
Abstract
In this study, we report for the first time a detailed evaluation of the phylogenetic history and molecular evolution of the major coleoid toxins: CAP, carboxypeptidase, chitinase, metalloprotease GON-domain, hyaluronidase, pacifastin, PLA2, SE-cephalotoxin and serine proteases, with the carboxypeptidase and GON-domain documented for the first time in the coleoid venom arsenal. We show that although a majority of sites in these coleoid venom-encoding genes have evolved under the regime of negative selection, a very small proportion of sites are influenced by the transient selection pressures. Moreover, nearly 70 % of these episodically adapted sites are confined to the molecular surface, highlighting the importance of variation of the toxin surface chemistry. Coleoid venoms were revealed to be as complex as other venoms that have traditionally been the recipient of the bulk of research efforts. The presence of multiple peptide/protein types in coleoids similar to those present in other animal venoms identifies a convergent strategy, revealing new information as to what characteristics make a peptide/protein type amenable for recruitment into chemical arsenals. Coleoid venoms have significant potential not only for understanding fundamental aspects of venom evolution but also as an untapped source of novel toxins for use in drug design and discovery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Venoms are key evolutionary innovations under-pinning the explosive radiation of many clades. Research to date has been heavily taxonomically biased, with cone snails, scorpions, snakes and spiders receiving a disproportionate amount of attention. The cephalopods are a conspicuously neglected area of venom research. The group as a whole has been the subject of scant research and even so, strongly biased towards the octopuses, with little attention devoted to other coleoids such as cuttlefish and squid. This narrow taxonomical view complicates the further study of venom molecular evolution (Fry et al. 2003). Recently, we have shown that octopus and cuttlefish share a common, venomous ancestor (Fry et al. 2009), confirming that cephalopods add new protein scaffolds to their arsenals via the duplication of body regulatory proteins and subsequent selective overexpression in the venom gland (Fry 2005; Fry et al. 2009), yet the number of species examined in detail remains low.
Toxicity of octopus saliva from the posterior pair of salivary glands to invertebrates was established as early as 1888 (Lo Bianco 1888). Ghiretti subsequently succeeded in isolating the crab toxic fraction from Sepia officinalis, Octopus vulgaris and Octopus macropus, which he termed cephalotoxin (Ghiretti 1959 1960). Ghiretti also notes that the initial hyper excitability observed in crabs is due to the amides present in coleoid venom, whilst the lethal phase was a result of cephalotoxin. Cariello and Zanetti (1977) isolated five proteins toxic to crabs from O. vulgaris PSG homogenate. However due to impurities in their samples only two components were further described: alpha and beta cephalotoxin. Both were found to consist of approximately 50 % carbohydrate, indicating heavy glycosylation. Tachykinins such as Eledoisin (Anastasi and Erspamer 1962), isolated from Eledone aldrovandi and Eledone moschata, OctTK 1 and 2 isolated from O. vulgaris (Kanda et al. 2003) and an OctTK 1 homologue from Octopus kaurna (Fry et al. 2009) have been isolated from octopus. SE-cephalotoxin, was described by Udea et al. (2008). Fry et al. (2009) described six novel putative toxins with no homology to any known peptide type, in addition to CriSP, and phospholipase A2, proteins from Hapalochlaena maculosa, O. kaurna and Sepia latimanus (Fry et al. 2009). Enzymes also play key roles in coleoid toxicity in addition to the small organic molecules, peptides, and non-enzymatic proteins. Indeed, large amounts of S1 peptidase gene transcripts from both H. maculosa and O. kaurna have been recently identified (Fry et al. 2009). Other studies also associate proteolytic activity with PSG extract of both Eledone cirrhosa (Grisley 1993; Grisley and Boyle 1987) and O. vulgaris (Morishita 1974). Hyaluronidase and Chitinase have also been identified from octopus venom (Fry et al. 2009; Grisley and Boyle 1990; Romanini 1952). Further evidence of functional diversification is that enzymes from the venoms of octopus species living in Antarctica have sub-zero temperature optimum efficiency, with decreased activity at higher temperatures (Undheim et al. 2010).
Whilst some toxic secretions of coleoids have been previously investigated, the literature to date shows high toxin diversity and offers little insight into the evolutionary aspects of coleoid toxicity. This leaves questions regarding the genetic origin and the strategies of venom recruitment of coleoid toxins entirely up to speculation. In this study, we present for the first time a glimpse into the coleoid venom phylogenetic history and molecular evolution. We report a particularly detailed evaluation of the major coleoid toxins (PLA2, CAP, pacifastin, and serine proteases) and highlight the prominent role of episodic diversifying selections in shaping some of these toxins.
Materials and Methods
Tissue Sampling and Taxon Selection
Posterior secretory glands were dissected from freshly euthanized specimens collected from tropical to polar waters, thus providing wide taxiconomical and ecological coverage: cuttlefish species included were S. latimanus (Osprey Reef, Coral Sea) and Sepia pharaonis (Hong Kong); octopus species include Abdopus aculeatus (Lizard Island, Queensland, Australia), Adeleiledone polymorpha [East Antarctica coastal waters off of George V’s Land (1398E to 1458E)], H. maculosa (Mornington Peninsula, Victoria Australia), O. cyanea (Lizard Island, Queensland, Australia), O. kaurna (Mornington Peninsula, Victoria, Australia) and Pareledone turqueti [East Antarctica coastal waters off of George V’s Land (1398E to 1458E)]; squid species included Loliolus noctiluca (Moreton Bay, Queensland, Australia) and Sepioteuthis australis (Moreton Bay, Queensland, Australia).
cDNA Library Construction and Analysis
Total RNA extracted using the standard TRIzol Plus method (Invitrogen). Extracts were enriched for mRNA using standard RNeasy mRNA mini kit (Qiagen) protocol. mRNA was reverse transcribed, fragmented, and ligated to a unique 10-base multiplex identifier (MID) tag prepared using standard protocols and applied to one PicoTitrePlate (PTP) for simultaneous amplification and sequencing on a Roche 454 GS FLX+ Titanium platform (Australian Genome Research Facility). 50,000 sequences for each sample were read. Automated grouping and analysis of sample-specific MID reads informatically separated sequences from the other transcriptomes on the plates, which were then post-processed to remove low quality sequences before de novo assembly into contiguous sequences (contigs) using v 3.4.0.1 of the MIRA software program. Assembled contigs were processed using CLC Main Work Bench (CLC-Bio) and Blast2GO bioinformatic suite (Gotz et al. 2011, 2008) to provide Gene Ontology, BLAST and domain/Interpro annotation. The above analyses assisted in the rationalisation of the large numbers of assembled contigs into phylogenetic ‘groups’ for the detailed phylogenetic analyses outlined below.
Sequences are available from Genbank with the Bioproject and Biosample retrieval numbers of: A. aculeatus PRJNA188569 SAMN01911391, A. polymorpha PRJNA188570 SAMN01911392, H. maculosa PRJNA188571 SAMN01911430, L. noctiluca PRJNA188572 SAMN01911444, O. cyanea PRJNA188574 SAMN01911445, O. kaurna PRJNA188658 SAMN01911449, P. turqueti PRJNA188575 SAMN01911446, S. latimanus PRJNA188659 SAMN01911447, S. pharaonis PRJNA188577 SAMN01911448 and S. australis PRJNA188576 SAMN01911450.
Bioinformatics
Phylogenetics
Phylogenetic analyses of the bioinformatically recovered transcripts were performed to allow reconstruction of the molecular evolutionary history of each toxin type. Toxin sequences were identified by comparison of the translated DNA sequences with previously characterised toxins using a BLAST search (Altschul et al. 1997) of the UniProtKB protein database. Molecular phylogenetic analyses of toxin transcripts were conducted using the translated amino acid sequences. Comparative sequences from physiological gene homologues identified from non-venom tissues were included in each dataset as outgroup sequences. To minimize confusion, all sequences obtained in this study are referred by their Genbank accession numbers (http://www.ncbi.nlm.nih.gov/sites/entrez?db=Nucleotide) and sequences from previous studies are labelled with their UniProtKB accession numbers (http://www.expasy.org/cgi-bin/sprot-search-ful). Resultant sequence sets were aligned using CLC Mainbench. When presented as sequence alignments, the leader sequence (as identified through use of SignalP) is shown in lowercase and cysteines are highlighted in black. > and < indicate incomplete N/5′ or C/3′ ends, respectively. Datasets were analysed using Bayesian inference implemented on MrBayes, version 3.0b4 (Ronquist and Huelsenbeck 2003). Two different run conditions were used to test for congruence: lset rates = invgamma with prset aamodelpr = fixed (WAG) and lset rates = gamma with prset aamodelpr = mixed. The analysis was performed by running a minimum of 1 × 107 generations in four chains, and saving every 100th tree. The log-likelihood score of each saved tree was plotted against the number of generations to establish the point at which the log-likelihood scores reached their asymptote, and the posterior probabilities for clades established by constructing a majority-rule consensus tree for all trees generated after completion of the burn-in phase. Trees shown are invgamma with WAG, which are identical in topology to gamma with mixed.
Test for Recombination
To overcome the effects of recombination on phylogenetic and evolutionary interpretations (Posada and Crandall 2002), we employed GARD and Single Breakpoint algorithms implemented in the HyPhy package and assessed recombination on all the toxin forms examined in this study (Delport et al. 2010; Kosakovsky Pond et al. 2006). When potential breakpoints were detected using the small sample Akaike information criterion (AICc), the sequences were compartmentalized before conducting the selection analyses.
Selection Analysis
We evaluated selection pressures using maximum-likelihood models (Goldman and Yang 1994; Yang 1998) implemented in CODEML of the PAML (Yang 2007). We first employed the one-ratio model that assumes a single ω for the entire phylogenetic tree. This model tends to be very conservative and can only detect positive selection if the ω ratio averaged over all the sites along the lineage is significantly >1. Because such lineage-specific models assume a single ω for the entire tree, they often fail to identify regions in proteins that might be affected by episodic selection pressures and ultimately, underestimate the strength of selection. Hence, we employed site-specific models which estimate positive selection statistically as a non-synonymous-to-synonymous nucleotide-substitution rate ratio (ω) significantly >1. We compared likelihood values for three pairs of models with different assumed ω distributions as no a priori expectation exists for the same: M0 (constant ω rates across all sites) versus M3 (allows the ω to vary across sites within ‘n’ discrete categories, n ≥ 3); M1a (a model of neutral evolution) where all sites are assumed to be either under negative (ω < 1) or neutral selection (ω = 1) versus M2a (a model of positive selection) which in addition to the site classes mentioned for M1a, assumes a third category of sites; sites with ω > 1 (positive selection) and M7 (Beta) versus M8 (Beta and ω), and models that mirror the evolutionary constraints of M1 and M2 but assume that ω values are drawn from a beta distribution (Nielsen and Yang 1998). Only if the alternative models (M3, M2a and M8: allow sites with ω > 1) show a better fit in likelihood ratio test (LRT) relative to their null models (M0, M1a and M8: do not show allow sites ω > 1), are their results considered significant. LRT is estimated as twice the difference in maximum-likelihood values between nested models and compared with the c2 distribution with the appropriate degree of freedom—the difference in the number of parameters between the two models. The Bayes empirical Bayes (BEB) approach (Yang et al. 2005) was used to identify amino acids under positive selection by calculating the posterior probabilities that a particular amino acid belongs to a given selection class (neutral, conserved or highly variable). Sites with greater posterior probability (PP ≥ 95 %) of belonging to the ‘ω > 1 class’ were inferred to be positively selected.
Single Likelihood Ancestor Counting (SLAC), fixed-effects likelihood (FEL) and random-effects likelihood models (Kosakovsky Pond et al. 2005) implemented in HyPhy (Kosakovsky Pond et al. 2005) were employed to provide additional support to the aforementioned analyses and to detect sites evolving under the influence of positive and negative selection. Mixed Effects Model Evolution (MEME) (Kosakovsky Pond et al. 2011) was also used to detect episodic diversifying selection. Since, the three domains of the coleoid pacifastin gene are expressed as multiple products, we also assessed the selection pressures influencing them simultaneously using Mgene (4) and option G test (Yang 1996) from Codeml. Further support for the results of the selection analyses was obtained using a complementary protein-level approach implemented in TreeSAAP (Woolley et al. 2003).
Structural Analyses
In order to depict the selection pressures influencing the evolution of venom components, we mapped the sites under positive selection on the homology models created using Phyre 2 webserver (Kelley and Sternberg 2009). Pymol 1.3 (DeLano 2002) was used to visualize and generate the images of homology models. Consurf webserver (Armon et al. 2001) was used for mapping the evolutionary selection pressures on the three-dimensional homology models. GETAREA (Fraczkiewicz and Braun 1998) was used to calculate the Accessible Surface Area (ASA)/solvent exposure of amino acid side chains. It uses the atom co-ordinates of the PDB file and indicates if a residue is buried or exposed to the surrounding medium by comparing the ratio between side-chain ASA and the ‘random coil’ values per residue. An amino acid is considered to be buried if it has an ASA <20 % and exposed if the ASA is more than or equal to 50 %.
Results
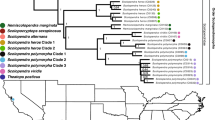
Analysis of coleoid posterior gland cDNA libraries, recovered sequences previously known from one or more coleoid lineages (Fry et al. 2009): CAP (CRiSP/Allergein/PR-1 protein family) (previously sequenced only from cuttlefish and octopus), chitinase (previously sequenced only from octopus), hyaluronidase (known only from its activity in octopus venom but never sequenced), PLA2 (previously sequenced only from cuttlefish), SE-cephalotoxin (previously sequenced only cuttlefish) and serine protease (previously sequenced only from cuttlefish and octopus) (Table 1). The phylogenetic history of these toxin types was previously unclear due to limited taxonomical sampling that had been done in earlier studies. Serine protease was shown to be present in the common coleoid ancestor whilst PLA2 was recovered only from decapodiforme lineages (cuttlefish and squid). In contrast, chitinase and hyaluronidase transcripts were only identified in octopodiformes. For each protein type, our phylogenetic analyses resolved all coleoid venom gland sequences into a monophyletic group to the exclusion of non-venom gland related sequences, thus demonstrating a shared history of the venom gland forms.
Variable degree of sequence conservation was evident between the different protein types. CAP cysteines were highly conserved whilst the prolines and charged residues were more variable. However, octopus CAP had a basic pI (~8.44) whilst squid and cuttlefish were acidic to neutral/slightly-acidic pI (5.72–7.40) (calculated using the Expasy pI/MW online service). The globular enzymatic hyaluronidase showed the least variation of all the recovered sequences. Like other type III PLA2, the coleoid sequences have a lengthy propeptide region, followed by a 10 cysteine arrangement in the processed final form (Fig. 1) The squid sequences, however, lacked the 9th ancestral cysteine, resulting in an odd number of cysteines, which may promote dimerization. The serine peptidase transcripts recovered were the most diverse and numerous of all toxin types recovered (Fig. 2). There is evidence for at least six gene duplication events prior to the divergence of octopus, cuttlefish and squid (Fig. 2). Cuttlefish and squid S1 proteases had basic pI values, including having the most basic sequences [S. australis SP1-SepT-4 (9.39)]. In contrast, in octopoids, the values ranged widely from strongly acidic to basic. Such extreme variations occurred within even a single species and were phylogenetically interleaved, e.g. A. aculeatus SP1-Abd-14 [8.07] and A. aculeatus SP1-Abd-7 [5.02] being phylogenetically sister sequences to each other. Although 10 cysteines were always conserved in the S1 proteases, there were considerable variation amongst both prolines and charged residues. Carboxypeptidease encoding transcripts were recovered included from all three coleoid lineages. The carboxypeptidase sequences feature a highly conserved enzymatic core region, with an increase in variability as either terminal is approached, including an uneven number of cysteines (Fig. 3). A number of recovered transcripts featured motifs characteristic of GON4 domains, alignments show a number of regions of high homology with major deletions in between, these deletions are highly variable in both length and location even within the same species (Fig. 4). However, despite the domain deletions, all GON4 were basic, with pI values ranging from 8.28 to 9.18.

Sequence alignment of venom type III PLA2 precursors from the coleoids (1) Sepioteuthis australis PLA2-SepT-1, (2) Loliolus noctiluca PLA2-Lol-2, (3) Sepia pharaonis PLA2-Sepea-1, (4) Sepia latimanus (B6Z1Y5), (5) the scorpion Hadrurus gertschi (P0C8L9), (6) the bee Apis mellifera (P00630) and (7) the lizard Abronia graminea (RL8c9). Propeptide sequence is shown underlined

Phylogenetic reconstruction of coleoid serine proteases. Bracket values indicate calculate pI

Sequence alignment of carboxypeptidase precursors from coleoid venom glands (1) Octopus cyanea Carb-Oct-1 and (2) Sepioteuthis australis Carb-SepT-1 and related non-venom sequences from (3) Homo sapiens (Q8IVL8) and (4) Mus musculus (P15089). Propeptide sequence is shown underlined

Sequence alignment of GON-domain sequences from coleoid venom glands (1) Sepioteuthis australis GON-SepT-1, (2) Sepioteuthis australis GON-SepT-2, (3) Loliolus noctiluca GON-Lol-1, (4) Sepia pharaonis GON-Sepea-1 and (5) Abdopus aculeatus GON-Abd-1
The greatest degree of variability was displayed by the multiple copies were recovered from all three coleoid lineages of a peptide type sequenced by us in an earlier publication which was phylogenetically unresolvable and simply referred to as ‘orphan 4’ (Fry et al. 2009). We were able to identify them as highly modified versions of the pacifastin peptide family. The pacifastin peptides were revealed to be encoded by a tri-product precursor, with each peptide post-translationally liberated from the others. The sequence previously obtained by us from H. maculosa (B6Z1Z0) was shown to contain only the first two domains whilst conversely the S. pharaonis sequence from this study was unique in having a fourth domain inserted between ancestral domains two and three (Fig. 5). Cysteines were highly conserved across the domains except domain 1 of O. cyanea c259 and domain 2 of A. aculeatus c6 and H. maculosa (B6Z1Z0). In contrast to the ancestral types, which are strongly basic in all domains (with the exception of the first domains of Q8MYK4, Q8MYK3 from Schistocerca gregaria), the coleoid sequences are shown to be highly variable in pI (isoelectric point) (Table 2). The acidic domain 1 forms are not monophyletic so this indicates convergent derivations. Domain 2 was also shown to be variable but to a lesser degree than Domain 1. Domain 3, however, remains in the ancestral basic state. In addition, S. pharaonis Paci-Sepia-1 had a fourth domain inserted after domains 2 and 3, which had a pI of 9.13 and a domain molecular weight of 4 kDa. The molecular weights of the coleoid forms (calculated between the first and last cysteines as the N- and C-terminal tails are variable in cleavage points) were consistently approximately 4 kDa for each domain. In contrast, the ancestral forms (also calculated between first and last cysteines) were approximately 3.1 kDa. The differences, however, may reflect a taxonomical trend, rather than a structural state impacting upon function. In contrast to the variability of the highly cysteine cross-linked scaffolds, the globular enzymes chitinase and hyaluronidase conversely showed extreme conservation.

Sequence alignment of pacifastin peptide precursors from coleoid venom glands (1) Hapalochlaena maculosa Paci-Hap-1, (2) Octopus kaurna Paci-Oct-1, (3) Loliolus noctiluca Paci-Lol-1, (4) Sepia pharaonis Paci-Sepea-1, (5) Abdopus aculeatus Paci-Abd-1, (6) H. maculosa (B6Z1Z0), (7) Octopus cyanea Paci-Oct-2, (8) Abdopus aculeatus Paci-Abd-2, (9) Abdopus aculeatus Paci-Abd-3, (10) Octopus cyanea Paci-Oct-3, (11) Octopus cyanea Paci-Oct-4 and (12) Sepioteuthis australis Paci-SepT-1. Pacifastin domains are shown in grey
In order to understand the molecular evolution of coleoid toxins, we first employed the conservative one-ratio model (ORM) which estimates a single omega value for the entire phylogenetic tree. It estimated an omega of 0.16, 0.15, 0.42 and 0.22 for the coleoid CAP, pacifastin, PLA2 and serine proteases, respectively, suggesting strong evolutionary conservation (Supplementary Tables 1–4). ORM can only detect positive selection if the average over all the sites along the lineage is significantly greater than one and hence fails to identify sites that are affected by episodic adaptations. We further employed the site-specific selection analyses. Site model 8 highlighted the strong influence of negative selection on the coleoid toxins and computed omega values that ranged widely (Table 3): 0.22, 0.36, 0.52 and 0.29 for the coleoid CAP, pacifastin, PLA2 and serine proteases, respectively. However, this model identified a single codon in the serine protease as evolving under the influence of positive selection. Single likelihood ancestral counting (SLAC), fixed-effects likelihood (FEL), random-effects likelihood (REL) and the evolutionary fingerprint analyses also supported the lack of variation in the coleoid toxins (Table 3 and Supplementary Fig. 1). The aforementioned models for identifying positive selection work best whilst detecting pervasive selection pressures. However, a large proportion of positively selected sites are often subjected to transient or episodic adaptations. When the majority of lineages in a phylogenetic tree follow the regime of negative selection, they mask the signal of positive selection that might be only influencing a small number of lineages. Hence, these analyses may fail to identify positive selection in such scenarios. To overcome this drawback, we employed the advanced mixed effects model evolution (MEME) (Kosakovsky Pond et al. 2011) which uses the fixed-effects likelihood (FEL) along the sites and random-effects likelihood (REL) across the branches to detect episodic diversifying selection and is capable of not only identifying the episodic adaptations but also the pervasive selection pressures. MEME identified 26 and 3 sites in coleoid serine proteases and pacifastins, respectively which were influenced by the diversifying selection pressures, whilst identifying a single site in PLA2 and CAP (Table 3 and Fig. 6). Assessment of selection pressures by partitioning different pacifastin domains revealed that they have been extremely constrained by negative selection (domain 1 ω = 0.15; domain 2 ω = 0.19 and domain 3 ω = 0.18).

This figure depicts the molecular evolution of different coleoid venom components. The homology models show the episodically adaptive sites in red (HyPhy, MEME approach). The total number of positively and negatively selected sites detected by HyPhy integrative approach (SLAC and FEL: 0.05 significance; REL 50 Bayes factor) are also indicated. Due to the lack of sequence information (n = 4), the molecular evolution of coleoid PLA gene could not be mapped onto its homology model, and hence it is shown in gray
Mutation of the surface chemistry is one of the prominent characteristics of venom evolution. Estimation of the accessible surface area ratio or the surface accessibility of amino acid side chains revealed that 85 % of the episodically adaptive sites in serine proteases were exposed, whilst only 15 % were buried (excluding the 6 sites that could not be assigned to buried or exposed class), suggesting that most mutations are focussed on the molecular surface (Fig. 7 and Supplementary Table 5). The mutation of the surface chemistry would not only increase the range of receptors these toxins can target but could also help in evading the host immune response. The remaining proportion of sites could not be significantly assigned to either buried or exposed class (Fig. 7 and Supplementary Table 5).

In this figure, a plot of amino acid positions (x-axis) against accessible surface area (ASA) ratio (y-axis) indicating the locations of amino acids (exposed or buried) in the crystal structure of coleoid serine protease is presented. Amino acids with an ASA ratio of more than or equal to 50 % are considered to be exposed to the surrounding solvent whilst those with a ratio lesser than 20 % are considered to be buried. Three-dimensional structure of coleoid serine protease is also presented and the episodically adaptive sites (HyPhy, MEME approach) with buried and exposed side chains are indicated by brown and blue labels, respectively. The sites which could not be assigned to the aforementioned classes are indicated with white labels
Discussion
The relative timing of recruitment of coleoid toxins has remained unclear due to the only limited sampling that has previously been undertaken combined with the lack of wide recognition that all coleoids share a single common venomous ancestor. Several of these toxins previously only known from one lineage were shown to be in fact basal when more lineages were characterised. We anticipate that with further sampling efforts, several other toxin types will also show to have an earlier evolutionary origin than recognised based on current data. Our results also revealed that coleoid venoms are much more diverse than previously anticipated, rivalling in complexity with more intensively studied venoms such as those of snakes. It was also strongly suggested that at least some of these toxins are actively evolving under selection, with the cysteine-rich pacifastin and kallikrein types being particularly abundant and diverse. This is consistent with other venoms, where the components with the greatest cysteine content are the scaffolds most amenable to structural and functional mutations (Casewell et al. 2011; Chang and Duda 2012; Fry et al. 2003; Kordis and Gubensek 2000; Weinberger et al. 2010; Wong and Belov 2012). In contrast, the globular enzymes such as carboxypeptidase and hyaluronidase showed extremely little variation, consistent with globular enzymes from other venoms (Fry 2005) that have a three-dimensional structure driven by non-covalent interactions and a single amino acid change could decimate the correct folding.
Site-specific algorithms can only detect positive selection, when its influence at each site is constant throughout time. Thus, they assume that the diversifying selection affects the majority of lineages in a phylogenetic tree. However, very rarely do we encounter scenarios where there is a constant influence of positive selection across all the lineages. Mixed effects model of evolution (MEME), allows omega to vary not only from site to site but also from branch to branch at a site. Site-specific models suggested that most coleoid venom components were extremely negatively selected, showing very little variation. However, MEME identified certain sites in most coleoid venom components as under the influence of episodic diversifying selection (Table 3; Figs. 6, 7). To provide further support to these results, we assessed the selective influence on the 31 biochemical/structural amino acid properties using TreeSAAP (Woolley et al. 2003). All the sites detected by MEME as episodically adaptive were also identified as positively selected for one or more of the biochemical/structural amino acid properties, providing significant support to the nucleotide-level selection analyses (Supplementary Table 5). Episodic nature of selection has ensured that the molecular scaffold of most coleoid toxins remains extremely conserved over time, whilst allowing subtle accumulation of advantageous changes in certain regions of the toxin.
Serine proteases are known for their diversified biological activities. They are involved in immune responses, cellular differentiation, digestion, complement activation, haemostasis, etc. The presence of serine proteases in the salivary secretions of coleoids might suggest for a digestive and/or prey envenoming role through proteolysis. Using the nucleotide and complementary protein-level selection analyses, we detected as many as 26 episodically adaptive sites in coleoid serine proteases that were accumulating rapid mutations (Table 3 and Supplementary Table 5; Figs. 6 and 7). These variations could enable these aquatic predators to feed on a diverse variety of prey types. In snakes and bees, serine proteases are known to prevent blood coagulation in the bite-victim, through fibrin(ogen)olysis, enabling the rapid spread of other venom components through the blood stream. Serine proteases could thus perform a similar role in coleoids by preventing the coagulation of the blood and enhancing the effects of other venom components.
The recovery of novel protein scaffolds from the glands studied here reinforces how little is known about the protein composition of coleoid venoms. This is underscored by the number and diversity of novel scaffolds recovered despite the relatively limited sampling employed. More extensive sampling will no doubt recover novel isoforms of types identified to date as well as entirely new toxin classes. Of particular, focus for follow-up research should be the structure–function relationships, particularly for the small cysteine knotted peptides such as the pacifastins. It is hoped that these results will stimulate further investigation of these neglected glands and their secretory proteins in an increasingly diverse range of coleoid species.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Anastasi A, Erspamer V (1962) Occurrence and some properties of eledoisin in extracts of posterior salivary glands of Eledone. Br J Pharmacol Chemother 19:326–333
Armon A, Graur D, Ben-Tal N (2001) ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J Mol Biol 307:447–463
Cariello L, Zanetti L (1977) Alpha- and beta-cephalotoxin: two paralysing proteins from posterior salivary glands of Octopus vulgaris. Comp Biochem Physiol C 57:169–173
Casewell NR, Wagstaff SC, Harrison RA, Renjifo C, Wuster W (2011) Domain loss facilitates accelerated evolution and neofunctionalization of duplicate snake venom metalloproteinase toxin genes. Mol Biol Evol 28:2637–2649
Chang D, Duda TF Jr (2012) Extensive and continuous duplication facilitates rapid evolution and diversification of gene families. Mol Biol Evol 29:2019–2029
DeLano WL (2002) The PyMOL molecular graphics system. DeLano Scientific, San Carlos
Delport W, Poon AF, Frost SD, Kosakovsky Pond SL (2010) Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457
Fraczkiewicz R, Braun W (1998) Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem 19:319–333
Fry BG (2005) From genome to “venome”: molecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res 15:403–420
Fry BG, Wüster W, Kini RM, Brusic V, Khan A, Venkataraman D, Rooney AP (2003) Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J Mol Evol 57:110–129
Fry BG, Roelants K, Norman JA (2009) Tentacles of venom: toxic protein convergence in the kingdom animalia. J Mol Evol 68:311–321
Ghiretti F (1959) Cephalotoxin: the crab-paralysing agent of the posterior salivary glands of cephalopods. Nature 183:1192–1193
Ghiretti F (1960) Toxicity of octopus saliva against crustacea. Ann N Y Acad Sci 90:726–741
Goldman N, Yang Z (1994) A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol 11:725–736
Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435
Gotz S, Arnold R, Sebastian-Leon P, Martin-Rodriguez S, Tischler P, Jehl MA, Dopazo J, Rattei T, Conesa A (2011) B2G-FAR, a species-centered GO annotation repository. Bioinformatics 27:919–924
Grisley MS (1993) Separation and partial characterization of salivary enzymes expressed during prey handling in the octopus eledone cirrhosa. Comp Biochem Physiol B 105:183–192
Grisley MS, Boyle PR (1987) Bioassay and proteolytic activity of digestive enzymes from octopus saliva. Comp Biochem Physiol B 88:1117–1123
Grisley MS, Boyle PR (1990) Chitinase, a new enzyme in octopus saliva. Comp Biochem Physiol B 95:311–316
Kanda A, Iwakoshi-Ukena E, Takuwa-Kuroda K, Minakata H (2003) Isolation and characterization of novel tachykinins from the posterior salivary gland of the common octopus Octopus vulgaris. Peptides 24:35–43
Kelley LA, Sternberg MJ (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371
Kordis D, Gubensek F (2000) Adaptive evolution of animal toxin multigene families. Gene 261:43–52
Kosakovsky Pond SL, Frost SDW, Muse SV (2005) HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679
Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD (2006) Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol 23:1891–1901
Kosakovsky Pond SL, Murrell B, Fourment M, Frost SD, Delport W, Scheffler K (2011) A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol 28:3033–3043
Lo Bianco S (1888) Notizie biologiche riguardanti specialmente il periodo di maturita sessuale degli animali del Golfo di Napoli. Mitth Zool Stat Neapel 8:385–440
Morishita T (1974) Participation in digestion by the proteolytic enzymes of the posterior salivary gland in octopus–II Isolation and purification of the proteolytic enzymes from the posterior salivary gland. Bull Jpn Soc Sci Fish 40:601–607
Nielsen R, Yang Z (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Posada D, Crandall KA (2002) The effect of recombination on the accuracy of phylogeny estimation. J Mol Evol 54:396–402
Romanini MG (1952) Osservazioni sulla ialuronidasi delle ghiandole salivari enteriorie posteriori degli Octopodi. Pubbl Staz Zool Napo 23:251–270
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Ueda A, Nagai H, Ishida M, Nagashima Y, Shiomi K (2008) Purification and molecular cloning of SE-cephalotoxin, a novel proteinaceous toxin from the posterior salivary gland of cuttlefish Sepia esculenta. Toxicon 52:574–581
Undheim EAB, Norman JA, Thoen HH, Fry BG (2010) Genetic identification of Southern Ocean octopod samples using mtCOI. CR Biol 333:395–404
Weinberger H, Moran Y, Gordon D, Turkov M, Kahn R, Gurevitz M (2010) Positions under positive selection—key for selectivity and potency of scorpion alpha-toxins. Mol Biol Evol 27:1025–1034
Wong ESW, Belov K (2012) Venom evolution through gene duplications. Gene 496:1–7
Woolley S, Johnson J, Smith MJ, Crandall KA, McClellan DA (2003) TreeSAAP: selection on amino acid properties using phylogenetic trees. Bioinformatics 19:671–672
Yang Z (1996) Maximum-likelihood models for combined analyses of multiple sequence data. J Mol Evol 42:587–596
Yang Z (1998) Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15:568–573
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang Z, Wong WSW, Nielsen R (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Acknowledgments
BGF was funded by the Australian Research Council, University of Queenland, Herman Slade Foundation and a Universitas 21 Edward Clarence Dyason Fellowship. BGF would also like to thank the University of Hong Kong’s Swire Institute of Marine Science for hosting him during Hong Kong fieldwork. SAA was the recipient of postdoctoral fellowship (PDRF Phase II Batch-V) from Higher Education Commission (HEC Islamabad) Pakistan. KS was funded by the PhD grant (SFRH/BD/61959/2009) from Fundação para a Ciência e a Tecnologia (FCT). AA was funded by the project PTDC/AACAMB/, 121301/2010 (FCOMP-01-0124-FEDER-019490) from FCT. EABU acknowledges funding from the University of Queensland (International Postgraduate Research Scholarship, UQ Centennial Scholarship, and UQ Advantage Top-Up Scholarship) and the Norwegian State Education Loans Fund.
Author information
Authors and Affiliations
Corresponding author
Additional information
Tim Ruder, Kartik Sunagar, Eivind A. B. Undheim, Syed A. Ali are joint first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Rights and permissions
About this article
Cite this article
Ruder, T., Sunagar, K., Undheim, E.A.B. et al. Molecular Phylogeny and Evolution of the Proteins Encoded by Coleoid (Cuttlefish, Octopus, and Squid) Posterior Venom Glands. J Mol Evol 76, 192–204 (2013). https://doi.org/10.1007/s00239-013-9552-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-013-9552-5