
Abstract
The internal transcribed spacer 2 (ITS2) of the eukaryotic ribosomal DNA (rDNA) cluster plays an essential role in processing of the ribosomal RNA, which is primarily accomplished by the secondary structures acquired by the molecule after transcription. Two possible structural conformation models have been proposed for the ITS2 region, the “ring model” and the “hairpin model,” and the former has been widely used in many molecular phylogenetic analyses incorporating structural information available to date. To evaluate the validity of this model, in vitro transcribed ITS2 molecules from species representing the three superfamilies of the Calyptratae clade (Diptera: Schizophora), namely Cochliomyia hominivorax, Musca domestica, and Glossina morsitans, were submitted to enzymatic digestion with single- and double-stranded specific nucleases (RNases I, A, T1, and V1). The resulting fragments were analyzed by capillary electrophoresis and digestion sites were mapped in the secondary structure models which were obtained by in silico prediction with further refinement by homology comparisons. The pattern of RNA fragments generated by these RNases show a high degree of correlation to most of the predicted helix-loop regions and structural motifs. Discrepancies to the models can be explained by alternative structural conformation dynamics (in M. domestica and G. morsitans) and by higher-order factors (such as tertiary interactions) that may stabilize thermodynamically unfavored structures (in C. hominivorax).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The internal transcribed spacer 2 (ITS2) is a small, non-coding nucleotide region located inside the ribosomal DNA (rDNA) nuclear cluster between the 5.8S (2S in Diptera—Jordan et al. 1976; Tautz et al. 1988) and 28S rRNA sequences (Veldman et al. 1981; Hillis and Dixon 1991) (Fig. 1a). ITS2 plays an essential role in the maturation of the pre-ribosomal RNA (pre-rRNA) as its secondary structures, acquired shortly after transcription, contain the cleavage sites, and secondary structure motifs recognized by the enzymatic complexes that act in the processing of the pre-rRNA (van der Sande et al. 1992; Mitchell et al. 1996, 1997; Peculis and Greer 1998; Geerlings et al. 2000; Côté et al. 2002). Therefore, the cleavage of the ITS2 region delimitates the 5′ and 3′ ends of the 5.8S/2S and 28S rRNAs, respectively. Factors or biochemical events that disrupt the correct assemblage of its secondary structure have been shown to cause a decrease in the amounts or complete absence of mature 28S rRNA (Côté et al. 2002). The essentiality of its function explains the widespread conservation of the ITS2 secondary structure among Eukaryotes (Joseph et al. 1999; Schultz et al. 2005; Wolf et al. 2005).

a Schematic representation of the nuclear ribosomal DNA cluster in Diptera (based on the sequence of D. melanogaster—genbank accession number M21017). b A detailed view of the ITS2 region with the four helix-loop domains highlighted. The sizes of each domain are proportional to the secondary structure proposed by Marinho et al. (2011) for the species Cochliomyia hominivorax (genbank accession number EF560181). Hybridization sites for the primers described on Table 1 are showed
The conservation of the ITS2 secondary structure, combined with the fast evolving nature of the underlying primary sequence, makes the ITS2 a potential molecular marker for phylogenetic inference, especially in lower taxonomic groups (i.e., species and genera) (Schlötterer et al. 1994; Coleman 2003, 2007, 2009), although it may be also suitable for resolving many divergence relationships dating to the Cenozoic era (~65 million years ago—present) (Hillis and Dixon 1991). This feature can be further enhanced and extended by the inclusion of structural information in the phylogenetic analyses (Telford et al. 2005; Wiemers et al. 2009; Letsch and Kjer 2011), mainly due to (1) the improvement in accuracy during positional homology determination (i.e., alignment) (Kjer 1995); (2) providing additional characters for phylogenetic estimation, such as the occurrence of compensatory base changes (CBCs) and information on the overall shape of the molecule (Coleman and Vacquier 2002; Wolf et al. 2005; Müller et al. 2007; Coleman 2009); and (3) the refinement of the phylogenetic analyses as a whole by formally taking into account the non-independence among sites in primary sequence evolution (Dixon and Hillis 1993; Schöniger and von Haeseler 1994; Muse 1995; Rzhetsky 1995; Tillier and Collins 1995; Gutell 1996; Savill et al. 2001; Yu and Thorne 2006).
In a previous study, our group has shown that the ITS2 region is an appropriate molecular marker for inferring phylogenetic relationships among species, genera, and subfamilies of the Calliphoridae family (Diptera: Calyptratae: Oestroidea) (Marinho et al. 2011). Interestingly, inclusion of structural information in the phylogenetic analyses led to improvements both in topology and in branch support estimation, thus producing better estimated phylogenies for this group (Marinho et al. 2011). The ITS2 secondary structure model adopted for Calliphoridae in this previous study is very similar to the structural model proposed for Drosophila melanogaster (Young and Coleman 2004), both conforming with the four-domain ring model currently accepted for Eukaryotes (Joseph et al. 1999; Schultz et al. 2006; Selig et al. 2008; Koetschan et al. 2010). Nevertheless, there is significant variation in local structural conformations when ITS2 secondary structures are compared at the familial (among Oestroidea families) and superfamilial (among Calyptratae superfamilies) levels (Marinho et al. 2012) (Fig. 2). This variation includes the presence/absence of accessory helices (e.g., helix IIa, present in the Drosophila and Hippoboscoidea structures, but not in the remaining Calyptratae) and of internal junctions in the established helix-loops domains (e.g., the junction located in the middle of domain III in Muscoidea and Oestroidea structures). Most of the ITS2 secondary structure models so far used in molecular phylogenetic analyses, including the ones proposed in our previous studies (Marinho et al. 2011, 2012), were based on computational predictions of individual secondary structures using Minimum Free Energy (MFE) computation algorithms with or without a further refinement by homologous comparison among the predicted structures and previously available ones.

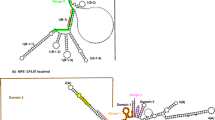
Secondary structure of the ITS2 region toward the Calyptratae superfamily phylogeny (according to McAlpine 1989). The four principal helix-loop domains (I–IV) are shown
Although the comparative approach can significantly increases the accuracy of the secondary structure prediction process (Pace et al. 1999; Reeder et al. 2006), especially when CBCs are observed in the corresponding sequence/structure alignment (Gutell et al. 1994; Mai and Coleman 1997; Coleman 2003), the initial MFE secondary structures may have a significant amount of mispredicted base pairings when compared to native structures (Eddy 2004; Gardner and Giegerich 2004), which can be maintained in the final secondary structure model proposed for a particular taxonomic group. This inaccuracy of the in silico prediction approach relies mostly in the fact that the biologically functional native structure does not necessarily corresponds to the MFE structure (Reeder et al. 2006; Shapiro et al. 2007; Schroeder 2009), as the finally acquired structural conformation may depend more strongly on other factors, such as folding kinetics, rather than thermodynamic free energies (Chen and Dill 2000; Schroeder 2009). Moreover, computationally predicted structure models are usually static representations of a most likely dynamic structure, including structures which are in the suboptimal space of the MFE, which may be possible in solution. In fact, the ITS2 region of Saccharomyces cerevisae has been shown to alternate between two possible secondary structures that are temporally determined, both of them containing structural motifs that are important during ITS2 processing (Côté et al. 2002). One of these secondary structures conforms with the “hairpin model” proposed by Yeh and Lee (1990), which was confirmed by chemical probing and enzymatic digestions analyses, and the other one was in conformation with the “ring model” proposed by Joseph et al. (1999), based on in silico prediction and homology modeling through Eukaryota taxa.
In this context, the present study was aimed to provide an evaluation of the “ring model” proposed for the secondary structure of the ITS2 region in Calyptrate (Diptera: Brachycera) by comparing it with data derived from nuclease digestions followed by primer extension and fragment analysis in capillary electrophoresis of transcribed ITS2 RNA molecules in solution from representative species of the three Calyptratae superfamilies.
Materials and Methods
PCR Amplification and Cloning
The complete ITS2 region of the species Cochliomyia hominivorax (Coquerel 1858), Musca domestica (Linnaeus 1758), and Glossina morsitans (Westwood 1851), including the 3′ side of the 2S rDNA and the 5′ side of the 28S rDNA, was PCR-amplified using the set of primers shown in Table 1. For C. hominivorax, the 5′ (helix-loop domains I and II) and 3′ (helix-loop domains III and IV) halves of the ITS2 region were also separately amplified, as shown on Fig. 1b and Table 1.
PCR reactions were conducted for a final volume of 50 μL with 20 mM Tris–HCl (pH 8.4), 50 mM KCl, 2 mM MgCl2, 0.2 mM dNTPs, 0.5 μM forward primer, 0.5 μM reverse primer, 1,25 U Taq DNA polymerase (Fermentas), and 1–2 μg of extracted total DNA. Amplification conditions included an initial denaturation step at 94 °C for 3 min, followed by 35 cycles at 94 °C for 1 min, 55 °C for 45 s, and 60 °C for 2 min with a final elongation step at 60 °C for 3 min. PCR products were visualized in 1.5 % agarose gels stained with ethidium bromide.
PCR amplicons were purified using the QIAquick PCR Purification Kit (QIAGEN) and then digested with the respective restriction enzymes (BglII/EcoRI or HindIII/BamHI) at 37 °C for 2 h. Digested fragments were cloned into a pHST7.0 plasmid, previously cleaved with the same set of enzymes, using a T4 DNA Ligase enzyme (Promega). Constructed vectors were inserted into competent DH5α E. coli cells by a chemical transformation protocol (Sambrook et al. 1989). Transformed cells were plated in solid LB media plates containing 50 μg/mL ampicillin and incubated overnight at 37 °C. Vectors were then extracted by an alkaline lyses protocol (Sambrook et al. 1989) and the cloned fragments were sequenced for confirmation in an ABI 3700 sequencer (Applied Biosystems) using the same set of primers used in the PCR reactions.
In vitro Transcription
Before the in vitro transcription reactions, the plasmids were linearized using the respective 3′-end restriction enzyme (EcoRI or BamHI). Digestions were visualized in 1 % agarose gels stained with ethidium bromide and the bands containing the linearized plasmids were excised from the gel and purified using the Invisorb DNA CleanUp Kit (Invitek). Linearized plasmids were used as templates for in vitro transcription reactions using the TranscriptAID T7 High Yield Transcription Kit (Fermentas). In vitro transcription reactions were set with 1 μg of linearized template DNA, 10 mM each NTP, 4 μL of the 5X TranscriptAid Reaction Buffer, 2 μL of the TranscriptAID Enzyme, and DEPC-treated water for a final volume of 20 μL. Reactions were incubated at 37 °C for 2 h. Template DNA was digested by the addition of 2 U of DNAse I followed by incubation at 37 °C for 15 min. Template digestion was interrupted by the addition of 2 μL of 0.5 M EDTA (pH 8.0) and incubation at 65 °C for 10 min. Transcribed RNAs were purified by a phenol/chloroform extraction followed by precipitation with ethanol/sodium acetate. The final RNA products were suspended in 20 μL DEPC-treated water, of which 1 μL was used for visualization of transcription reactions in a 2 % agarose gel stained with ethidium bromide.
Denaturation and Refolding of Transcribed RNA and RNA Structure Analyses with Nucleases
For the denaturation/refolding protocol, a solution containing 10 μg of transcribed RNA, 16 μg of sheared yeast RNA (Ambion—Applied Biosystems), 16 μL of 10× RNA Structure Buffer (100 mM Tris pH 7.0; 1 M KCl; 100 mM MgCl2) and DEPC-treated water for 144 μL was incubated at 70 °C for 10 min in a MJ-Research PTC-200 thermocycler and then cooled down 1 °C per min until 25 °C, temperature in which the solution was stabilized for 5 min, and then stored on ice. The solution was then aliquoted in 16 tubes (9 μL per tube, ~600 ng of transcribed RNA) and 1 μL of different serial dilutions of the RNases I (cleaves 3′ of single-stranded As, Cs, Gs, and Us), A (cleaves 3′ of single-stranded Cs and Us), T1 (cleaves 3′ of single-stranded Gs), and V1 (cleaves double-stranded RNA) was added as follows: [1] RNase I (initial concentration: 100 U/μL): 1:10, 1:100, 1:1000; [2] RNase A (initial concentration: 1 ng/μL): 1, 1:10, 1:100, 1:1000; [3] RNase T1 (initial concentration: 1 U/μL): 1, 1:10, 1:100, 1:1000; [4] RNase V1 (initial concentration: 0.1 U/μL): 1, 1:10, 1:100, 1:1000. Nuclease digestion reactions were incubated at room temperature for 15 min and then interrupted by the addition of 20 μL of the inactivation/precipitation buffer (Ambion). A solution in which no nucleases were added was kept as control. Nuclease-digested RNAs were ethanol precipitated, washed, and then suspended in 10 μL of DEPC-treated water.
Primer Extension
Primer extension reactions were carried out with ~600 ng of digested template RNA, 0.5 mM dNTPs, 2 pmol of VIC-labeled reverse primer (Applied Biosystems), 50 mM Tris–HCl (pH 8.3), 75 mM KCl, 5 mM MgCl2, 10 mM DTT, 40 U RNase OUT (Invitrogen), and 200 U of the M-MLV Reverse Transcriptase enzyme (Invitrogen). The reaction was incubated at 37 °C for 50 min and then stopped by heating at 70 °C for 15 min. VIC-labeled reverse primers used for each one of the five different RNA products (Table 1) were as follows: [1] Ch ITS2I,II,III,IV; Ch ITS2III,IV, and Gm ITS2I,II,III,IV: 28S-R-VIC (5′-[VIC]GGTAATCCCATATGAGTTGAGG-3′); [2] Md ITS2I,II,III,IV: 28S-R-Md-VIC (5′-[VIC]GGTAGTCCCATATGAGTTGAGG-3′); [3] Ch ITS2I,II: ITS2-R-Ch-VIC (5′-[VIC]GAGGTTTTGTATCTTTAGC-3′). The resulting cDNA fragments were precipitated with 80 % ethanol, followed by washing with 70 % ethanol, and then dried at 37 °C for 5 min in a dry bath.
Capillary Electrophoresis and Fragment Analyses
The dried cDNA was suspended in 19 μL of Hi-Di formamide (Applied Biosystems), followed by the addition of 1 μL of the GeneScan 600 LIZ size standard (Applied Biosystems). Capillary electrophoresis for fragment analysis was conducted in an ABI 3130 XL Genetic Analyzer (Applied Biosystems). The resulting chromatograms were analyzed (fragment sizing and intensity determination—peak’s height) by the Peak Scanner 1.0 software (Applied Biosystems). Fragment sizes were then mapped in the primary sequence and predicted secondary structure model of the ITS2 region for the considered species, which were taken from Marinho et al. (2011, 2012). These structures were modeled based on a combined approach including in silico prediction and homology-based modeling using the ITS2 secondary structure of Drosophila melanogaster (Young and Coleman 2004). Alternative conformations for these models were obtained by raw in silico prediction using the software Mfold v2.3 (Zuker 2003) [default options; folding temperature = 25 °C] and RNAfold (Hofacker 2003) [minimum free energy and partition function option]. Predicted RNA structures were drawn and visualized using the program VARNA v3.7 (Darty et al. 2009).
Results
Capillary Electrophoresis and Fragment Analyses: Cochliomyia hominivorax
The control (non-treated RNA) chromatogram for the C. hominivorax ITS2 RNA (Fig. S1, Online Resource 1) showed no significant signals of RNA degradation, while the experimental chromatograms showed a consistent pattern among the distinct nucleases considered.
Mapping the fragments into the predicted secondary structure of the ITS2 molecule (Fig. 3; Fig. S2, Online Resource 2) corroborates most of the in silico predicted structural conformations and motifs. The ITS2 proximal stem, formed by the pairing of the 3′ region of the 2S rRNA, and the 5′ region of the 28S rRNA (as present in the rRNA structural model for D. melanogaster—Cannone et al. 2002), is probably stably formed, as suggested by the consistent cleavage by RNase V1 of all nucleotides in the region between bases U379 and U386, including the nucleotides in the G25-C385 (possibly paired), G26-C384 and U27-A383 pairs.

a Cutting sites of RNases V1 (squares) and I (triangles) mapped on the predicted secondary structure of the ITS2 region of C. hominivorax. Digestion intensity scale is showed on the upper-right corner. b, c Alternative conformations for the distal portion of domains I and II, respectively. d Alternative conformations predicted for the forth domain of the ITS2 molecule with their respective ΔG values
In the central-ring region, most nucleotides appear to be in a single-stranded (s.s.) conformation with exception of some isolated paired nucleotides (e.g., G39-C138/C40-G137 and U45-G92/A46-U91). The 3′ end of the ITS2 region (bases U375UUA378) is probably in a double-stranded (d.s.) conformation although both regions that were predicted in silico to base pair (nucleotides U30AAG33 in mfold and U139AAA142 in RNAfold) were not cleaved by RNase V1.
The cleavage pattern of domain I indicates that it is stably formed and that its proximal region is most probably delimited by the pairs G48-C89 and U49-A88 (or U50-A88) instead of G51-C87 and C52-G86, as considered by just theoretical prediction (Marinho et al. 2011). However, the former base pairs are probably not stable enough to be present simultaneously in all structures in solution, while the G51C52/G87C86 configuration is one of the most stable in terms of base pairing and base-stacking energies (Svozil et al. 2010; Wu et al. 2009). Fragment analyses of the remaining regions are consistent with the predicted secondary structure with exception of its distal portion, in which the cleavage pattern suggests the existence of a bulge (U63AUU66) that displaces the predicted terminal pentaloop U66UAAU70 to a triloop A69UA71 even though this conformation is thermodynamically less stable (Fig. 3b).
The cleavage pattern for domain II is also consistent with the in silico predicted secondary structure, except for its distal portion. The G95-C131 pair seems to enclose the helix in its proximal end, while in the distal end the formation of the bulge U116UA118 displaces the predicted terminal hexaloop U111UUUAU116 to a triloop U112UU114. In fact, the whole region between bases U112 and U121 may be in a s.s. conformation in most of the structures in solution, as the base pairs closing this bulge are weak, specially the pair U108-G119. As in domain I, the formation of this bulge next to the terminal loop region is thermodynamically less favored (Fig. 3c), but it seems to be preferred over the formation of a longer terminal loop. The pyrimidine mismatch found in this domain (C101 in C. hominivorax), characteristic of the ITS2 region of Schizophora species, is probably in a s.s. conformation in most structures (as suggested by the RNase A digestion, Fig. S2), although the RNase V1 digestion (Fig. 3) suggests that this base may be possibly paired in some structures in solution.
The proximal portion of domain III, comprising the predicted double-helix region between base pairs A141-U338 and A146-U333 is probably in a s.s. conformation in the majority of the ITS2 secondary structures acquired in solution, as nucleotides U337 and U338 (possibly paired with nucleotides A142 and A141, respectively) are the only ones cleaved by RNase V1, while the remaining bases were consistent cleaved by the s.s. specific RNases, including a high intensity cleavage pattern by RNase A in the region adjacent to the less stable G143-U336 base pair. Nevertheless, the three consequent d.s. regions (between base pairs C147-G331/C153-G325, A161-U324/A167-U318, and A168-U314/A174-U308) are more stably formed, despite of the existence of two destabilizing bulge regions (5′-U154CAUAAA160-3′ and 5′-U315UA317-3′), as suggested by the digestion pattern of RNase V1. In fact, the presence of the base pairs C152-G326, C153-G325 and G163-C322 seems to be essential to insure the formation of the d.s. helix regions.
The existence of the central junction in domain III, from which both sub-domains IIIa and IIIc extend (Fig. 3) and that is absent in the ITS2 secondary structure predicted for D. melanogaster (Young and Coleman 2004), is in agreement with the observed cleavage pattern of the RNases used (Fig. 4a). Even though mfold also predicted a non-branched structure among the suboptimal structures for this domain (ΔG 25°C = −54.29 kcal/mol, against ΔG 25°C = −61.29 kcal/mol of the optimal structure, Fig. 4b), the cleavage pattern observed for the RNases I and T1 on nucleotides U191, U192, U195, G196, U197, G293, U294, G295, U296, and A297 (predicted in d.s. regions) and for the RNase V1 on nucleotides U268, U269, U285, U286, U289, U290, A302, and A303 (predicted as s.s.) are inconsistent with the non-branched structure shown on Fig. 4b. However, the proximal portion of subdomain IIIb in the non-branched structure (Fig. 4b) is thermodynamically more stable than the conformation predicted in the branched structure (ΔG 25°C = –26.26 kcal/mol, compared to ΔG 25°C = −22.13 kcal/mol). The base pairing rearrangements involved in this alternative conformation, between the poly-U (5′ side) and poly-A (3′ side) regions located at the proximal portion of this subdomain, result in the formation of four G-C/C-G pairs in the structure (C213-G279, G224-C267, G226-C265 and G227-C264), instead of only one (G227-C267) in the branched structure. The branched structure also comprises three unstable U-G/G-U pairs (U215-G279, G224-U270, and G226-U268), thus contributing to its lower stability. While the RNase V1 cleavage pattern between nucleotides A279 and A284 suggests the existence of a fairly stable d.s. region, which is more likely to correspond to the alternative structural conformation (Fig. 4b), cleavage of nucleotides U271 and U272 by the same enzyme corroborates the initially predicted structure (Fig. 4a). Nevertheless, formation of the bulge 5′-G263CC265-3′, which occurs solely in the branched structure, is not consistent with the cleavage pattern of the s.s. specific RNases, with exception of the weak cleavage observed for RNase T1 (Fig. S2). This probably indicates that both conformations may occur in solution.

Alternative conformations for the mid portion of domain III in C. hominivorax. a Branched structure, thermodynamically more stable, and b non-branched structure. Nucleotides showed in gray are in the same structural conformation in both structures. Sites cleaved with high intensity (>2,000) by the single-stranded specific RNases I and/or T1 are indicated by black stars, while sites cleaved by the double-stranded specific RNase V1 with low (<2,000) and high (>2,000) intensities are indicated by white and black squares, respectively
In the distal portion of this subdomain, the formation of the bulge 5′-A252AA254-3′ and of the terminal loop 5′-A243UA245-3′ are consistent with the RNase I cleavage pattern, while RNase V1 cleavage sites suggest that base pairs C233-G258, G236-C255, and C237-G251 are the most stable regions in this portion of subdomain IIIb.
The digestion pattern observed for domain IV is the most inconsistent with the predicted secondary structure shown in Fig. 3, probably explained by the variety of alternative conformations with similar ΔGs that can be adopted by this region (Fig. 3d) and that may co-exist in solution. The digestion pattern for RNase T1 is in agreement with the primary sequence showed on Fig. 3 and Fig. S2, as cleavage sites match the regions near the nucleotides G343, G346, G347, and G355. G347 is probably in a s.s. conformation, while G355 is likely to be in s.s. or d.s. conformation in different structures present in vitro. RNase V1 cleavage pattern suggests the existence of two stable d.s. regions, namely the nucleotides between bases A351 and A356 (which is consistent only with the structure showed on Fig. 3a) and between bases U367 and A370 (which is consistent with all predicted structures, Fig. 3a–d). The terminal loop regions predicted in all possible conformations for this domain were not digested with high intensity by none of the s.s. specific RNases used.
Capillary Electrophoresis and Fragment Analyses: Musca domestica and Glossina morsitans
Control chromatograms for both M. domestica (Fig. S3, Online Resource 3) and G. morsitans (Fig. S4, Online Resource 4) species did not show significant signals of RNA degradation. While the observed digestion patterns were consistent among the distinct RNases for G. morsitans, for M. domestica both the number and intensity of fragments generated were significantly lower, although still allowing for the evaluation of some of the predicted structural motifs for this species.
In M. domestica, the 2S-28S proximal stem is probably formed in vitro, as suggested by RNase V1 cleavage sites on nucleotides A14, A16, A23, G24, A399, C400, and A401 (Fig. 5). Nevertheless, both proximal and distal helices in this stem region could also be in a s.s. conformation in solution, as showed by the cleavage of the regions between nucleotides A16 and U20 (5′ side) and U398 and C404 (3′ side) by the s.s. specific RNases (Fig. 5; Fig. S5, Online Resource 5). The s.s. central-ring region is probably not formed in vitro, as the low number of fragments generated by the s.s. specific RNases, along with the sites observed to be digested by RNase V1 (which are consistent with the d.s. regions predicted in silico G33ACUAU38/A163UAGUC168 and A43AUA46/U94GUU97), suggest that this region is in a d.s. conformation.

a Cutting sites of RNases V1 (squares) and I (triangles) mapped on the predicted secondary structure of the ITS2 region of M. domestica. Digestion intensity scale is showed on the upper-left quadrant. b Alternative conformations for the lateral helix-loop regions IIIa and IIIc that can be formed in the mid portion of domain III with their respective ΔG values
Both domains I and II seems to be present in solution although the weak cleavage pattern observed renders it difficult to interpretation. Domain I is probably enclosed by the base pair G48-C92 in its proximal portion, but, as in C. hominivorax, the first two base pairs predicted in the structure presented on Fig. 5 may be unstable and the domain is more likely to be enclosed by the stable base pair configuration G51-C90/C52-G89. Domain II is more likely to be enclosed by the base pairs U105-A154 and G106-C153 rather than by the predicted pairs A98-U160 and U99-A159, as there are no cleavage sites for RNase V1 in the first helix in this domain.
The proximal portion of domain III seems also to be stably formed, as suggested by the RNase V1 digestion pattern, even in its most proximal region, which is enclosed by the unstable base pair configuration A170-U364/G171-U363. The asymmetrical internal loop G187/U337CAUUUGAA345, although not consistently cleaved by any of the s.s. specific RNases (except for RNase T1 in G187), is the only region in this stem which is not cut by RNase V1, thus suggesting a s.s. conformation. The central region of domain III (showed as an internal loop on Fig. 5a) can adopt three possible branched conformations (Fig. 5b). However, the observed cleavage pattern on this region suggests that it is in a s.s. conformation in most of its extension. Cleavage sites for RNase V1 indicate that some nucleotides are likely to be involved in base pairing in this region, such as A221, A297, U310, A312 and A326 (Fig. 5b). This pattern may indicate that, although in most structures in solution this region is in a s.s. conformation, the helix-loop structures comprising both lateral subdomains IIIa and IIIc may be present at low levels in the in vitro structures.
The predicted secondary structure for subdomain IIIb is in agreement with the cleavage pattern observed for the enzyme RNase V1. Formation of both symmetrical internal loops present in this subdomain (U236U237/U287U288 and A242AGG245/A279UUG282), though suggested by a weak cleavage pattern by the s.s. specific RNases, is confirmed by the absence of RNase V1 cuts in these regions. Formation of both the terminal hexaloop (U258GAUUU263) and the bulge (A271AA273) is confirmed by the high intensity cleavage pattern observed for RNase T1 (probably referring to G259 and G270—the last one in the vicinity of the bulge).
The digestion pattern observed in the region comprising the 3′ end of the ITS2 molecules indicates the existence of at least two stretches in a s.s. conformation (U371AACU375 and U385AUCUG390) and two in a d.s. conformation (C374UACU378 and G390UUUU394). This is consistent with the in silico predicted subdivided domain IV (Fig. 5), except for a small displacement (two nucleotides) in the terminal loop and in the 3′ side of the helix on subdomain IVb.
In the G. morsitans secondary structure (Fig. 6; Fig. S6, Online Resource 6), the formation of the 2S-28S proximal stem in vitro is suggested only by its helix more close to the central-ring, as it is the only d.s. region cleaved by RNase V1. Nevertheless, the remaining helices presented only a weak cleavage pattern for the s.s. specific RNases (RNase T1 and A), thus indicating that these regions might be in a d.s. conformation.

a Cutting sites of RNases V1 (squares) and I (triangles) mapped on the predicted secondary structure of the ITS2 region of G. morsitans. Digestion intensity scale is showed on the upper-left quadrant. b Alternative conformation for the region contained between domains II and III with the possible formation of subdomain IIa, as predicted by mfold and considered in the ITS2 secondary structure of D. melanogaster (Young and Coleman 2004)
The ITS2 central-ring was consistently cleaved by all s.s. specific RNases, possibly indicating its formation in most molecules in solution. However, the RNase V1 cleavage sites in this region indicate the existence of some d.s. stretches, which are in agreement with alternative conformations predicted in at least one of the two programs considered in our analyses (mfold/RNAfold). Bases U38 and C40 were predicted by mfold as interacting with the 5′ side of the 2S-28S proximal stem helix (U27-A42, U28-A41, G29-C40, U30-G39, and A31-U38) while bases A43 and C45 were predicted by RNAfold as interacting, respectively, with bases U145 and G143 (in a stretch also including the base pairs U44-A144, A46-U142 and A47-U141). On the opposite side of the central-ring, bases C139, A140 and U141 were predicted by mfold as interacting with the poly-U stretch located in the 5′ side of the proximal helix of domain III, composing the subdomain IIa (Fig. 6b), while base A151 was predicted by RNAfold as pairing with nucleotide U264 (showed on Fig. 6 as part of domain IV). Thus, the formation of the subdomain IIa, which is also present in the ITS2 secondary structure proposed for D. melanogaster (Young and Coleman 2004—though with no sequence homology), is probably accomplished in some molecules in solution.
Both domains I and II seem to be stably formed, as indicated by the digestion pattern of RNase V1. The proximal helix of domain II, though enclosed by the unstable base pair U85-G130, presented high intensities of cleavage by this enzyme. Some RNase V1 cleavage sites on regions predicted as in s.s. conformation (such as the nucleotides on the symmetrical internal loop U98A99/A117U118) probably indicate tertiary or intra-loop interactions. This pattern was also observed in some predicted s.s. motifs on domain III (e.g., the symmetrical internal loop A187AUG190/A220UUG223 and the bulge A244AUAA248). Helix regions on domain III seems to be stably formed with exception of some less stable regions near sparse s.s. motifs. On domain IV, digestion patterns of both RNases I and V1 are in agreement with the predicted secondary structure.
Discussion
Capillary Electrophoresis and Fragment Analyses: Overall View
Ribonuclease digestion and chemical modification experiments followed by fragment analyses by electrophoresis has been a rapid and relatively cheap method recurrently used to provide further insights into the secondary structure of RNA molecules (e.g., Yeh and Lee 1990; Alkemar and Nygard 2004, 2006; Nygard et al. 2006; Weeks 2010). In this scenario, the observed digestion patterns of the complementary s.s. and d.s. specific RNases used here corroborate most of the motifs and helix-loop domains presented in the previously proposed secondary structure models for the ITS2 region in the considered species (Marinho et al. 2011, 2012). However, some differences/discrepancies to the theoretical models can be seen, e.g., in the distal portion of domains I and II in C. hominivorax. In these regions, the predicted terminal loops do not match the observed nuclease digestion patterns, which indicate the formation of a bulge next to the terminal portion of the helix region, reducing the predicted terminal loops. This result indicates that other factors rather than base pairing and base-stacking energies alone influence the folding of the molecule in solution. These factors, which may include folding dynamics (Chen and Dill 2000; Chen 2008) and tertiary interactions (Tinoco and Bustamante 1999; Holbrook 2005, 2008), may stabilize thermodynamically disfavored structures and thus restrict the accuracy of MFE predictions for RNA secondary structure modeling.
Another discrepancy, observed in M. domestica, comprises the very core of the “ring model” for the secondary structure of the ITS2 region proposed for eukaryotes. Although the adoption of a s.s. central-ring region was confirmed by the observed digestion patterns in both C. hominivorax and G. morsitans species, in M. domestica, they suggested the formation of at least two helix regions in this portion of the molecule, thus incompatible with the formation of a central ring. However, as demonstrated by Côté et al. (2002), both models proposed for the secondary structure of the ITS2 regions (i.e., the “ring model” and the “hairpin model”) seem to be present in vivo in yeasts, though temporally apart, thus revealing a necessary conformation dynamics for the correct processing of the RNA molecule. As this temporal determination of the structural conformation may involve the action of additional factors, it is possible that neither models need to be thermodynamically favored. Thus, assuring the formation of the central ring region by thermodynamic properties alone is not necessarily enforced by selection and there are no contradictions to this in the observed results. It is, however, noteworthy that d.s. regions equivalent to the ones predicted in the central-ring region of M. domestica were also predicted in C. hominivorax, but their formation was not observed in vitro.
The mid portion of domain III in M. domestica also comprises an additional discrepancy. In silico-predicted structures suggested the existence of at least three different combinations of structural conformations, both with the adoption of two lateral helix-loop subdomains (IIIa and IIIc). Nevertheless, none of them is in agreement with the observed RNases cleavage sites, indicating that even though these helix-loops are formed in solution, they are not stable. This is consistent with the mapping of base pair probabilities calculated both in the MFE and in the centroid in silico predicted structures by RNAfold (Fig. S7B, Online Resource 7), which shows an instability zone in the mid portion of domain III in M. domestica that is actually more pronounced in two close-related species of the Muscidae family, Haematobia irritans and Stomoxys calcitrans (Fig. S7B). This unstable area, though also present in C. hominivorax (Fig. S7C), is less pronounced in this species, in close-related species of the same family (Calliphoridae, Fig. S7D) and in some of the other families in the Oestroidea superfamily (specially in Rhiniidae and Sarcophagidae, Fig. S7E).
A final discrepancy can be seen in the portion encompassing domains II and III in the ITS2 secondary structure of G. morsitans, where there is the formation of a possible accessory helix-loop region, the subdomain IIa. Although present in the ITS2 secondary structure described for D. melanogaster (Young and Coleman 2004) and in the secondary structure predicted for G. morsitans by the mfold program, the nuclease digestion pattern does not supports its existence, as its formation is impaired by the base pairs formed in the initial portion of domain III. Nevertheless, as it can be seen on the base pairing probabilities calculated by RNAfold (Fig. S7A), the initial helix region of domain III in G. morsitans is formed with low stability even in the MFE structure and it is possible that subdomain IIa is formed at some extent in solution, possibly involving dynamic conformational changes.
In fact, the combination of both experimental nuclease digestion patterns and in silico calculated base pairing probabilities provides important insights about which regions of the putative secondary structure present a dynamic configuration of conformational changes, such as the ones observed in the mid portion of domain III in M. domestica, in the possible formation of subdomain IIa in G. morsitans and in the initial portion of subdomain IIIb in C. hominivorax (Figs. 4, S7). These dynamic conformational changes are of difficult interpretation based solely on nuclease digestion patterns, but can be seen as inconsistencies when compared to the secondary structures predicted by in silico methodologies, which are static representations of actually very dynamical processes.
Finally, though not necessarily an exception to the validity of the in silico predicted structural models, the domain IV of C. hominivorax was also incongruent with the observed digestion pattern. The great plasticity of secondary structures with similar thermodynamic stabilities coupled with the low overall conservation of both primary sequence and secondary structure in this domain among calliphorids (Marinho et al. 2011), which is even missing in some species, indicates the existence of very low or even absent selective constraints in the evolution of this particular domain and suggest a small role, if any, in the processing of the ITS2 molecule.
Based on all these findings, the experiments described here provide further evidence for the validity, at least in thermodynamic aspects, of the proposed “ring model” for the secondary structure of the ITS2 region in eukaryotes, which is currently the most accepted and used model. Although with some particular discrepancies, the structural model currently proposed for the ITS2 molecule in the considered species seems to contain all the major helix-loop regions and structural motifs present in the secondary structures acquired in vitro, and thus seems a good approximation to it. Nevertheless, it is not possible to insure with absolute certainty that these structures actually correspond to the ones acquired in vivo since, besides the already mentioned additional factors that influence the RNA folding process (i.e., folding dynamics and tertiary interactions), structures acquired in vivo are also influenced and determined by other factors, such as transcription speed (Pan and Sosnick 2009) and the action of transacting factors that function in pre-rRNA processing (e.g., the snoRNA U8 in mammals, which directly interacts with the 3′ and 5′ ends of the 5.8S and 28S rRNAS, respectively—Michot et al. 1999; reviewed by Kressler et al. 2010).
Conclusions
The inclusion of structural information in molecular phylogenetic analyses has been widely recognized as having positive effects on both resolution and accuracy of the inferred phylogenetic trees (e.g., Telford et al. 2005; Wiemers et al. 2009), even though in some cases only minor overall improvements are observed in terms of tree topologies and branches supports (Letsch and Kjer 2011; Marinho et al. 2012). Nevertheless, a real improvement in the inferred phylogenies can only be expected if the proposed structural models incorporated in the analyses are indeed accurate. In this context, results presented here corroborate, although with some limitations, the secondary structure models previously proposed for the considered species in the Calyptratae clade and incorporated in the molecular phylogenetic analyses conducted by Marinho et al. (2011, 2012), which were based on the general “ring model” proposed for Eukaryotes. Furthermore, we here emphasize the importance of empirical evaluations of in silico predicted RNA secondary structures to both validate and refine major and minor predicted structural motifs, thus conferring more robustness and accuracy to the structural models incorporated in molecular phylogenetic analyses.
References
Alkemar G, Nygard O (2004) Secondary structure of two regions in expansion segments ES3 and ES6 with the potential of forming a tertiary interaction in eukaryotic 40S ribosomal subunits. RNA 10:403–411
Alkemar G, Nygard O (2006) Probing the secondary structure of expansion segment ES6 in 18S ribosomal RNA. Biochemistry 45:8067–8078
Cannone JJ, Subramanian S, Schnare MN, Collett JR, D’Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Müller KM, Pande N, Shang Z, Yu N, Gutell RR (2002) The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform 3:2
Chen SJ (2008) RNA Folding: conformational statistics, folding kinetics and ion electrostatics. Annu Rev Biophys 37:197–214
Chen SJ, Dill KA (2000) RNA folding energy landscapes. Proc Natl Acad Sci USA 97(2):646–651
Coleman AW (2003) ITS2 is a double-edged tool for eukaryote evolutionary comparisons. Trends Genet 19:370–375
Coleman AW (2007) Pan-eukaryote ITS2 homologies revealed by RNA secondary structure. Nucleic Acids Res 35:3322–3329
Coleman AW (2009) Is there a molecular key to the level of “biological species” in eukaryotes? A DNA guide. Mol Phylogenet Evol 50:197–203
Coleman AW, Vacquier VD (2002) Exploring the phylogenetic utility of its sequences for animals: a test case for abalone (haliotis). J Mol Evol 54:246–257
Côté CA, Greer CL, Peculis BA (2002) Dynamic conformational model for the role of ITS2 in pre-rRNA processing in yeast. RNA 8:786–797
Darty K, Denise A, Ponty Y (2009) VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics 25:1974–1975
Dixon MT, Hillis DM (1993) Ribosomal RNA secondary structure: compensatory mutations and implications for phylogenetic analysis. Mol Biol Evol 10:256–267
Eddy SR (2004) How do RNA folding algorithms work? Nat Biotechnol 22(11):1457–1458
Gardner PP, Giegerich R (2004) A comprehensive comparison of comparative RNA structure prediction approaches. BMC Bioinform 30(5):140–158
Geerlings TH, Vos JC, Raué HA (2000) The final step in the formation of 25S rRNA in Saccharomyces cerevisiae is performed by 5′ ⇒ 3′ exonucleases. RNA 6:1698–1703
Gutell RR (1996) Comparative sequence analysis and the structure of 16S and 23S rRNA. In: Zimmermann RA, Dahlberg AE (eds) Ribosomal RNA: structure, evolution, processing, and function in protein biosynthesis. CRC Press, New York, pp 111–128
Gutell RR, Larsen N, Woese CR (1994) Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol Rev 58:10–26
Hillis DM, Dixon MT (1991) Ribosomal DNA: molecular evolution and phylogenetic inference. Q Rev Biol 66:411–453
Hofacker IL (2003) Vienna RNA secondary structure server. Nucleic Acids Res 31(13):3429–3431
Holbrook SR (2005) RNA structure: the long and the short of it. Curr Opin Struct Biol 15:302–308
Holbrook SR (2008) Structural principals from large RNAs. Annu Rev Biophys 37:445–464
Jordan BR, Jourdan R, Jacq B (1976) Late steps in the maturation of Drosophila 26 S ribosomal RNA: generation of 5.8 S and 2 S RNAs by cleavages occurring in the cytoplasm. J Mol Biol 101:85–105
Joseph N, Krauskopf E, Vera MI, Michot B (1999) Dynamic conformational model for the role of ITS2 in pre-rRNA processing in yeast. RNA 8:786–797
Kjer KM (1995) Use of rRNA secondary structure in phylogenetic studies to identify homologous positions: an example of alignment and data presentation from the frogs. Mol Phylogenet Evol 4(3):314–330
Koetschan C, Förster F, Keller A, Schleicher T, Ruderisch B, Schwarz R, Müller T, Wolf M, Schultz J (2010) The ITS2 Database III: sequences and structures for phylogeny. Nucleic Acids Res 38(Database issue):D275–D279
Kressler D, Hurt E, Bassler J (2010) Driving ribosome assembly. Biochim Biophys Acta 1803(6):673–683
Kutty SN, Pape T, Wiegmann BM, Meier R (2010) Molecular phylogeny of the Calyptratae (Diptera: Cyclorrhapha) with an emphasis on the superfamily Oestroidea and the position of Mystacinobiidae and McAlpine’s fly. Syst Entomol 35:614–635
Letsch HO, Kjer KM (2011) Potential pitfalls of modelling ribosomal RNA data in phylogenetic tree reconstruction: evidence from case studies in the Metazoa. BMC Evol Biol 11:146
Mai JC, Coleman AW (1997) The internal transcribed spacer 2 exhibits a common core secondary structure in greens algae and flowering plants. J Mol Evol 44:258–271
Marinho MAT, Junqueira ACM, Azeredo-Espin AML (2011) Evaluation of the internal transcribed spacer 2 (ITS2) as a molecular marker for phylogenetic inference using sequence and secondary structure information in blow flies (Diptera: Calliphoridae). Genetica 139(9):1189–1207
Marinho MAT, Junqueira ACM, Paulo DF, Esposito MC, Villet MH, Azeredo-Espin AML (2012) Molecular phylogenetics of Oestroidea (Diptera: Calyptratae) with emphasis on Calliphoridae: insights into the inter-familial relationships and additional evidence for paraphyly among blowflies. Mol Phylogenet Evol 65(3):840–854
McAlpine JF (1989) Manual of Nearctic Diptera 3. Agriculture Canada Monograph 32. Canadian Government Publishing Centre, Hull
Michot B, Joseph N, Mazan S, Bachellerie JP (1999) Evolutionarily conserved structural features in the ITS2 of mammalian pre-rRNAs and potential interactions with the snoRNA U8 detected by comparative analysis of new mouse sequences. Nucleic Acids Res 27(11):2271–2282
Mitchell P, Petfalski E, Tollervey D (1996) The 3′ end of yeast 5.8S rRNA is generated by an exonuclease processing mechanism. Genes Dev 10:502–513
Mitchell P, Petfalski E, Shevchenko A, Mann M, Tollervey D (1997) The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′ ⇒ 5′ exoribonucleases. Cell 91:457–466
Müller T, Philippi N, Dandekar T, Schultz J, Wolf M (2007) Distinguishing species. RNA 13:1469–1472
Muse SV (1995) Evolutionary analyses of DNA sequences subject to constraints of secondary structure. Genetics 139:1429–1439
Nygard O, Alkemar G, Larsson SL (2006) Analysis of the secondary structure of expansion segment 39 in ribosomes from fungi, plants and mammals. J Mol Biol 357:904–916
Pace NR, Thomas BC, Woese CR (1999) Probing RNA structure, function and history by comparative analysis. In: Gesteland RF, Cech TR, Atkins JF (eds) The RNA World, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 113–141
Pan T, Sosnick T (2009) RNA folding during transcription. Annu Rev Biophys Biomol Struct 35:161–175
Peculis BA, Greer CL (1998) The structure of the ITS2-proximal stem is required for pre-rRNA processing in yeast. RNA 4(12):1610–1622
Reeder J, Höchsmann M, Rehmsmeier M, Voss B, Giegerich R (2006) Beyond Mfold: recent advances in RNA bioinformatics. J Biotechnol 124(1):41–55
Rzhetsky A (1995) Estimating substitution rates in ribosomal RNA genes. Genetics 141:771–783
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning—a laboratory manual, 2nd edn. Cold Spring Habour Laboratory Press, New York
Savill NJ, Hoyle DC, Higgs PG (2001) RNA sequence evolution with secondary structure constraints: comparison of substitution rate models using maximum-likelihood methods. Genetics 157:399–411
Schlötterer C, Hauser MT, von Haeseler A, Tautz D (1994) Comparative evolutionary analysis of rDNA ITS regions in Drosophila. Mol Biol Evol 11:513–522
Schöniger M, von Haeseler A (1994) A stochastic model for the evolution of autocorrelated DNA sequences. Mol Phylogenet Evol 3:240–247
Schroeder SJ (2009) Advances in RNA structure prediction from sequence: new tools for generating hypotheses about viral RNA structure-function relationships. J Virol 83(13):6326–6334
Schultz J, Maisel S, Gerlach D, Muller T, Wolf M (2005) A common core of secondary structure of the internal transcribed spacer 2 (ITS2) throughout the Eukaryota. RNA 11:361–364
Schultz J, Müller T, Achtziger M, Seibel PN, Dandekar T, Wolf M (2006) The internal transcribed spacer 2 database: a web server for (not only) low level phylogenetic analyses. Nucleic Acids Res 34(Web Server issue):W704–W707
Selig C, Wolf M, Müller T, Dandekar T, Schultz J (2008) The ITS2 Database II: homology modelling RNA structure for molecular systematics. Nucleic Acids Res 36(Database issue):D377–D380
Shapiro BA, Yingling YG, Kasprzak W, Bindewald E (2007) Bridging the gap in RNA structure prediction. Curr Opin Struct Biol 17(2):157–165
Svozil D, Hobza P, Sponer J (2010) Comparison of intrinsic stacking energies of ten unique dinucleotide steps in A-RNA and B-DNA duplexes. Can we determine correct order of stability by quantum-chemical calculations? J Phys Chem B 114:1191–203; Erratum in: J Phys Chem B (2010) 114:2547
Tautz D, Hancock JM, Webb DA, Tautz C, Dover GA (1988) Complete sequences of the rRNA genes of Drosophila melanogaster. Mol Biol Evol 5:366–376
Telford MJ, Wise MJ, Gowri-Shankar V (2005) Consideration of RNA secondary structure significantly improves likelihood-based estimates of phylogeny: examples from the bilateria. Mol Biol Evol 22:1129–1136
Tillier E, Collins R (1995) Neighbor-Joining and maximum likelihood with RNA sequences: addressing the inter-dependence of sites. Mol Biol Evol 12:7–15
Tinoco I Jr, Bustamante C (1999) How RNA folds. J Mol Biol 293:271–281
van der Sande CA, Kwa M, van Nues RW, van Heerikhuizen H, Raué HA, Planta RJ (1992) Functional analysis of internal transcribed spacer 2 of Saccharomyces cerevisiae ribosomal DNA. J Mol Biol 223:899–910
Veldman GM, Klootwijk J, van Heerikhuizen H, Planta RJ (1981) The nucleotide sequence of the intergenic region between the 5.8S and 26S rRNA genes of the yeast ribosomal RNA operon. Possible implications for the interaction between 5.8S and 26S rRNA and the processing of the primary transcript. Nucleic Acids Res 9:4847–4862
Weeks KM (2010) Advances in RNA secondary and tertiary structure analysis by chemical probing. Curr Opin Struct Biol 20(3):295–304
Wiemers M, Keller A, Wolf M (2009) ITS2 secondary structure improves phylogeny estimation in a radiation of blue butterflies of the subgenus Agrodiaetus (Lepidoptera: Lycaenidae: Polyommatus). BMC Evol Biol 9:300
Wolf M, Achtziger M, Schultz J, Dandekar T, Müller T (2005) Homology modeling revealed more than 20,000 rRNA internal transcribed spacer 2 (ITS2) structures. RNA 11:1616–1623
Wu JC, Gardner DP, Ozer S, Gutell RR, Ren P (2009) Correlation of RNA secondary structure statistics with thermodynamic stability and applications to folding. J Mol Biol 391:769–783
Yeh LCC, Lee JC (1990) Structural analysis of the internal transcribed spacer 2 of the precursor ribosomal RNA from Saccharomyces cerevisiae. J Mol Biol 211:699–712
Young I, Coleman AW (2004) The advantages of the ITS2 region of the nuclear rDNA cistron for analysis of phylogenetic relationships of insects: a Drosophila example. Mol Phylogenet Evol 30:236–242
Yu J, Thorne JL (2006) Dependence among sites in RNA evolution. Mol Biol Evol 23(8):1525–1537
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415
Acknowledgments
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, Grant 06/61217-3). The authors would like to thank Patrícia P. Coltri for technical training and assistance and Rosângela A. Rodrigues, Tereza Cristina L. Silva, and Wanderley P. da Graça for technical support.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Fig. S1
Control and experimental chromatograms of the fragment analyses conducted in the ITS2 region of Cochliomyia hominivorax. Enzymes and concentrations used are given in the correspondent chromatograms. Peaks are showed relative to a scaled schematic representation of the ITS2 secondary structure (as showed on Fig. 3 and Fig. S2) (TIFF 5947 kb)
Fig. S2
(A) Cutting sites of RNases A (circles) and T1 (hexagons) mapped on the predicted secondary structure of the ITS2 region of C. hominivorax. Digestion intensity scale is showed on the upper-right corner. (B) and (C) Alternative conformations for the distal portion of domains I and II, respectively. (D) Alternative conformations predicted for the forth domain of the ITS2 molecule with their respective ΔG values (TIFF 5437 kb)
Fig. S3
Control and experimental chromatograms of the fragment analyses conducted in the ITS2 region of Musca domestica. Enzymes and concentrations used are given in the correspondent chromatograms. Peaks are showed relative to a scaled schematic representation of the ITS2 secondary structure with two alternative conformations for the helix-loop regions IIIa and IIIc (as showed on Fig. 5; Fig. S5) (TIFF 6580 kb)
Fig. S4
Control and experimental chromatograms of the fragment analyses conducted in the ITS2 region of Glossina morsitans. Enzymes and concentrations used are given in the correspondent chromatograms. Peaks are showed relative to a scaled schematic representation of the ITS2 secondary structure (as showed on Fig. 6; Fig. S6) (TIFF 6632 kb)
Fig. S5
(A) Cutting sites of RNases A (circles) and T1 (hexagons) mapped on the predicted secondary structure of the ITS2 region of M. domestica. Digestion intensity scale is showed on the upper-left quadrant. (B) Alternative conformations for the lateral helix-loop regions IIIa and IIIc that can be formed in the mid portion of domain III with their respective ΔG values (TIFF 5128 kb)
Fig. S6
(A) Cutting sites of RNases A (circles) and T1 (hexagons) mapped on the predicted secondary structure of the ITS2 region of G. morsitans. Digestion intensity scale is showed on the upper-left quadrant. (B) Alternative conformation for the region contained between domains II and III with the possible formation of subdomain IIa, as predicted by mfold and considered in the ITS2 secondary structure of D. melanogaster (Young and Coleman 2004) (TIFF 3660 kb)
Fig. S7
Base pair probability maps calculated by RNAfold in domain III of (A) Glossina morsitans; (B) Musca domestica and two close related species of Muscidae (Haematobia irritans and Stomoxys calcitrans); (C) Cochliomyia hominivorax, and (D) four other representatives of the family Calliphoridae (Calliphora vicina, Lucilia eximia, Hemigymnochaeta unicolor, and Mesembrinella bicolor); (E) representatives of other families in the Oestroidea superfamily (Rhiniidae: Rhinia sp., Sarcophagidae: Sarcophaga bullata, Tachinidae: Prophorostoma pulchrum). *The subfamily Mesembrinellinae is formally considered inside Calliphoridae, though this status is doubtful (Kutty et al. 2010; Marinho et al. 2012) (TIFF 6831 kb)
Rights and permissions
About this article
Cite this article
Marinho, M.A.T., Azeredo-Espin, A.M.L. & Zanchin, N.I.T. Structural Characterization of the Internal Transcribed Spacer 2 (ITS2) of the Ribosomal DNA (rDNA) Cluster in Calyptratae (Diptera: Schizophora) and its Implications for Molecular Phylogenetic Analyses. J Mol Evol 76, 158–171 (2013). https://doi.org/10.1007/s00239-013-9548-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-013-9548-1