergent substitutions that were prevalent germline genotypes in some species but in humans appeared only as somatic cancerous mutations. Better understanding the resistance to cancer may lead to new methods of cancer prevention in humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer is a dramatic consequence of somatic evolution within the body, posing fundamental challenges to clinical interventions and therapy (Nowell 1976). A cancer cell accumulates mutations, many of which can give the cell a growth advantage over neighboring noncancerous cells. Cancer development is a multistage process that involves the activation of oncogenes and deactivation of tumor suppressor genes (TSGs). To date, there have been about 120 genes found to induce tumorigenesis when mutated (Vogelstein et al. 2013). Typically somatic cells need to accumulate enough mutations to cause cancer, and about 95 % of mutations in cancer-related genes are single-base substitutions (Vogelstein et al. 2013).
Although the idea of cancer as a Darwinian process within an organism is by no means new, relatively little attention has been paid to the evolution of cancer-related genes between species, primarily due to the unavailability of genome sequences from non-model organisms in the pre-genomics era. Since tumors decrease fitness, selection for anticancer adaptations that prevent or delay their occurrence is expected in populations (Burt and Trivers 2006). Indeed, as humans are about 3,000 times larger and live 20–30 times longer than mice, we would not make it out of the womb alive, let alone reach puberty, did human cells have the same probability of becoming cancerous as mouse cells (Peto et al. 1975). Similarly, blue whales (Balaenoptera musculatus) must be capable of suppressing cancer 1,000 times better than humans, given the body size difference (Caulin and Maley 2011; Lichtenstein 2005).
Comparative genomics has already begun to provide examples of divergence in anticancer genetic mechanisms between mammalian species. One of the most striking includes mole rats from genera Heterocephalus and Spalax, the longest living rodents (>20 years), remarkably resistant to both congenital and experimentally induced cancerogenesis (Edrey et al. 2011; Manov et al. 2013). The naked mole rat (Heterocephalus glaber) has undergone an expansion of seven cancer-related gene families relative to rat (Yang et al. 2013), as well as accumulated unique non-synonymous mutations in hyaluronan synthase 2 (HAS2) (Tian et al. 2013) and several DNA repair genes (Kim et al. 2011). Intriguingly, the cancer resistance in subterranean blind mole rats (Spalax) occurs in spite of an amino acid substitution in TP53 that normally induces tumorigenesis in mice and humans (Avivi et al. 2007). While in rodents and primates TP53 is present in one copy only, the gene seems to have multiple paralogs in elephants (Loxodonta africana) (Caulin and Maley 2011).
Thomas et al. 2003 analyzed the relative substitution rate Ka/Ks for 311 human disease genes between human and mouse, and demonstrated a significant purifying selection on cancer genes compared to other disease genes, a result later confirmed on a human-macaque comparison (Blekhman et al. 2008). However, a more accurate and complete view of cancer, as well as the evolution of oncogenes and TSGs is expected to emerge from the new genomes (including human cancer genomes) being added to the public domain. To examine a possible selection pressure operating on cancer-related genes at a finer evolutionary time scale, we compared genomic sequences of 120 oncogene and TSG orthologs among seven hominoid species, including two extinct hominids, Neanderthal (Green et al. 2010) and Denisovan (Meyer et al. 2012). Using the number of non-synonymous substitutions per non-synonymous site (Ka), the number of synonymous substitutions per synonymous site (Ks), and their ratio (Ka/Ks), we found opposing selection pressures exerted on oncogenes and TSGs. Notably, as many as 108 substitutions (SNPs) present in humans only as somatic cancerous mutations occurred in other hominoid species as prevalent germline genotypes different from human.
Materials and Methods
Oncogenes and TSGs
A total of 120 genes classified as either oncogenes or TSGs were used. Genes were classified as oncogenes if they had an oncogene score > 20 % and classified as a TSG if the TSG Score was >20 %, using the 20/20 rule (Vogelstein, et al. 2013). The oncogene score was defined as the number of clustered mutations (i.e., missense mutations at the same amino acid or identical in-frame insertions or deletions) divided by the total number of mutations, while the TSG score was defined as the number of truncating mutations (e.g., nonsense mutations, insertions, deletions, and splice-site mutations that alter reading frame or the protein product length) divided by the total number of mutations (Vogelstein et al. 2013).
Genome Sources and Tools
Genome sequences and annotation files for chimpanzee (CHIMP2.1.4.74), human (GRCh37.74), gibbon (Nleu1.0.74), gorilla (gorGor3.1.74), and orangutan (PPYG2.74) were downloaded from Ensembl (www.ensembl.org), and transcripts with the longest CDS regions were chosen to represent corresponding genes for each species. Alignment results for Neanderthal and Denisovan against the human (hg19) genome were downloaded from Neanderthal genome project (www.eva.mpg.de/neandertal/) and Denisovan genome project (www.eva.mpg.de/denisova). Samtools (Li et al. 2009) was used to generate pileup outputs for Neanderthal and Denisovan, with Phred scores lower than 20 filtered out, and the major allele set to be the genotype for each nucleotide position. Positions with coverage depth lower than five were excluded and marked as missing to avoid an impact on Ka/Ks calculations.
Ka/Ks Ratio Calculations
Sequences of same ortholog sets for all species were grouped together, and Clustal-omega (Sievers et al. 2011) was used to conduct the multiple sequence alignments. Nucleotide sequences were parsed to amino acid sequences before carrying multiple-sequence alignments to avoid a possible frame-shift, and the amino acid sequences were changed back to nucleotide sequences for Ka/Ks calculations. PAML (version 4.7) (Yang 2007) was used to calculate the Ka/Ks ratio (ω) values, setting the model = 0 in the control file of codeml, and applying a single tree to all Ka/Ks estimates.
Results and Discussion
A total of 53 oncogenes and 67 TSGs were used, along with their oncogene/TSG scores (see “Materials and Methods” section, Table S1). The average Ka (0.019) and Ks (0.107) for all genes were lower than those from a comparison of 153 orthologs between human and mouse (0.065 and 0.830, respectively) (Nekrutenko et al. 2002), reflecting closer phylogenetic distances between species in our study. No significant difference in Ka and Ks was found between oncogenes and TSGs (t test, P value = 0.388 and 0.849 for Ka and Ks, respectively, Table 1). However, when we compared average Ka/Ks between oncogenes and TSGs, we found a higher Ka/Ks in TSGs (0.212) than in oncogenes (0.151), an indication that TSGs were presumably under weaker purifying selection compared to oncogenes (t test, P value = 0.039). BRCA1, an important tumor suppressor in breast and ovarian cancers, had the second highest Ka/Ks value (0.670). We then tested for a correlation between Ka/Ks and the TSG score or oncogene score (Vogelstein et al. 2013). Interestingly, genes with higher TSG scores tended to have higher Ka/Ks values (Spearman’s r = 0.223, P value = 0.014), while genes with higher oncogene scores tended to have lower Ka/Ks values (Spearman’s r = −0.269, P value = 0.003), a possible hallmark of opposing selection pressures operating on oncogenes and TSGs.
Why would selection pressures differ between oncogenes and TSGs? To answer this question, we note that the main distinction between oncogenes and TSGs refers to dominant effects of the former and recessive effects of the latter. The concept of a TSG emerged from a statistical analysis of retinoblastoma incidence in children, leading to the 2-hit hypothesis that inactivating mutations (hits) in both alleles of the retinoblastoma gene are required for this particular cancer to develop (Knudson 1971). Unlike TGSs, oncogenes can contribute to tumorigenesis even only one allele is mutated, amplified, or inappropriately expressed (Vogelstein and Kinzler 2002). As oncogenes confer an immediate disadvantage to the individual even when in heterozygosity, purifying selection will be more efficient in purging mutations across the population. TSG mutations will only start being discerned by purifying selection when in homozygosity, which means that the mutation has to reach considerable frequency in the population. The purging process can be further slowed down or even reversed if the first hit within a TSG has selectively advantageous effects (Iwasa et al. 2005).
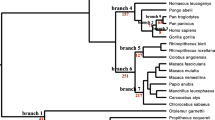
We then asked a question whether any substitution different between humans and the other six species in the 120 cancer-related genes have been observed in humans as a cancer-specific somatic mutation, similar to the TP53 genotype that occurs as wild-type variant in Spalax mole rats, but induces tumorigenesis when present in mice and humans (Avivi et al. 2007). We mapped interspecies substitutions against the Catalogue of Somatic Mutations in Cancer (COSMICS, v68), and to minimize chances of the substitutions being rare human alleles, we further excluded all alleles that matched dbSNP database (Build 139) with allelic frequency greater than 0.01. Out of 7,600 non-human genomic substitutions (SNPs), we found a total of 108 substitutions that in humans are present only as somatic cancerous mutations, but in other species can be a germline wild-type (Fig. 1 and Table S2). Among them, 62 were non-synonymous substitutions, out of which 38 were shared by multiple species as wild-type, presumably as ancestral genotype. For example, a human stomach cancer mutation in APC (3512G > A) occurs as a wild genotype among four ape species (chimpanzee, gibbon, gorilla, and orangutan, Fig. 2). It thus seems that a common ancestor of human and all apes carried the “cancer” APC genotype “A”, while the common ancestor of modern humans, Neanderthals, and Denisovans acquired “G” that mutates back to “A” only in cancer. A mutation in TP53 (245C > T or P82L, Fig. 1) found in many types of human tumors (e.g., haematopoietic, intestine, salivary, and upper aerodigestive tract), and shown to have a higher transcriptional activity than wild-type TP53 in adenocarcinoma cell lines (Bisio et al. 2013), was present in gibbons as a wild-type genotype (Figs. 1 and 2). Among the 108 differential SNPs, 59 of them were found in gibbon, 36 in orangutan, 25 in gorilla, 13 in chimpanzee, and only one in Neanderthal and one in Denisovan (Fig. 1 and Table S2), consistent with the evolutionary distances among the hominoid species. Neanderthal had a BRAF variant (1781A > C) typical of many human cancers (skin, stomach, ovary, lung, and intestine), whereas Denisovan had a variant TNFAIP3 (322A > G) found in humans only as a somatic mutation in tumor samples from haematopoietic and lymphoid tissues (Zhang et al. 2013). These results show that there may be alternative genetic routes to cancer and cancer suppression even among relatively closely related species of primates. As a cautionary note, however, we should add that although the substitutions are cancer specific, they do not necessarily need to be cancer causative, or driver mutations.

Numbers (in parentheses next to species names) and examples of non-human genomic variants matching cancer somatic mutations in humans in relation to phylogeny of hominoids. Distance between branches approximately based on estimated divergence time (Chatterjee et al. 2009). Asterisk indicates a substitution present in multiple types of cancer

Two examples of interspecies genomic substitutions with the non-human variant present in humans only as somatic cancer mutations. a Gibbon shows a wild-type T at chr17: 7,579,442 (TP53:245), while other species show a wild-type C, with TP53:245C > T being a somatic mutation found in tumor samples from haematopoietic, intestine, salivary, and other tumors; b Chimpanzee, gorilla, orangutan, and gibbon all have a wild-type A at chr5:112,174,803 (APC:3512) while Denisovan, Neanderthal and human have G as wild-type; a human somatic mutation in stomach tumor occurs in same position (APC:3512G > A)
The challenge of suppressing cancer increases with larger bodies and longer lifespan, as each proliferating cell within the body is at risk of becoming malignant. For example, osteosarcomas inflict large dog breeds 200 times more frequently than small- and medium-sized breeds (Dobson 2013). However, this principle does not hold above the species level, since large animals do not seem to suffer from cancer more often than small animals, a phenomenon known as Peto’s paradox (Caulin and Maley 2011). If natural selection operating on cancer-related genes is a solution to Peto’s paradox, one could predict that (1) there is substantial genetic divergence between species in the genes and (2) unlike oncogenes, TSGs exhibit signatures of either positive or at least relaxed purifying selection. We showed that both of the predictions were valid with respect to hominoids, despite relatively close phylogenetic distances and small body size differences in the primate group.
References
Avivi A et al (2007) P53 in blind subterranean mole rats—loss-of-function versus gain-of-function activities on newly cloned Spalax target genes. Oncogene 26:2507–2512. doi:10.1038/sj.onc.1210045
Bisio A, De Sanctis V, Del Vescovo V, Denti MA, Jegga AG, Inga A, Ciribilli Y (2013) Identification of new p53 target microRNAs by bioinformatics and functional analysis. BMC Cancer 13:552. doi:10.1186/1471-2407-13-552
Blekhman R et al (2008) Natural selection on genes that underlie human disease susceptibility. Curr Biol 18:883–889
Burt A, Trivers R (2006) Genes in conflict : the biology of selfish genetic elements. Belknap Press of Harvard University Press, Cambridge
Caulin AF, Maley CC (2011) Peto’s Paradox: evolution’s prescription for cancer prevention. Trends Ecol Evol 26:175–182
Chatterjee HJ, Ho SY, Barnes I, Groves C (2009) Estimating the phylogeny and divergence times of primates using a supermatrix approach. BMC Evol Biol 9:259
Dobson JM (2013) Breed-predispositions to cancer in pedigree dogs. ISRN Vet Sci 2013:941275
Edrey YH, Hanes M, Pinto M, Mele J, Buffenstein R (2011) Successful aging and sustained good health in the naked mole rat: a long-lived mammalian model for biogerontology and biomedical research. ILAR J 52:41–53
Green RE et al (2010) A draft sequence of the Neandertal genome. Science 328:710–722
Iwasa Y, Michor F, Komarova NL, Nowak MA (2005) Population genetics of tumor suppressor genes. J Theor Biol 233:15–23
Kim EB et al (2011) Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479:223–227
Knudson AG Jr (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 68:820–823
Li H et al (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079
Lichtenstein AV (2005) On evolutionary origin of cancer. Cancer Cell Int 5:5
Manov I et al (2013) Pronounced cancer resistance in a subterranean rodent, the blind mole-rat, Spalax: in vivo and in vitro evidence. BMC Biol 11:91
Meyer M et al (2012) A high-coverage genome sequence from an archaic Denisovan individual. Science 338:222–226
Nekrutenko A, Makova KD, Li WH (2002) The K(A)/K(S) ratio test for assessing the protein-coding potential of genomic regions: an empirical and simulation study. Genome Res 12:198–202
Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194:23–28
Peto R, Roe FJ, Lee PN, Levy L, Clack J (1975) Cancer and ageing in mice and men. Br J Cancer 32:411–426
Sievers F et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
Thomas MA, Weston B, Joseph M, Wu W, Nekrutenko A, Tonellato PJ (2003) Evolutionary dynamics of oncogenes and tumor suppressor genes: higher intensities of purifying selection than other genes. Mol Biol Evol 20:964–968
Tian X et al (2013) High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 499:346–349
Vogelstein B, Kinzler KW (2002) The genetic basis of human cancer, 2nd edn. McGraw-Hill, New York
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339:1546–1558
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang Z, Zhang Y, Chen L (2013) Investigation of anti-cancer mechanisms by comparative analysis of naked mole rat and rat. BMC Syst Biol 7(Suppl 2):S5
Zhang J et al (2013) Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA 110:1398–1403
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kang, L., Michalak, P. The Evolution of Cancer-Related Genes in Hominoids. J Mol Evol 80, 37–41 (2015). https://doi.org/10.1007/s00239-014-9649-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-014-9649-5