
Abstract
Antifolate antimalarials, such as pyrimethamine, have experienced a dramatic reduction in therapeutic efficacy as resistance has evolved in multiple malaria species. We present evidence from one such species, Plasmodium vivax, which has experienced sustained selection for pyrimethamine resistance at the dihydrofolate reductase (DHFR) locus since the 1970s. Using a transgenic Saccharomyces cerevisiae model expressing the P. vivax DHFR enzyme, we assayed growth rate and resistance of all 16 combinations of four DHFR amino acid substitutions. These substitutions were selected based on their known association with drug resistance, both in natural isolates and in laboratory settings, in the related malaria species P. falciparum. We observed a strong correlation between the resistance phenotypes for these 16 P. vivax alleles and previously observed resistance data for P. falciparum, which was surprising since nucleotide diversity levels and common polymorphic variants of DHFR differ between the two species. Similar results were observed when we expressed the P. vivax alleles in a transgenic bacterial system. This suggests common constraints on enzyme evolution in the orthologous DHFR proteins. The interplay of negative trade-offs between the evolution of novel resistance and compromised endogenous function varies at different drug dosages, and so too do the major trajectories for DHFR evolution. In simulations, it is only at very high drug dosages that the most resistant quadruple mutant DHFR allele is favored by selection. This is in agreement with common polymorphic DHFR data in P. vivax, from which this quadruple mutant is missing. We propose that clinical dosages of pyrimethamine may have historically been too low to select for the most resistant allele, or that the fitness cost of the most resistant allele was untenable without a compensatory mutation elsewhere in the genome.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The development of effective clinical therapeutics relies on an understanding of the molecular evolution of drug resistance. Factors which may affect evolutionary pathways of resistant alleles are drug concentration, epistatic interactions between mutations within the same gene, mutation bias in the genome, and adaptive conflict over resistance and function (Brown et al. 2010; Chou et al. 2011; Lozovsky et al. 2009; Weinreich et al. 2006). The malaria parasite presents a unique opportunity for studying the constraints that a fitness landscape may impose on the evolution of an organism as it has historically undergone waves of strong selection for drug resistance. These waves have been marked by the introduction, and eventual compromised efficacy, of a changing arsenal of first-line malaria drugs, including chloroquine, atovaquone, and pyrimethamine (Looareesuwan et al. 1996; Peterson et al. 1988; Snewin et al. 1989; Wellems 2002). Artemisenin-combination therapies (ACTs) are the last line of defense for the treatment of malaria, but signs of resistance building in small pockets of Southeast Asia demonstrate pervasive selection for resistance, and highlights the importance of understanding the molecular basis of resistance evolution (Breman 2012).
Drug concentration is considered critical for the selection of drug resistance in infectious agents such as HIV (Kepler and Perelson 1998) and malaria (White et al. 2009). Varying concentrations of drugs have varying potential to facilitate resistance: there is no selection for resistance if drug concentrations are high enough to kill all sensitive and resistant parasites, or too low to kill either. Thus, the window of drug concentrations that allows evolution of resistant variants is relatively narrow (Brown et al. 2010; Chou et al. 2011; Kepler and Perelson 1998; Lozovsky et al. 2009; Weinreich et al. 2006). In addition, diminishing returns in enzyme optimization suggest that for an enzyme to be fully optimized for a new activity, high selective pressure (in this case, drug concentration), must be sustained and later mutations tend to have progressively smaller impacts (Tokuriki et al. 2012). A second factor to constrain evolutionary trajectories of resistant phenotypes is epistasis among multiple mutant sites in the same gene: the effect of any amino acid replacement may be dependent on the genetic context in which it finds itself, for example the same mutation may increase resistance in some backgrounds but decrease it in others. This type of non-additive interaction is termed sign epistasis, and has been reported in multiple instances, including beta-lactamase in Escherichia coli (Looareesuwan et al. 1996; Peterson et al. 1988; Snewin et al. 1989; Weinreich et al. 2006; Wellems 2002) and dihydrofolate reductase (DHFR) in P. falciparum (Breman 2012; Brown et al. 2010). Another factor, mutation bias, can restrict the likelihood of fixation of a mutation (however favorable it may be) by limiting the likelihood of de-novo mutations to a particular base. For example, the genome of P. falciparum is strongly biased toward AT, and AT-rich codons are thus favored over others (Depristo et al. 2006; Kepler and Perelson 1998). Finally, adaptive conflict can constrain evolutionary pathways because amino acid replacements which confer high resistance can come at a fitness cost by impairing endogenous functioning of the protein in the absence of drug (Sirawaraporn et al. 1997; White et al. 2009), although such negative trade-offs are not always necessary (Aharoni et al. 2005; Kondrashov 2005).
In this study, we focus on the malaria parasite Plasmodium vivax and the evolution within this species of drug resistance to pyrimethamine, a drug that competitively inhibits DHFR. DHFR is a parasite enzyme required for the synthesis of tetrahydrofolate, an essential precursor of purines and several amino acids (Kompis et al. 2005). Although P. vivax is not normally treated with pyrimethamine directly, the high frequency of mixed infections with P. falciparum (which has been treated by the combination drug sulfadoxine–pyrimethamine since the 1970s), has exposed a large number of P. vivax parasites to selection for drug resistance in many areas. P. vivax is remotely related to P. falciparum, the two having diverged approximately 100 million years ago (Ayala et al. 1999; Carter 2003). The life cycle of P. vivax features a latent liver infection responsible for relapses months or years following the initial infection. Although seldom fatal, P. vivax elicits incapacitating clinical symptoms and recurrent severe relapses. Recent work suggests that the morbidity and mortality associated with these cycles of reinfection are greater than previously believed (Mendis et al. 2001; Sina 2002).
The combination of a long history of pyrimethamine use and the common co-infection of P. vivax and P. falciparum has facilitated the evolution of drug resistance in both species. Point mutations in the dhfr gene and their impact on sensitivity of P. falciparum to pyrimethamine have been studied extensively in vitro and in vivo (Cortese and Plowe 1998; Randrianasolo et al. 2004; Wooden et al. 1997). However, similar data for P. vivax is lacking due to the difficulties associated with culturing this species in the laboratory.
The active site regions of DHFR in P. vivax and P. falciparum are strongly conserved, despite the two proteins preserving only ~66 % sequence identity (Kongsaeree et al. 2005). The dhfr coding region is encoded by about 700 nucleotides in both species. However, work in P. falciparum shows that only a limited number of mutations are associated with drug resistance. Mutations at amino acid residues 16, 50, 51, 59, 108, and 164 have been observed to affect resistance to pyrimethamine, or the related antifolate drugs cycloguanil or chlorcycloguanil, in P. falciparum isolates from around the world (Gregson and Plowe 2005; Hyde 2005; Sibley et al. 2001). Four of these mutations (N51I, C59R, S108N, and I164L) were found to be most commonly associated with drug resistance, and were extensively studied by Lozovsky et al. (2009) to predict favored pathways of drug resistance based on empirical estimates of mutational spectrum and probabilities of fixation based on relative levels of resistance. Although far fewer isolates of P. vivax have been studied, numerous alleles of dhfr have been identified, which is in contrast to the lack of diversity in P. falciparum (Hawkins et al. 2008).
Sequence alignment indicates that the four mutations important to pyrimethamine resistance in P. falciparum DHFR (N51I, C59R, S108N, and I164L) correspond to mutations N50I, S58R, S117N, and I173L in P. vivax DHFR. Interestingly, mutations at these exact residues have been observed to be important for pyrimethamine resistance in P. vivax DHFR in vivo and in vitro studies (Hastings et al. 2004; Hawkins et al. 2008), suggesting that P. falciparum and P. vivax DHFR may experience similar functional and selective constraints despite approximately 100 million years of divergence. However, the story may not be so simple, as mutations which are common and convey high resistance in P. falciparum, such as N51L and I164L, are rarely observed in P. vivax (where the homologous mutations are N50I and I173L). Therefore, evolutionary trajectories of mutation acquisition by the two proteins may differ more than expected.
The goal of this experiment was to assay whether these four point mutations in DHFR, which in P. falciparum have been demonstrated to convey drug resistance and mold the evolutionary landscape of mutation acquisition, have similar effects in P. vivax. The mutational landscape in DHFR differs between P. falciparum and P. vivax quite extensively, with far greater polymorphism in the latter species. Some mutations that are commonly observed in P. vivax, which are not included in the set we have studied, include F57I, T61M, and S117T. Due to this, the emphasis of our study is not on practical applications of the results in a clinical setting, but rather to test whether orthologous amino acid replacements in orthologous proteins have parallel effects. And to apply a forward-time approach (rather than traditional coalescent models) to simulate evolutionary trajectories that use fitness landscapes informed by multiple parameters such as drug concentration, direct selection coefficients, and drift (rather than IC50 alone).
Using a transgenic yeast system, we explored the mutational landscape of pyrimethamine resistance in P. vivax DHFR. Following others (Brown et al. 2010; Lozovsky et al. 2009; Weinreich et al. 2006), we genetically engineered all 16 possible combinations of four amino acid replacements. Each of these mutations has been observed in various combinations to be associated with drug resistance, even rare ones such as N50I and I164L (Hawkins et al. 2008). Fitness landscapes and stepwise mutational trajectories can be predicted by analyzing the phenotypes of these alleles in the presence of the drug. Importantly, trade-offs between novel function (resistance) and existing function (activity) can be gauged simultaneously by translating growth rates at various drug concentrations into proportional fitness peaks and valleys in the resulting landscape.
Overall, our results provide the first predicted pathways of mutation acquisition in the dhfr gene in P. vivax. Importantly, these pathways are given for a gradient of drug concentrations, which can represent a temporally and spatially heterogeneous drug reservoir within the population created by sporadic noncompliance to antibiotic regimes, varying dosage regimes, or compartmentalization of drugs in different tissues. We show that varying drug concentrations affect the fitness of the most resistant alleles. In other words, adaptive fitness landscapes are strongly molded by drug concentrations. The landscape is dominated by the single peak of the non-mutant allele at the low end of the drug concentration spectrum, and is effectively flat (and without a single peak) for very high drug concentrations. Medium-range drug concentrations have distinctly different projected evolutionary trajectories, and the selectivity window for the most resistant quadruple mutant is constrained within a narrow range. Second, P. vivax DHFR shows sign epistasis at multiple mutation sites, which explains why many evolutionary pathways are rendered inaccessible to selection in our scenarios. Finally, to address the ability of adaptive conflict to influence evolution, we used a composite measure of resistance and endogenous function, both of which determine fitness, to represent the nodes in our fitness landscapes. This is in contrast to previous studies that primarily relied on resistance only to construct adaptive landscapes (Brown et al. 2010; Lozovsky et al. 2009; Weinreich et al. 2006).
Materials and Methods
E. coli and Yeast Strain Construction
Plasmodium vivax DHFR coding sequence (Genbank accession AB547458) was isolated and cloned into the vector pET17 (Novagen). The resulting plasmid was transformed into E. coli strain HMS174(DE3) (Novagen). All 16 possible combinations of mutations at four sites (N50I, S58R, S117N, I173L) were introduced by QuikChange Site-Directed Mutagenesis Kit (Strategene). Each mutagenized plasmid was sequenced to confirm the presence of expected mutations, and the absence of any other mutations. Plasmids containing all mutated alleles of DHFR were then introduced into E.coli strain LH18, thyA Δfol:kan donated by Howell et al. (1988). This strain lacks DHFR, as well as thymidylate synthase, and requires plating on full LB medium supplemented with 200 μg/mL of thymine for growth. In addition, this strain carries a kanamycin-resistance selective marker. Once transformed, this strain can be grown on Bonner–Vogel minimal medium supplemented with 200 μg/mL of thymine.
To create yeast strains with the same mutated alleles, we cloned each allele into the GR7 shuttle vector, a derivative of the pRS314 yeast shuttle vector (Sikorski and Hieter 1989). We used S. cerevisiae strain TH5 (MATa leu2-3,112 trp1 ura3-53, dhfr1::URA3 tup1, provided by Carol Sibley) to assay pyrimethamine resistance conferred by each DHFR allele. TH5 lacks DFR1, the yeast orthologue of the DHFR gene, and when not transformed with functional DHFR, requires media supplemented with 100 μg/mL deoxythymidine monophosphate (dTMP) for growth. TH5 was transformed with each of the 16 alleles of DHFR on tryptophan drop-out medium (SC trp-) to select for the presence of the GR7 vector carrying DHFR. The SC trp-medium represents “minimal medium” for yeast strains in all future references.
We used both yeast and bacterial transgenic systems for our assays to rule out possible artifacts of background mutations in the vector to ensure that the results represented biologically relevant behavior of the parasite DHFR alleles themselves. In this paper, we focus on the results from the yeast transgenic system, but results from both systems are strongly correlated.
Growth Rate Calculations
For each strain, we picked two to five colonies from a solid media plate and inoculated the appropriate liquid minimal medium culture with our transformed E. coli and yeast strains. After overnight culture, cultures were diluted to an OD600 of 0.01 (or approximately 6 × 104 cells/mL) in a series of concentrations of pyrimethamine in minimal medium, and then dispensed into microtiter plates. These plates were transferred to a Bioscreen C microbiological workstation (Thermo Labsystems), which recorded OD600 readings every 15 min for 2 days. Culturing temperatures used were 37 °C for E. coli and 30 °C for S. cerevisiae. Growth rate was calculated by taking a least squares linear regression of log absorbance versus time for a 3 h sliding window over the length of the growth curve (Brown et al. 2010; Joseph and Hall 2004). Growth rates represent the maximum regression coefficient among all sliding windows.
IC50 Calculations
We calculated the resistance of each strain using inhibitory concentration 50 (IC50) measurements, which represent the pyrimethamine concentration at which growth rate is 50 % of what it is in the absence of pyrimethamine. IC50 values were obtained as follows. For each strain, we fit the following logarithmic curve to our growth rate versus pyrimethamine concentration:
where G i is the growth rate of strain i, A i is the maximum growth rate in the absence of pyrimethamine, x is the natural log of (pyrimethamine concentration + 1), b i is the pyrimethamine concentration at which G i is half of A i , and c i is a scaling parameter determining the shape of the logistic curve. This curve gave us predicted growth rates at a range of pyrimethamine concentrations. Nonlinear least squares regressions were used to determine the value of b i , which represents the IC50 values for each strain.
Calculation of Possible Evolutionary Trajectories
To investigate properties of the adaptive landscape, we conducted forward-time simulations using simuPOP (Peng and Kimmel 2005). Each simulation began with a population of individuals that were fixed for the ancestral haplotype. We drew the population size for each simulation from an e unif(ln(1000),ln(100000) distribution, or a natural-log scaled uniform distribution from 1,000 to 100,000 individuals. Mutations were then added according the relative rate matrix for P. vivax that we computed from the data presented in Neafsey et al. (2012). To convert the relative substitution rate matrix to more realistic per-generation rate matrix, we divided all relative rates by a factor of 103. Ultimately, we were interested in the rate of amino acid substitutions; hence to compute the per-generation substitution rates, we summed the rates of all nucleotide substitutions that can produce the amino acid substitutions of interest. We performed 500 simulations at concentrations 0, 2, 4, 6, 8 as described in the Supplemental Information. Concentrations above 8 were not explored because for most population sizes in this range, substitutions are “nearly neutral”, i.e. Ns < 1 (over the range of population sizes investigated), thus the adaptive landscape is effectively flat. Maximum growth rate for each haplotype at each concentration was computed as explained above. We then assigned selective coefficients to each haplotype based on the relative maximum growth rates estimated for each concentration. Simulations were run until the population fixed the haplotype of optimal fitness or until 1,000,000 generations had passed. The script that we used to simulate the adaptive landscape is available as Supplemental File 2.
We used these simulations to determine the adaptive trajectory for each simulation as follows. We recorded the frequency of each haplotype every ten generations during the simulation. If, at one time step, any haplotype had a frequency greater than 0.5, we considered the population to be in this haplotype state. We then recorded the transitions between haplotypes and removed loops (e.g., a transition from haplotype 3–4 and immediately back again to 3 is recorded as just 3) to determine the adaptive trajectory for that simulation. These were then compiled for each concentration and the four most likely adaptive trajectories determined from the aggregate.
Results
In order to assess how various mutational combinations in the coding sequence of P. vivax DHFR affected resistance to pyrimethamine, a drug which competitively inhibits this protein’s activity, we created all 16 possible combinations of the mutants N50I, S58R, S117N, and I173L. The mutagenized DHFR coding sequences were cloned into a yeast vector and transformed into a yeast strain lacking DFR1, the yeast orthologue of the DHFR gene.
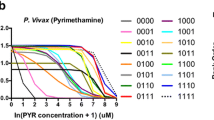
We estimated the level of pyrimethamine resistance as the concentration of drug that inhibited cell growth by half, a metric known as the IC50 (see “Materials and Methods” section). The results are shown in the barplot in Fig. 1. The DHFR alleles indicated along the horizontal axis are given in the form of a vector of 0’s and 1’s corresponding, from left to right, the amino acid residues 50, 58, 117, and 173. Each 0 indicates a non-mutant codon, and each 1 indicates a mutant codon. Therefore, 0000 indicates the wildtype, nonmutated allele, and 1111 indicates the quadruple mutant. Figure 1 shows that of the four single mutants, 0010 is much more resistant to pyrimethamine, and may likely be favored as the first mutational step. Consistent with data from previous yeast or bacterial complementation systems for P. falciparum DHFR (Brown et al. 2010; Lozovsky et al. 2009), the quadruple mutant (N50I, S58R, S117N, I173L) was the most resistant, with triple mutants also exhibiting high levels of resistance. Although the quadruple mutant we studied (N50I, C59R, S108N, and I164L) is missing from natural isolates, clinical data from malarial patients treated with a sulfadoxine–pyrimethamine drug regimen showed higher failure rates with a different quadruple mutant combination in P. vivax DHFR (at positions 57, 58, 61, 117) compared to wildtype (Tjitra et al. 2002).

Boxplot showing resistance phenotypes of 16 DHFR alleles mutated at four possible sites. Alleles are shown in order of increasing resistance
Figure 2 shows estimated growth rates of all alleles across a range of drug concentrations. Alleles vary not only at the point at which they cross the Y-axis (growth in the absence of pyrimethamine), or concentration of drug at which growth is reduced by half (IC50), but also by the shape of the logistic growth curve (whether growth falls sharply with increasing dosage of drug). Beyond the obvious low resistance of the wildtype 0000 allele and the very high resistance of the quadruple mutant 1111 allele, the evolutionary landscape is complex. Epistatic interactions among the mutant sites are common (Table 1). For example, the S58R mutation increases resistance in four backgrounds, has a negligible effect on resistance in three backgrounds, and actually decreases resistance in one allelic background.

Fitted logarithmic growth rates of all alleles at increasing concentration of pyrimethamine. Concentration of pyrimethamine (x) is transformed as ln(x + 1). Black line represents wildtype (0000) allele, which is least resistance to pyrimethamine. Single mutants (1000, 0100, 0010, 0001) are represented by blue lines. Double (0011, 0101, etc.) and triple mutants (1110, 1011, etc.) are represented by green and orange lines, respectively. Red line quadruple mutant (1111) is the most resistant allele
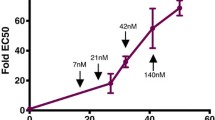
Correlation between IC50 values for P. vivax and P. falciparum dhfr alleles were strongly significant and positive (rho = 0.8607, P = 2.2e−16, Spearman rank correlation test, P. falciparum DHFR data from Lozovsky et al. (2009, Supplementary Fig. 1). Allele 0011 was removed from this calculation because this allele failed to grow in P. falciparum DHFR, most likely due to adaptive conflict between resistance to inhibition and maintenance of enzyme activity (Lozovsky et al. 2009). In contrast, the P. vivax 0011 DHFR allele maintained functional enzyme activity, and had high resistance to pyrimethamine.
The development of a new function often comes at a cost of a previous function, thus proteins evolving for increased drug resistance are often thought to be under adaptive conflict for endogenous function. In our case, the evolution of resistance to pyrimethamine may compromise the catalytic activity of DHFR. However, this kind of trade-off may not be necessary, as was suggested by Brown et al. (2010), who observed no association between resistance level and growth rate. In our data, we also see no clear association between these two phenotypes, as correlation analysis reveals (Pearson’s correlation between log-transformed IC50 and maximum growth rate in the absence of drug, P = 0.3416). However, maximum growth rate in the absence of drug and IC50 are only two metrics, which taken individually paint an incomplete picture of fitness. As Fig. 2 shows, drug concentration has a strong effect on relative fitness of alleles, with landscapes dominated by the wildtype (0000) allele in the absence of drug (pyrimethamine concentration equal to 0) and very flat at high drug concentrations. The flat fitness landscape at high drug concentrations means there are no pronounced peaks or valleys, and the population is fixing alleles mainly by drift. Simulations for evolutionary pathways in such landscapes can give us little insight into the likely evolutionary pathway of new mutations, and were excluded from analysis. Gene-by-environment interactions are important, as moderate dosages of pyrimethamine have greater power to select for resistant alleles. This is because, in contrast to the flat landscapes at either dosage extreme, moderate dosages induce greater variation in fitness differences between the alleles. In addition, effective population size of the malaria species and mutation bias in the genome can also affect the path of evolution (Chang et al. 2012; Depristo et al. 2006). In order to simulate an evolutionary pathway for DHFR evolution, we incorporated all these elements in a forward-time simulation (see “Materials and Methods” section).
The results of the simulation are displayed in Fig. 3. In the absence of drug, the fitness landscape is dominated by the fitness peak of the wildtype (0000) allele. Despite random mutations and sometimes small population sizes, no simulations ever wandered off this peak. At higher drug concentrations (log-transformed pyrimethamine concentrations 2–6), the quadruple mutant does not have the highest fitness (Fig. 2), likely reflecting adaptive conflict between resistance and catalytic activity. The triple mutant 1110 allele fixes in the vast majority of cases, and populations that remain segregating for two or more polymorphic alleles at the end of 1 million generations make up a small percentage of the runs (2.4, 1.0, and 0 % for log-transformed concentrations 2, 4, and 6, respectively). At a log-transformed pyrimethamine concentration of 8, which translates into an actual pyrimethamine concentration of ~3,000 μM, the quadruple mutant 1111 allele fixes in 99.8 % of simulated runs. At this concentration, the 1111 allele resides at the highest peak of the fitness landscape. Again, since we use growth rates to estimate fitness at different drug dosages, this fitness is informed by both resistance and catalytic activity. Taken together, these results suggest that despite being the most resistant allele, the quadruple mutant is not selectively favored unless very high dosages of drugs are administered. These simulations also reinforce the idea that effective population size plays a large role in the time to fixation of favorable alleles, with larger population sizes leading to faster fixation of favorable mutations (Fig. S2). In addition, smaller effective population sizes were more likely to fix alleles which were not the absolute fitness peak in the landscape because drift pushed them more frequently onto sub-optimal peaks (Table S1).

Proportion of simulation runs which fixed for any particular allele, at varying concentrations of pyrimethamine. Concentration of pyrimethamine (x) is transformed as ln(x + 1). “Other” represents alleles which fixed, but with frequencies of less than 1 %. “Polymorphic” represents simulations which remained segregating for polymorphic alleles at the end of 1 million generations
This result is consistent with amino acid replacements observed in worldwide surveys of P. vivax (Table 2). The majority of P. vivax samples isolated from patients in Southeast Asia and India were mutated in at least one of these four positions. In contrast to the prevalence of the 1111 allele in P. falciparum samples (Ahmed et al. 2006; Anderson et al. 2005; van den Broek et al. 2004), this allele has not yet been observed in P. vivax, perhaps because clinical dosages of pyrimethamine never reach high enough levels for the quadruple mutant to have a fitness advantage. Another possibility is that long-term maintenance of the quadruple allele requires the presence of a compensatory mutation elsewhere in the genetic background, as has been shown in P. falciparum (Nair et al. 2008). In addition, mutations not investigated in this study may present more resistant alleles in natural isolates of P. vivax, or these mutations could carry out the resistance function of the rare N50I and I164L mutations, thus making allele 1111 unlikely to fix.
Other polymorphisms commonly found in natural isolates (such as the alleles 0110 and 0111) do coincide with several of the intermediates predicted by our simulated most likely pathways (Fig. 4). Two things are notable from these results. First, despite the striking similarity between IC50 of alleles between P. falciparum and P. vivax, the trajectories differ. For example, at the highest pyrimethamine concentration we examined, where the quadruple mutant fixed in most simulations, the first evolutionary step was most often 0100 in our simulations (Fig. 4d), but 0010 in P. falciparum (Lozovsky et al. 2009). However, at lower pyrimethamine concentrations, 0010 was also the first evolutionary step in P. vivax. This highlights the subtle differences in fitness landscapes produced by using IC50 alone (as in the case of P. falciparum simulations) and by using a multi-parameter model incorporating drug concentration, selection, and drift, as we did with P. vivax. The second notable point is that the allele 1110, which is an important intermediate step in our evolutionary pathways, has not yet been isolated in natural populations. The overall increased nucleotide diversity in P. vivax compared to P. falciparum is reflected in the variation found in P. vivax DHFR alleles, where mutations other than the ones studied here are sometimes found at high frequencies. Epistatic interactions between these multitudes of mutations could result in evolutionary pathways that were unexplored in our simulations. For example, the 1110 background may stand as an unfavorable background for an otherwise highly favored mutation. Such sign epistasis is quite common, both within genes, as demonstrated by amino acid replacement S58R and I173L in our results (Table 1) and in β-lactamase in E. coli (Weinreich et al. 2006); as well as between genes, such as in Methylobacterium (Chou et al. 2011).

Preferred evolutionary pathways of pyrimethamine resistance in P. vivax DHFR. a–d Represent log-transformed pyrimethamine concentrations of x = 2, 4, 6, 8, respectively, where the transformation is evaluated at ln(x + 1). The top four major pathways for each concentration are shown, except when probabilities fall below 0.01. Widths of lines in pathways correspond to their probabilities. The major pathway is shown in red, and given with an estimated probability
Discussion
We describe the resistance of mutant DHFR proteins in P. vivax to the anti-malarial drug pyrimethamine. Correlation analysis reveals that the adaptive landscape of the P. vivax DHFR alleles is highly correlated with the adaptive landscape of the P. falciparum DHFR alleles (Lozovsky et al. 2009). This suggests that orthologous mutations in the active sites of related proteins have similar functional significance for relatively distantly related species. This hypothesis is supported by more recent computational analysis of binding between malaria DHFR and anti-folates in four Plasmodium species—P. falciparum, P. vivax, P. malariae, and P. ovale—which found binding to be broadly similar, and determined by an analogous set of seven residues (Choowongkomon et al. 2010). In a way, this result is surprising, since P. vivax nucleotide diversity far surpasses that of P. falciparum nucleotide diversity, and mutations which are rare or missing in P. falciparum have been found to be important for resistance in P. vivax (Hawkins et al. 2007). Thus, we would not necessarily expect analogous mutations to have similar functional consequences. However, this result is also intuitively congruent with our current understanding of function in orthologous proteins that share common catalytic sites.
The evolutionary pathways we identified to be important in the evolution of DHFR in P. vivax have been previously identified by in vivo studies of therapeutic efficacies of ACT-pyrimethamine combination drugs: natural isolates from Indonesia found mutations at residues 58 and 117 common in P. vivax isolates of malaria patients, as well as quadruple mutations (at residues 57, 58, 61, 117) (Tjitra et al. 2002). These mutations were associated with increased resistance to sulfadoxine–pyrimethamine treatments. Based on these results, the authors suggested a stepwise drug selection process in mutations favored mutation at residue 117 first, followed by mutations at 50 and 58, which aligns with our results. Additional mutations favored by natural selection, but which were not assessed by us, included mutations at residues 57 and 61.
The serine to asparagine mutation in codon 117 (which corresponds to position 108 in P. falciparum) has repeatedly been demonstrated to be a major determinant of antifolate resistance (de Pécoulas et al. 1998; Sibley et al. 2001; Tjitra et al. 2002). The importance of the S117N mutation for resistance in P. vivax is even more stark than the S108N mutation in P. falciparum: in P. vivax, the S117N mutation confers a ~4,000 fold increased resistance to pyrimethamine (Leartsakulpanich et al. 2002), whereas in P. falciparum, the S108N mutation confers only a ~100 fold increased resistance (Cowman et al. 1988; Peterson et al. 1988). Analysis of crystal structures of the DHFR protein reveals that a steric conflict arising from the side chain of a S117N mutant enzyme, accompanied by loss of binding to the serine at residue 120 is mainly responsible for the reduction in binding of pyrimethamine (Kongsaeree et al. 2005).
In addition to amino acid replacement S117N, in vitro assays of pyrimethamine sensitivity in mutated P. vivax isolates also identified S58R to be important for resistance (de Pécoulas et al. 1998). The importance of these mutations for resistance in Plasmodium DHFR has also been observed in P. malariae, where natural isolates resistant to pyrimethamine contained mutations S58R and S114N, corresponding to S58R and I173L in P. vivax and C59R and S108N in P. falciparum (Khim et al. 2012).
Some additional mutations which are associated with pyrimethamine resistance in P. vivax include F57L and T61M. Two amino acid replacements which have been found to be important for evolution of resistance in P. falciparum DHFR (N51I and I164L), are analogous to mutations N50I and I173L in P. vivax, but were very rarely observed in natural isolates in the latter species.
Pyrimethamine binds directly to the active site of DHFR, and competitively prevents the binding of dihydrofolates (Yuvaniyama et al. 2003). DHFR evolves resistance to pyrimethamine through acquiring mutations which sterically inhibit binding to the drug, which consequently increase substrate specificity for dihydrofolate (Rastelli et al. 2000). Such modifications to the active site of an enzyme is thought to impose greater trade-offs in native enzyme function than drugs that bind externally (Berkhout 1999; Tawfik 2005). Kinetic parameters of protein function are the pyrimethamine dissociation constant (K i ) and the catalytic turnover rate (k cat). The relative fitness of parasites is highly informed by these two parameters, the first of which indicates level of resistance while the second indicates maintenance of endogenous function. In the case of malaria DHFR, adaptive conflict is in play as improved substrate specificity (K i ) often comes at the expense of catalytic activity (k cat) (Lozovsky et al. 2009; Sirawaraporn et al. 1997). In other cases, the trade-offs may be absent, as Brown et al. (2010) showed that DHFR proteins are capable of evolving resistance without compromising existing endogenous catalytic function. In addition, stability of the protein encoded by different alleles could dramatically impact which ones are selectively favored. For example, a particular allele may not be selectively favored even if it improved its substrate specificity to dihydrofolate, and thus improve resistance phenotype, if stability of the protein was severely impaired (Wang et al. 2002). We directly analyzed how adaptive conflict can shape evolution at various drug concentrations by using relative growth rates (which incorporate both substrate specificity and catalytic capacity) into our simulations of evolutionary pathways across fitness landscapes.
Our simulations showed that fitness landscapes could vary dramatically across drug concentrations. The wildtype allele (0000) shows the highest fitness in a drug-free environment, supporting the adaptive conflict theory that trade-offs can occur between resistance and growth. Although the quadruple mutant (1111) has the highest IC50 value, it is not the most favored allele at most drug concentrations. In fact, at lower drug concentrations, endogenous enzyme activity is compromised to an extent that limits the fitness benefits derived from higher drug resistance. A small window of drug concentrations can favor the fixation of the quadruple mutant, which in our assays was around 3,000 μM pyrimethamine. However, it is difficult to predict how this concentration translates into an analogous dosage in a human patient, outside of a yeast transgenic system. Nevertheless, the incorporation of drug concentrations as a factor for predicted evolutionary pathways is a novel approach in tracking drug resistance in malaria research. This has important ramifications for human health. Since fitness landscapes are so strongly shaped by drug dosages, clinicians could modify dosage procedures over the course of treatment to subvert the evolution of resistance. This could be done perhaps by drawing populations into repeated cycles of suboptimal fitness using varying concentrations of drug, which induce previously highly fit alleles to reside at suboptimal fitness peaks in the current dosage regime.
Mathematical modeling based on another anti-malarial drug, mefloquine, suggest that drug dosage plays a strong role in selection of resistance (Simpson et al. 2000). They suggest that the initial deployment of lower doses provides an opportunity for selection of resistant alleles. This resistance would spread more rapidly than the de-novo application of maximal doses. Our simulations show that maximal doses that do not allow the growth of any alleles can circumvent resistance (in our yeast vector system, this dose was ~3,000 μM), however such high doses are probably eschewed as clinical treatments because of intolerable toxic side effects. Lower dosages of drugs can achieve adequate levels of parasitemia clearance with unmutated alleles, but also provides a breeding ground for resistance.
Ranked IC50 values for S. cerevisiae and E. coli strains carrying the same DHFR alleles are significantly correlated (rho = 0.7323, P value = 0.001812, Table S2). The similarity of resistance phenotypes in two different species rules out experimental artifacts such as mutations in the genetic background of the yeast strains. Despite these similarities, the yeast and bacterial systems differ in some notable respects. For example, in yeast, a higher concentration of pyrimethamine is needed to inhibit DHFR activity sufficiently to measure IC50 than in bacterial cells: mean ln(IC50 + 1) values for E. coli is 3.58, versus 5.40 for S. cerevisiae. This may reflect a lower requirement for DHFR activity in yeast relative to E. coli, as has been previously noted (Brown et al. 2010), or else an over-expression of DHFR in the yeast system.
An in-depth understanding of the molecular pathways of drug resistance evolution is necessary because infections by P. vivax pose a serious challenge to global health in Asia, South America, Central America, the Middle East, and parts of Africa (Hawkins et al. 2007; Parekh and Moorthy 2011). Our results lend novel insights into the evolution of anti-malarial resistance in the DHFR protein of P. vivax. The larger than expected agreement between major pathways identified in our analysis, and those observed in natural populations of P. vivax (Table 1), supports the use of model organisms in studies of drug resistance, particularly in organisms, such as P. vivax, that do not lend themselves to easy continuous culture in the laboratory. However, differences between the laboratory setting and the field are manifold, and may explain why the most resistant quadruple mutant has not yet been found in nature. For example, we can create artificially high drug dosages in the laboratory that are avoided in the clinic, leading us to over-estimate the fitness of very resistant alleles, which likely evolve in a naturally lower drug dosage environment where their fitness advantage is reduced by negative trade-offs over endogenous function. Further areas of research could use SDS-PAGE to directly assay expression level of DHFR proteins encoded by these alleles, and to test the four mutations we investigated with other known polymorphisms in the P. vivax DHFR gene. Such molecular characterizations are outside the scope of our paper, but would provide excellent insights into the molecular mechanisms of this system.
Nonetheless, two results strengthen the validity of using model systems to predict evolutionary examples. The first is the congruence of growth rates for P. vivax alleles expressed in the E. coli and S. cerevisiae systems, which seems to rule out experimental artifacts. And the second is the highly significant correlation between the fitness of corresponding alleles in P. vivax and P. falciparum. Both these observations strongly support the use of model organisms as a helpful system for studying the evolution of the malaria parasite.
References
Aharoni A, Gaidukov L, Khersonsky O et al (2005) The ‘evolvability’ of promiscuous protein functions. Nat Genet 37:73–76
Ahmed A, Das MK, Dev V et al (2006) Quadruple mutations in dihydrofolate reductase of Plasmodium falciparum isolates from Car Nicobar Island, India. Antimicrob Agents Chemother 50:1546–1549
Anderson TJC, Nair S, Sudimack D et al (2005) Geographical distribution of selected and putatively neutral SNPs in Southeast Asian malaria parasites. Mol Biol Evol 22:2362–2374
Ayala FJ, Escalante AA, Rich SM (1999) Evolution of Plasmodium and the recent origin of the world populations of Plasmodium falciparum. Parasitologia 41:55–68
Berkhout B (1999) HIV-1 evolution under pressure of protease inhibitors: climbing the stairs of viral fitness. J Biomed Sci 6:298–305
Breman JG (2012) Resistance to artemisinin-based combination therapy. Lancet Infect Dis 12:820–822
Brown KM, Costanzo MS, Xu W et al (2010) Compensatory mutations restore fitness during the evolution of dihydrofolate reductase. Mol Biol Evol 27:2682–2690
Carter R (2003) Speculations on the origins of Plasmodium vivax malaria. Trends Parasitol 19:214–219
Chang H-H, Park DJ, Galinsky KJ et al (2012) Genomic sequencing of Plasmodium falciparum malaria parasites from senegal reveals the demographic history of the population. Mol Biol Evol 29:3427–3439
Choowongkomon K, Theppabutr S, Songtawee N et al (2010) Computational analysis of binding between malarial dihydrofolate reductases and anti-folates. Malar J 9:65
Chou H-H, Chiu H-C, Delaney NF et al (2011) Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science 332:1190–1192
Cortese JF, Plowe CV (1998) Antifolate resistance due to new and known Plasmodium falciparum dihydrofolate reductase mutations expressed in yeast. Mol Biochem Parasitol 94:205–214
Cowman AF, Morry MJ, Biggs BA et al (1988) Amino acid changes linked to pyrimethamine resistance in the dihydrofolate reductase–thymidylate synthase gene of Plasmodium falciparum. Proc Natl Acad Sci USA 85:9109–9113
de Pécoulas PE, Tahar R, Ouatas T et al (1998) Sequence variations in the Plasmodium vivax dihydrofolate reductase–thymidylate synthase gene and their relationship with pyrimethamine resistance. Mol Biochem Parasitol 92:265–273
Depristo MA, Zilversmit MM, Hartl DL (2006) On the abundance, amino acid composition, and evolutionary dynamics of low-complexity regions in proteins. Gene 378:19–30
Gregson A, Plowe CV (2005) Mechanisms of resistance of malaria parasites to antifolates. Pharmacol Rev 57:117–145
Hastings MD, Porter KM, Maguire JD et al (2004) Dihydrofolate reductase mutations in Plasmodium vivax from Indonesia and therapeutic response to sulfadoxine plus pyrimethamine. J Infect Dis 189:744–750
Hawkins VN, Joshi H, Rungsihirunrat K et al (2007) Antifolates can have a role in the treatment of Plasmodium vivax. Trends Parasitol 23:213–222
Hawkins VN, Auliff A, Prajapati SK et al (2008) Multiple origins of resistance-conferring mutations in Plasmodium vivax dihydrofolate reductase. Malar J 7:72
Howell EE, Foster PG, Foster LM (1988) Construction of a dihydrofolate reductase-deficient mutant of Escherichia coli by gene replacement. J Bacteriol 170:3040–3045
Hyde JE (2005) Drug-resistant malaria. Trends Parasitol 21:494–498
Joseph SB, Hall DW (2004) Spontaneous mutations in diploid Saccharomyces cerevisiae: more beneficial than expected. Genetics 168:1817–1825
Kepler TB, Perelson AS (1998) Drug concentration heterogeneity facilitates the evolution of drug resistance. Proc Natl Acad Sci USA 95:11514–11519
Khim N, Kim S, Bouchier C et al (2012) Reduced impact of pyrimethamine drug pressure on Plasmodium malariae dihydrofolate reductase gene. Antimicrob Agents Chemother 56:863–868
Kompis IM, Islam K, Then RL (2005) DNA and RNA synthesis: antifolates. Chem Rev 105:593–620
Kondrashov FA (2005) In search of the limits of evolution. Nat Genet 37:9–10
Kongsaeree P, Khongsuk P, Leartsakulpanich U et al (2005) Crystal structure of dihydrofolate reductase from Plasmodium vivax: pyrimethamine displacement linked with mutation-induced resistance. Proc Natl Acad Sci USA 102:13046–13051
Leartsakulpanich U, Imwong M, Pukrittayakamee S et al (2002) Molecular characterization of dihydrofolate reductase in relation to antifolate resistance in Plasmodium vivax. Mol Biochem Parasitol 119:63–73
Looareesuwan S, Viravan C, Webster HK et al (1996) Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am J Trop Med Hyg 54:62–66
Lozovsky ER, Chookajorn T, Brown KM et al (2009) Stepwise acquisition of pyrimethamine resistance in the malaria parasite. Proc Natl Acad Sci USA 106:12025–12030
Mendis K, Sina BJ, Marchesini P, Carter R (2001) The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg 64:97–106
Nair S, Miller B, Barends M et al (2008) Adaptive copy number evolution in malaria parasites. PLoS Genet 4:e1000243
Neafsey DE, Galinsky K, Jiang RHY et al (2012) The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet 44:1046–1050
Parekh F, Moorthy V (2011) Plasmodium vivax vaccine research: insights from Colombian studies. Am J Trop Med Hyg 84:1
Peng B, Kimmel M (2005) simuPOP: a forward-time population genetics simulation environment. Bioinformatics 21:3686–3687
Peterson DS, Walliker D, Wellems TE (1988) Evidence that a point mutation in dihydrofolate reductase–thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc Natl Acad Sci USA 85:9114–9118
Randrianasolo L, Randriamanantena A, Ranarivelo L et al (2004) Monitoring susceptibility to sulfadoxine–pyrimethamine among cases of uncomplicated, Plasmodium falciparum malaria in Saharevo, Madagascar. Ann Trop Med Parasitol 98:551–554
Rastelli G, Sirawaraporn W, Sompornpisut P et al (2000) Interaction of pyrimethamine, cycloguanil, WR99210 and their analogues with Plasmodium falciparum dihydrofolate reductase: structural basis of antifolate resistance. Bioorg Med Chem 8:1117–1128
Sibley CH, Hyde JE, Sims PF et al (2001) Pyrimethamine–sulfadoxine resistance in Plasmodium falciparum: what next? Trends Parasitol 17:582–588
Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27
Simpson JA, Watkins E, Price RN et al (2000) Mefloquine pharmacokinetic–pharmacodynamic models: implications for dosing and resistance. Antimicrob Agents Chemother 44:3414–3424
Sina B (2002) Focus on Plasmodium vivax. Trends Parasitol 18:287–289
Sirawaraporn W, Sathitkul T, Sirawaraporn R et al (1997) Antifolate-resistant mutants of Plasmodium falciparum dihydrofolate reductase. Proc Natl Acad Sci USA 94:1124–1129
Snewin VA, England SM, Sims PF, Hyde JE (1989) Characterisation of the dihydrofolate reductase–thymidylate synthetase gene from human malaria parasites highly resistant to pyrimethamine. Gene 76:41–52
Tjitra E, Baker J, Suprianto S et al (2002) Therapeutic efficacies of artesunate–sulfadoxine–pyrimethamine and chloroquine–sulfadoxine–pyrimethamine in vivax malaria pilot studies: relationship to Plasmodium vivax dhfr mutations. Antimicrob Agents Chemother 46:3947–3953
Tokuriki N, Jackson CJ, Afriat-Jurnou L, Wyganowski KT, Tang R, Tawfik DS (2012) Diminishing returns and tradeoffs constrain the laboratory optimization of an enzyme. Nat Commun 3:1257
van den Broek IVF, van der Wardt S, Talukder L et al (2004) Drug resistance in Plasmodium falciparum from the Chittagong Hill Tracts, Bangladesh. Trop Med Int Health 9:680–687
Wang X, Minasov G, Shoichet BK (2002) Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol 320:85–95
Weinreich DM, Delaney NF, Depristo MA, Hartl DL (2006) Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 312:111–114
Wellems TE (2002) Plasmodium chloroquine resistance and the search for a replacement antimalarial drug. Science 298:124–126
White NJ, Pongtavornpinyo W, Maude RJ et al (2009) Hyperparasitaemia and low dosing are an important source of anti-malarial drug resistance. Malar J 8:253–271
Wooden JM, Hartwell LH, Vasquez B, Sibley CH (1997) Analysis in yeast of antimalaria drugs that target the dihydrofolate reductase of Plasmodium falciparum. Mol Biochem Parasitol 85:25–40
Yuvaniyama J, Chitnumsub P, Kamchonwongpaisan S et al (2003) Insights into antifolate resistance from malarial DHFR-TS structures. Nat Struct Biol 10:357–365
Acknowledgments
This research was supported by FQEB Grant #RFP-12-09 to DLH from the John Templeton Foundation program on Foundational Questions in Evolutionary Biology (FQEB). We’d also like to thank Hsiao-Han Chang for her helpful insights during data analysis and write-up.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Jiang, PP., Corbett-Detig, R.B., Hartl, D.L. et al. Accessible Mutational Trajectories for the Evolution of Pyrimethamine Resistance in the Malaria Parasite Plasmodium vivax . J Mol Evol 77, 81–91 (2013). https://doi.org/10.1007/s00239-013-9582-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-013-9582-z