
Abstract
The phenotypes of biological systems are to some extent robust to genotypic changes. Such robustness exists on multiple levels of biological organization. We analyzed this robustness for two categories of amino acids in proteins. Specifically, we studied the codons of amino acids that bind or do not bind small molecular ligands. We asked to what extent codon changes caused by mutation or mistranslation may affect physicochemical amino acid properties or protein folding. We found that the codons of ligand-binding amino acids are on average more robust than those of non-binding amino acids. Because mistranslation is usually more frequent than mutation, we speculate that selection for error mitigation at the translational level stands behind this phenomenon. Our observations suggest that natural selection can affect the robustness of very small units of biological organization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Two computational approaches to characterize functionally important amino acids of a protein are widespread. The first focuses on the accessible surface area, which describes the accessibility of an amino acid by the solvent surrounding a protein (Lee and Richards 1971). Amino acids that are involved in binding ligands commonly occur in large and deep clefts on a protein’s surface with low accessible surface area, which may help to increase the specificity and stability of binding (Bartlett et al. 2002; Laskowski et al. 1996). The analysis of solvent accessibility requires detailed knowledge of ligand binding sites, which is limited to proteins with known ligand-bound structures. The second approach uses evolutionary conservation of amino acids (Capra et al. 2009; Lichtarge and Sowa 2002). For example, amino acids in catalytic sites of enzymes are more conserved on average (Bartlett et al. 2002). However, because evolutionary conservation is influenced by multiple factors, such as the divergence time between orthologs, the background rate of amino acid substitutions, and mutational biases (Sasidharan and Chothia 2007), information on conservation alone is not enough to characterize functional sites. Many studies thus combine these two approaches to improve the characterization of binding sites (Bartlett et al. 2002; Capra et al. 2009). Here we suggest a third, complementary approach that may help characterize specifically those amino acids that bind ligands. It focuses on their robustness to mutation or mistranslation. Because such amino acids are especially important for the function of a protein, they can be subject to selection increasing their robustness relative to non-ligand-binding amino acids.
Biological systems on multiple levels of organization are to some extent robust to genetic or environmental change. Examples include the genetic code of extant organisms, which is more robust to nucleotide changes than the vast majority of hypothetical alternative codes (Freeland and Hurst 1998); proteins, which can continue to function when many of their amino acids are mutated (Bowie et al. 1990; Guo et al. 2004; Huang et al. 1996; Loeb et al. 1989; Markiewicz et al. 1994; Suckow et al. 1996); gene regulatory circuits, whose phenotypes are to some extent robust to changes in regulatory interactions (Von Dassow and Odell 2002; Ingolia 2004; Isalan et al. 2005, 2008; Li et al. 2006); and genome-scale metabolic networks, which can tolerate deletions of multiple enzyme-coding genes without detectable phenotypic effects in standard laboratory environments (Hillenmeyer et al. 2008). Such robustness may reflect intrinsic system properties that may not have been shaped by natural selection. Alternatively, it may be the result of evolutionary adaptation, either to ameliorate the detrimental effects of DNA mutations, of environmental change, or of both.
Among the four principal ways in which random change in a codon can occur—DNA mutation, mistranscription, mRNA alteration, and mistranslation—we focus on mutation and the mistranslation of mRNA, which are well-documented and probably most frequent. Such mistranslation occurs when a ribosome incorporates incorrect amino acids when synthesizing a protein from an mRNA template. There are at least three non-exclusive classes of evolutionary mechanisms by which the cost of mistranslation can be minimized. The first is selection of translational accuracy. Akashi (1994) suggested that such selection causes genes or specific sites in genes to be encoded by codons that correspond to abundant tRNAs. Such high fidelity codons have higher chances of being accurately translated. The second is selection of translational robustness, which has been proposed by Drummond and Wilke (2008, 2006). According to these authors, proteins (and especially highly expressed proteins) show evolved tolerance in their fold to missense translational errors (Zhou et al. 2009). The third involves error mitigation. Among those synonymous codons that encode the same amino acid, some are more robust to changes in individual nucleotides than others. That is, even though a random change in a robust codon may change the encoded amino acid, the new amino acid has, on average, similar physicochemical properties or does not perturb protein folding strongly Archetti (2006, 2004). In error mitigation, codons that are likely to be mistranslated into radically different amino acids are avoided. Previous studies (Archetti 2004; Najafabadi et al. 2007) showed that amino acids in eukaryotic and prokaryotic proteins are often encoded by codons whose mistranslation leads to the substitution of amino acids with limited deleterious effects. The most important of these three causes for our work is error mitigation.
Here we ask whether selection helps shape the codon usage of ligand-binding amino acids. To this end, we analyze the robustness of codons to mutation or mistranslation for two classes of codons in a protein, that is, codons that encode amino acids which are or are not involved in the binding of a small molecular ligand. We use an estimator of robustness that incorporates the likely effects of an amino acid change on the physicochemical properties of an amino acid, and on protein folding. Our analysis shows that ligand-binding amino acids are on average more robust to mutation or mistranslation than non-binding amino acids, which is consistent with selection pressure for error mitigation.
Methods
We use a codon robustness score φ(c) derived from the weighted average load function of Ardell (1998). This score aims to capture the predicted effect that a particular amino acid change has on the folding free energy of a protein and on physicochemical amino acid properties. Specifically,
where summation is applied over all nine 1-mutant neighbors of a codon c. In this expression, p(c’|c) is the probability of changing a codon c for another codon c’, which is computed by multiplying the position-specific transition-transversion bias of mistranslations with the relative mistranslation frequency of a given nucleotide position. \( g\left[ {a\left( c \right),a\left( {c^{'} } \right)} \right] \) is the physicochemical effect or “cost” of substituting the amino acid encoded by codon c, a(c), with that encoded by codon c’, a(c’). We used the mutation matrix generated by Gilis and colleagues (2001), to calculate the cost of such an amino acid change (see Online Resource 1a for the matrix). This matrix uses information on changes in folding free energy and physiochemical properties of amino acid features, such as hydrophobicity, after an amino acid change. We note that the use of different substitution matrices would not strongly affect codon robustness scores (Najafabadi et al. 2005).
To give an example of how we calculated robustness scores, consider the codon tta that encodes leucine. It has nine 1-mutant neighbors, one of them being tca. Mistranslation from tta to tca corresponds to a transition at a nucleotide in the second position of a codon. With a relative mistranslation frequency of second position nucleotides of 0.1 and a transition transversion bias at second position nucleotides of 5, we computed a value of p(tca|tta) = 0.5, by multiplying these numbers. Because tca encodes serine, we multiply this value by −1, which is the cost of mutation from leucine to serine based on the mutation matrix. We perform an exactly analogous calculation for all other 1-mutant neighbors of tta to arrive at \( \varphi \left( {tta} \right) \). We then normalize this score by dividing it by the mean codon robustness of all leucine-encoding codons to eliminate the possible effects of amino acid biases. Finally, we normalize the scores of all codons to the interval (0,1). The resulting scores are shown in Online Resource 2.
We used proteins in our analysis (Online Resource 3) that (i) have a reviewed (non-putative) 3D structure deposited in the protein data bank (PDB) (Bourne et al. 2004), (ii) exert their biological function as monomers, and (iii) bind to one of the small ligands in Online Resource 4. Binding to large molecules, such as other proteins, RNA and DNA involve highly divergent interaction types, and large interface areas, which might decrease functional importance of amino acids that contact a molecule (Lichtarge and Sowa 2002). As Clackson and Wells (1995) showed, only a fraction of those residues actually contribute to binding. We therefore excluded those larger molecules. From this data set, we eliminated proteins that bind to multiple ligands, as well as proteins with more than 90 % sequence identity to other proteins, thus arriving at a final data set of 275 proteins. We extracted a protein’s coding exons by aligning the encoding gene (obtained from NCBI (Benson et al. 2004)) and the amino acid sequence (Bourne et al. 2004) with the tool Exonerate (Slater and Birney 2005).
Results and Discussion
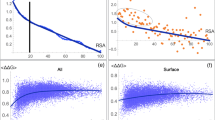
We subdivided all amino acids of the proteins in our data set into two categories, those not involved in the binding of small ligands, and those involved in the binding of small ligands, which we defined to be lying within a 5 Ångstrom radius of a ligand in the published tertiary structure. We then computed the robustness scores of codons using a wide range of mistranslation parameters. We varied two key parameters at each nucleotide position, the transition-transversion bias for which we used five different values between one and five, and the mistranslation frequency, for which we used ten different values between 0.1 and 1. We then asked for each of 50 different parameter combinations whether robustness scores encoding the binding and the non-binding amino acids differ. We found that codons encoding ligand-binding amino acids are significantly more robust in all cases, with either the same or very similar P values (greatest P < 10−30, smallest P < 10−36, Wilcoxon Rank Sum test used throughout, unless otherwise mentioned). For the sake of simplicity, we thus used one particular parameter combination for all subsequent analyses, which is that of Freeland and Hurst (1998, see Online Resource 1b). Even though it may not be universally accurate (Kramer et al. 2010), it has also been employed by several other studies similar to ours and on a wide range of organisms (Archetti 2004, 2006; Drummond and Wilke 2008; Najafabadi et al. 2007). Figure 1a indicates the distribution of robustness scores based on these mistranslation biases (P < 10−35, thick horizontal lines indicate medians).

Box-plot of a robustness, b accessible surface area (ASA) and c amino acid conservation scores. Thick black horizontal lines in the middle of each box mark the median. The edges of the boxes correspond to the 25th and 75th percentiles. Data is based on a sample of n = 49,133 non-binding amino acids (left box in each panel) and of n = 5,552 ligand-binding amino acids (right box in each panel)
We then repeated our analysis using an estimator of codon robustness by Archetti, which takes only the physicochemical effects of changed amino acids into account (Clackson and Wells 1995), but not the likely effect on protein misfolding, as does our estimator. Again, ligand-binding amino acids are significantly more robust when using this estimator (P < 10−8). We also compared the Z-statistic, which is the standardized value of U, the Wilcoxon ranked sum statistic. For large samples like ours, U is normally distributed (Rice 1995), and thus Z follows a standard-normal (N(0,1)) distribution. We found that the Z-statistic is much greater for our own robustness estimator (Z = 12.38) than for Archetti’s estimator (Z = 3.77). This means that taking effects on misfolding into account, binding and non-binding amino acids differ to a much greater extent in their robustness.
We next compared differences in codon robustness to differences between more conventional indicators of functionally important binding amino acids. The first of them is the accessible surface area. We obtained the accessible surface area scores of each amino acid from (Kabsch and Sander 1983) and normalized them, so that they range between 0 and 1. As previous studies did (Bartlett et al. 2002; Laskowski et al. 1996), we found that ligand-binding amino acids indeed have significantly smaller accessible surface area (P < 10−27) (Fig. 1b). The difference becomes more significant (P < 10−300), when we remove the residues in the hydrophobic core, that is the residues with normalized accessible surface area values less than 0.25 from the analysis.
The second indicator is the extent of evolutionary conservation. We compared amino acid conservation scores (obtained from (Goldenberg et al. 2009)) for ligand-binding and non-binding amino acids in our data set. In line with previous studies (Bartlett et al. 2002; Capra et al. 2009), we found that the binding amino acids are significantly more conserved (P < 10−258) (Fig. 1c). We next asked whether codon robustness discriminates to a similar extent between binding and non-binding amino acids as do these two quantities. To this end, we examined again the Z-statistic of the Wilcoxon test, and found that evolutionary conservation differs most between binding and non-binding amino acids (Z = 34.35), the accessible surface area differs least (Z = 10.89) and codon robustness lies in between them (Z = 12.38). These observations suggest that robustness, while not as informative as evolutionary conservation, may have similar value as accessible surface area to characterize functionally important amino acids. Finally, we calculated the association of codon robustness with the other two indicators, and found that neither accessible surface area (r 2 = 0.15, P < 10−300) nor evolutionary conservation (r 2 = −0.06, P < 10−48) are strongly correlated with robustness. These weak correlations suggest that robustness is complementary to the two other two quantities in characterizing ligand-binding amino acids.
We next asked whether different genes also differ in the robustness scores of the ligand-binding amino acids they encode. To this end, we ranked proteins according to the mean robustness score and displayed the corresponding data as a rank plot (Fig. 2a). Specifically, the plot shows the rank-ordered codon robustness scores of ligand-binding amino acids (orange dots), together with the robustness of the non-binding amino acids (grey dots). For 63 percent or 173 proteins, the mean codon robustness of the binding amino acids was greater than the mean codon robustness of the non-binding amino acids, where the mean is taken over all non-binding amino acids in all proteins. A minority of 37 percent of proteins had a lower robustness of binding amino acids than those of non-binding amino acids. To investigate these differences in robustness further, we focused on two classes of proteins, the 28 proteins in the bottom 10th-percentile (blue shading in Fig. 2a), and the 28 proteins in the top 10th-percentile (pink shading in Fig. 2a). We refer to them as the proteins with the lowest and highest robustness of ligand-binding amino acids.

a Plot of mean robustness scores, ranked based on the mean robustness of ligand-binding amino acids for each of our 275 study proteins. Grey dots correspond to mean robustness scores of non-binding amino acids, orange dots correspond to mean robustness scores of ligand binding amino acids. The pink and blue regions correspond to the upper and lower 10th percentiles in robustness, respectively. b Box-plot of RNA expression levels in the brain for the proteins with lowest (left box) and highest (right box) robustness of ligand-binding amino acids. The edges of the boxes correspond to the 25th and 75th percentiles. Data is based on a sample of gene expression values n = 168 for the left box, n = 56 for the right box. c Gene ontology (Ashburner et al. 2000) functional annotations of proteins with lowest (left bar) and highest (right bar) robustness of ligand-binding amino acids. Legend displays the major functional classes presented in the bars
We first asked whether the genes encoding these two classes of proteins differ substantially in their expression. To this end, we used a gene expression data set (Brawand et al. 2011) from six humans and five different organs (brain, heart, kidney, liver and testis), obtained through high throughput RNA sequencing (RNA seq). The genes in the highest robustness category did not show significantly higher expression when we analyzed pooled data from all organs, nor when we analyzed data from four of the five organs. The only exception were expression data from the brain, where these proteins were significantly more highly expressed (P = 0.029) (Fig. 2b). Although the signal becomes insignificant after a correction for false discovery rate (FDR, Benjamini and Hochberg 1995), this pattern is consistent with an earlier analysis (Drummond and Wilke 2008), which showed that selection for translationally robust codons is strongest in brain and other neural tissues. The likely reason is the extreme sensitivity of neuronal functions to protein misfolding and dysfunction, which is associated with neurodegenerative diseases and neurotoxic effects (Bucciantini et al. 2002; Lee et al. 2006). Using the gene ontology (Ashburner et al. 2000) classification of gene functions, we also found that proteins with highest robustness are significantly more enriched in functions related to development, differentiation (Exact Binomial Test, P < 10−3), whereas proteins with lowest robustness are significantly more enriched in metabolic functions (Fig. 2c). In sum, these analyses reveal differences between proteins whose ligand-binding amino acids differ most in their robustness, although they fall short of explaining the low robustness we observe for these amino acids in some proteins.
That functionally or structurally important amino acids or codons are subject to special constraints has been proposed by previous work and in contexts different from ours. First, Bartlett and colleagues provided evidence that catalytic sites harbor certain classes of amino acids. Specifically, charged amino acids are more often found in catalytic sites, whereas hydrophobic amino acids are more often found in the structure-stabilizing hydrophobic core (Bartlett et al. 2002). Second, Pakula and Sauer (1989) showed that such sites are highly constrained in the substitutions they can tolerate. Third, Zhou and colleagues (2009) provided evidence in several eukaryotes and prokaryotes that some parts of proteins are more sensitive to misfolding, and show a more constrained codon usage, the phenomenon that different synonymous codons for the same amino acid are not used equally frequently in protein coding genes (Akashi 1994; Akashi and Eyre-Walker 1998; Comeron and Aguadé 1998; Duret 2002; Gouy and Gautier 1982; Ikemura 1981; 1985; Moriyama and Hartl 1993; Plotkin et al. 2004; Sharp et al. 1986; Sharp and Li 1987; Stoletzki and Eyre-Walker 2007). In general, the strength of this bias varies within genes and becomes stronger at functionally important sites (Akashi 1994; Stoletzki and Eyre-Walker 2007). In sum, our observations that robust codons are favored at ligand-binding amino acids are consistent with a broad range of related evidence.
Limitations of our analysis include the moderate number of 275 proteins we could study, as well as a small number of binding amino acids (18 on average) per protein, which renders rigorous statistical analysis of individual proteins infeasible. Despite these limitations, our joint analysis of multiple proteins showed a significant preference of robust codons in ligand-binding pockets of proteins, exactly where amino acid changes can have a highly detrimental effect on protein function.
Another limitation is that codon robustness alone-like accessible surface area and conservation—does not have much power to predict ligand binding sites. To predict such sites, more complex models incorporating multiple characterizing elements are necessary (see for example, Capra et al. 2009; Lichtarge and Sowa 2002; Wass et al. 2011). Because codon robustness differs more than accessible surface area between ligand binding and non-binding amino acids, our approach can help improve such models and their predictive power.
Selection may have favored robust codons in ligand-binding amino acids because they are robust to mutation or to translation. Although mistranslation is not genetic change –it leaves the DNA encoding a mRNA unchanged—it does alter the encoded protein randomly (Drummond and Wilke 2009). Translational error rates in microbes have been estimated at 10−3–10−4 per codon. This number is at least five orders of magnitude higher than typical mutation rates (Kramer et al. 2010; Kramer and Farabaugh 2007; Ogle and Ramakrishnan 2005). At this error rate, 15 percent of protein molecules would contain at least one mistranslated amino acid. Translation errors can induce protein misfolding, aggregation, toxicity and cell death, which underlie a broad array of neurodegenerative diseases (Bucciantini et al. 2002; Lee et al. 2006). Also, mistranslation at functionally important sites can disrupt protein function (Guo et al. 2004; Markiewicz et al. 1994). For these reasons, we speculate that selection for error mitigation at the translational level is the prevalent driving force of high robustness in codons that encode ligand-binding amino acids. Why a minority of ligand-binding amino acids has especially low codon robustness remains an open question for future work.
References
Akashi H (1994) Synonymous codon usage in Drosophila Melanogaster: natural selection and translational accuracy. Genetics 136:927–935
Akashi H, Eyre-Walker A (1998) Translational selection and molecular evolution. Curr Opin Genet Dev 8:688–693
Archetti M (2004) Selection on codon usage for error minimization at the protein level. J Mol Evol 59:400–415. doi:10.1007/s00239-004-2634-7
Archetti M (2006) Genetic robustness and selection at the protein level for synonymous codons. J Evol Biol 19:353–365. doi:10.1111/j.1420-9101.2005.01029.x
Ardell DH (1998) On error minimization in a sequential origin of the standard genetic code. J Mol Evol 47:1–13
Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25–29. doi:10.1038/75556
Bartlett GJ, Porter CT, Borkakoti N, Thornton JM (2002) Analysis of catalytic residues in enzyme active sites. J Mol Biol 324:105–121
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 57:289–300. doi:10.2307/2346101
Benson DA, Karsch-Mizrachi I, Lipman DJ et al (2004) GenBank: update. Nucleic Acids Res 32:D23–D26
Bourne PE, Addess KJ, Bluhm WF et al (2004) The distribution and query systems of the RCSB protein data bank. Nucleic Acids Res 32:D223–D225
Bowie JU, Reidhaar-Olson JF, Lim WA, Sauer RT (1990) Deciphering the message in protein sequences: tolerance to amino acid substitutions. Science 247:1306–1310
Brawand D, Soumillon M, Necsulea A et al (2011) The evolution of gene expression levels in mammalian organs. Nature 478:343–348. doi:10.1038/nature10532
Bucciantini M, Giannoni E, Chiti F et al (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416:507–511
Capra JA, Laskowski RA, Thornton JM et al (2009) Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput Biol 5:18
Clackson T, Wells JA (1995) A hot spot of binding energy in a hormone-receptor interface. Science 267:383–386
Comeron JM, Aguadé M (1998) An evaluation of measures of synonymous codon usage bias. J Mol Evol 47:268–274
Drummond DA, Wilke CO (2008) Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134:341–352. doi:10.1016/j.cell.2008.05.042
Drummond DA, Wilke CO (2009) The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet 10:715–724
Duret L (2002) Evolution of synonymous codon usage in metazoans. Curr Opin Genet Dev 12:640–649
Freeland SJ, Hurst LD (1998) The genetic code is one in a million. J Mol Evol 47:238–248
Gilis D, Massar S, Cerf NJ, Rooman M (2001) Optimality of the genetic code with respect to protein stability and amino-acid frequencies. Genome Biol 2: RESEARCH0049
Goldenberg O, Erez E, Nimrod G, Ben-Tal N (2009) The ConSurf-DB: pre-calculated evolutionary conservation profiles of protein structures. Nucleic Acids Res 37:D323–D327
Gouy M, Gautier C (1982) Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res 10:7055–7074. doi:10.1093/nar/gkn1031
Guo HH, Choe J, Loeb LA (2004) Protein tolerance to random amino acid change. Proc Natl Acad Sci USA 101:9205–9210
Hillenmeyer ME, Fung E, Wildenhain J et al (2008) The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science (New York, NY) 320:362–365. doi:10.1126/science.1150021
Huang W, Petrosino J, Hirsch M et al (1996) Amino acid sequence determinants of beta-lactamase structure and activity. J Mol Biol 258:688–703
Ikemura T (1981) Correlation between the abundance of E. coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J Mol Biol 151:389–409
Ikemura T (1985) Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol 2:13–34
Ingolia NT (2004) Topology and Robustness in the Drosophila Segment polarity network. PLoS Biol 2:e123
Isalan M, Lemerle C, Serrano L (2005) Engineering gene networks to emulate Drosophila Embryonic pattern formation. PLoS Biol 3:e64
Isalan M, Lemerle C, Michalodimitrakis K et al (2008) Evolvability and hierarchy in rewired bacterial gene networks. Nature 452:840–845
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577–2637
Kramer EB, Farabaugh PJ (2007) The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA Ny 13:87–96
Kramer EB, Vallabhaneni H, Mayer LM, Farabaugh PJ (2010) A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA NY 16:1797–1808
Laskowski RA, Luscombe NM, Swindells MB, Thornton JM (1996) Protein clefts in molecular recognition and function. Protein Sci 5:2438–2452
Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55:379–400
Lee JW, Beebe K, Nangle LA et al (2006) Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 443:50–55
Li S, Assmann SM, Albert R (2006) Predicting essential components of signal transduction networks: a dynamic model of guard cell abscisic acid signaling. PLoS Biol 4:17
Lichtarge O, Sowa ME (2002) Evolutionary predictions of binding surfaces and interactions. Curr Opin Struct Biol 12:21–27
Loeb DD, Swanstrom R, Everitt L et al (1989) Complete mutagenesis of the HIV-1 protease. Nature 340:397–400
Markiewicz P, Kleina LG, Cruz C et al (1994) Genetic studies of the lac repressor. XIV. Analysis of 4000 altered E. coli lac repressors reveals essential and non-essential residues, as well as “spacers” which do not require a specific sequence. J Mol Biol 240:421–433
Moriyama EN, Hartl DL (1993) Codon usage bias and base composition of nuclear genes in Drosophila. Genetics 134:847–858
Najafabadi HS, Goodarzi H, Torabi N (2005) Optimality of codon usage in E. coli due to load minimization. J Theor Biol 237:203–209
Najafabadi HS, Lehmann J, Omidi M (2007) Error minimization explains the codon usage of highly expressed genes in E. coli. Gene 387:150–155
Ogle JM, Ramakrishnan V (2005) Structural insights into translational fidelity. Annu Rev Biochem 74:129–177
Pakula AA, Sauer RT (1989) Genetic analysis of protein stability and function. Annu Rev Genet 23:289–310
Plotkin JB, Robins H, Levine AJ (2004) Tissue-specific codon usage and the expression of human genes. Proc Natl Acad Sci USA 101:12588–12591
Rice JA (1995) Mathematical statistics and data analysis. High Educ 72:330. doi:10.2307/3619963
Sasidharan R, Chothia C (2007) The selection of acceptable protein mutations. Proc Natl Acad Sci USA 104:10080–10085
Sharp PM, Li WH (1987) The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15:1281–1295
Sharp PM, Tuohy TM, Mosurski KR (1986) Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res 14:5125–5143
Slater GSC, Birney E (2005) Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31
Stoletzki N, Eyre-Walker A (2007) Synonymous codon usage in E. coli: selection for translational accuracy. Mol Biol Evol 24:374–381. doi:10.1093/molbev/msl166
Suckow J, Markiewicz P, Kleina LG et al (1996) Genetic studies of the Lac repressor. XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J Mol Biol 261:509–523
Von Dassow G, Odell GM (2002) Design and constraints of the Drosophila segment polarity module: robust spatial patterning emerges from intertwined cell state switches. J Exp Zool 294:179–215
Wass MN, David A, Sternberg MJE (2011) Challenges for the prediction of macromolecular interactions. Curr Opin Struct Biol 21:382–390
Wilke CO, Drummond DA (2006) Population genetics of translational robustness. Genetics 173:473–481
Zhou T, Weems M, Wilke CO (2009) Translationally optimal codons associate with structurally sensitive sites in proteins. Mol Biol Evol 26:1571–1580. doi:10.1093/molbev/msp070
Acknowledgments
AW and TB acknowledge support through Swiss National Science Foundation grants 315230-129708, as well as through the YeastX project of SystemsX.ch, and the University Priority Research Program in Systems Biology at the University of Zurich. TB acknowledges support through TUBITAK BIDEB 2209 and thanks Turkan Haliloglu and Niv Sabath for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Bilgin, T., Kurnaz, I.A. & Wagner, A. Selection Shapes the Robustness of Ligand-Binding Amino Acids. J Mol Evol 76, 343–349 (2013). https://doi.org/10.1007/s00239-013-9564-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-013-9564-1