
Abstract
Although some α-glucosidases from the α-amylase family (glycoside hydrolase family GH13) have been studied extensively, their exact number, organization on the chromosome, and orthology/paralogy relationship were unknown. This was true even for important disease vectors where gut α-glucosidase is known to be receptor for the Bin toxin used to control the population of some mosquito species. In some cases orthologs from related species were studied intensively, while potentially important paralogs were omitted. We have, therefore, used a bioinformatics approach to identify all family GH13 α-glucosidases from the selected species from Metazoa (including three mosquito species: Aedes aegypti, Anopheles gambiae, and Culex quinquefasciatus) as well as from Fungi in an effort to characterize their arrangement on the chromosome and evolutionary relationships among orthologs and among paralogs. We also searched for pseudogenes and genes coding for enzymatically inactive proteins with a possible new function. We have found GH13 α-glucosidases mostly in Arthropoda and Fungi where they form gene families, as a result of multiple lineage-specific gene duplications. In mosquito species we have identified 14 α-glucosidase (Aglu) genes of which only five have been biochemically characterized so far, two are putative pseudogenes and the rest remains uncharacterized. We also revealed quite a complex evolutionary history of the eukaryotic α-glucosidases probably involving multiple losses of genes or horizontal gene transfer from bacteria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
α-1,4-Glucosidase (EC 3.2.1.20) is a glucosidase acting upon α-1,4-glucosidic bonds. The α-glucosidases are presently found in five families of glycoside hydrolases (GHs): GH4, GH13, GH31, GH97, and GH122, based on the Carbohydrate-Active enZymes (CAZy) classification system (Cantarel et al. 2009). Those from families GH13, GH31, and GH97 share a (β/α)8-barrel fold of their catalytic domain. Interestingly tertiary structure of α-glucosidases from the family GH4 is different. It is more similar to NAD-dependent dehydrogenases (2-hydroxyacid dehydrogenases), with typical Rossman fold of their NAD+-binding site (Lodge et al. 2003).
The family GH13 is also known as the α-amylase family (MacGregor et al. 2001) and together with GH70 and GH77 it forms the clan GH-H (Cantarel et al. 2009). To be classified in the clan GH-H, enzymes have to share following features: (i) catalytic domain (domain A) formed by the (β/α)8-barrel fold (i.e., TIM-barrel), with a small domain (domain B) inserted between the strand β3 and the helix α3; (ii) catalytic machinery consisting of an aspartate (catalytic nucleophile) situated at the β4-strand, a glutamate (proton donor) at the β5-strand, and another aspartate (transition-state stabilizer) located at the β7-strand; (iii) retaining reaction mechanism; and (iv) four up to seven conserved sequence regions (CSRs) positioned mainly at the β-strands of the catalytic domain A (Matsuura et al. 1984; Janecek et al. 1997; Kuriki and Imanaka 1999; MacGregor et al. 2001; Janecek 2002).
Family GH13, with its ~30 different enzyme specificities and more than 12,000 sequenced members belongs to the largest GH families (Cantarel et al. 2009). Whereas different specificities could be clustered together due to their sequence similarities, the entire family could be divided into subfamilies (Janecek 1995, 2002). Initially two GH13 subfamilies were established. The oligo-1,6-glucosidase and neopullulanase subfamilies were based on a specific sequence of the fifth CSR (Oslancova and Janecek 2002). According to this classification α-glucosidases belong to the oligo-1,6-glucosidase subfamily (Oslancova and Janecek 2002). Presently the family GH13 consists of 37 curator-established subfamilies (Stam et al. 2006).
To date there is a large amount of proteins predicted to be α-glucosidases based on the sequence comparison, but only a small fraction of them was actually biochemically characterized. Three α-glucosidase izoenzymes I, II, and III from Apis mellifera were most extensively studied. They are encoded by the genes hbg1, hbg2, and hbg3, respectively, and they differ in substrate and tissue specificities and pH optima (Huber and Thompson 1973; Takewaki et al. 1980, 1993; Kimura et al. 1990; Nishimoto et al. 2001; Kubota et al. 2004). Also two mosquito α-glucosidases were characterized. It was reported that these two α-glucosidases use maltose as a substrate and are expressed in the midgut of Anopheles gambiae (Zheng et al. 1995). Putative maltase genes were identified also in the salivary glands of adult Aedes aegypti (James et al. 1989) and Aedes albopictus (Marinotti et al. 1996). Initially only small α-glucosidase (maltase) gene clusters consisting of three genes (lvpH, lvpD, and lvpL) in Drosophila melanogaster (Snyder and Davidson 1983; Henikoff and Wallace 1988) and composed of two genes (mav1 and mav2) in Drosophila virilis (Vieira et al. 1997) were known. Based on a bioinformatics approach seven more putative maltase genes were recently identified in 12 Drosophila species (Gabrisko and Janecek 2011). Particular genes are situated next to each other forming two clusters on two chromosomes and they are likely a result of ancient series of duplications, which all took place in a common ancestor of recent Drosophila species (Gabrisko and Janecek 2011).
In fungi there are two types of α-glucosidases: (i) α-1,4-glucosidase (EC 3.2.1.20) or maltase which hydrolyzes α-1,4 glucosidic bonds; and (ii) α-1,6-glucosidase (EC 3.2.1.10) known as isomaltase acting upon α-1,6 glucosidic bonds. In Saccharomyces cerevisiae five MAL loci situated near the telomeres were identified: MAL1 (chromosome VII), MAL2 (chromosome III), MAL3 (chromosome II), MAL4 (chromosome XI), and MAL6 (chromosome VIII) (Winge and Roberts 1950; Barnett 1976), each of them consisting of one or more copies of three different genes situated next to each other. Beside maltase (MALx2/MALS) there is a maltose permease (MALx1/MALT) used for maltose transport across the plasma membrane and a positive trans-acting regulatory protein (MALx3/MALR) (Cohen et al. 1984; Charron et al. 1986; Novak et al. 2004). Similar MAL clusters were identified in the genus Aspergillus. Aspergillus flavus possesses three, Aspergillus oryzae two; Aspergillus clavatus, Aspergillus fumigatus, and Aspergillus fischeri one copy of MAL. No MAL cluster was found in Aspergillus niger (Vongsangnak et al. 2009). A. niger uses another regulatory system based on the AmyR regulator which activates two α-glucosidases (AgdA and a putative AgdB), the α-amylase (AamA) and the glucoamylase (GlaA) (Yuan et al. 2008; van der Kaaij et al. 2007). One copy of MAL cluster consisting of maltase (HPMAL1), maltase permease (HPMAL2), and the putative MAL activator gene was identified also in Hansenula polymorpha (Viigand et al. 2005). Maltase gene CAMAL2 homologous to MAL62 and gene CASUC1 homologous to MAL63 of S. cerevisiae were found in human pathogen Candida albicans (Geber et al. 1992).
For isomaltose utilization in yeasts the isomaltase gene cluster IMA consisting of five genes (IMA1-IMA5) is responsible (Teste et al. 2010). Alike maltase, isomaltase is intracellularly acting enzyme and, therefore, a transporter to move isomaltose inside the cell is needed. For this purpose, the AGT1 transporter with a broad substrate specificity, which is part of MAL1 locus is used (Han et al. 1995).
Although α-glucosidase primary role is to participate in the saccharide metabolism, in process of evolution it gained other important functions. The product of hemoglobin digestion in hematophagous insect is cytotoxic heme. In Rhodnius prolixus it is detoxified in a process of biomineralization, where reactive heme is transformed into a nonreactive, insoluble crystalline form called hemozoin (β-hematin), the α-glucosidase being responsible here for the hemozoin nucleation (Mury et al. 2009).
Another example of secondary function gain is the utilization of α-glucosidase as a toxin receptor in some mosquito species. For the so-called binary toxin, a two-domain protein product of Bacillus sphaericus, to initiate its deadly action on larvae of C. pipiens, C. quinquefasciatus, and Anopheles gambiae, it needs to be recognized by the α-glucosidase bound to midgut membrane with glycosylphosphatidylinositol (GPI) anchor (Silva-Filha et al. 1999; Darboux et al. 2001; Opota et al. 2008; Ferreira et al. 2010). Although both the above-mentioned proteins acquired a new function, they still retained the original enzymatic activity. But there are proteins that not only evolved new function but also changed their enzymatic properties as well. Heavy subunits of heterodimeric amino acid transporter (HAT) proteins, homologous to enzymes from the α-amylase family, likely lost enzymatic activity completely and became the auxiliary unit of HATs (Wells and Hediger 1992; Janecek et al. 1997; Chillaron et al. 2001; Broer and Wagner 2002; Gabrisko and Janecek 2009).
Although no α-glucosidase from the family GH13 has been reported in most eukaryotic lineages so far, this important enzymatic specificity is still present there. Where GH13 α-glucosidases are missing, α-glucosidases from the family GH31 play that role instead. They have been identified and characterized in protozoa (Alam et al. 1996; Jones et al. 2005), plants (Tibbot and Skadsen 1996; Matsui et al. 1997), nematodes (Sikora et al. 2010), and vertebrates (Kunita et al. 1997; Dennis et al. 2000), some of them endowed with a special function. There are five α-glucosidases from this family in humans. Two of them deserve particular attention. One is responsible for degradation of glycogen in lysosomes (Hermans et al. 1991; Hoefsloot et al. 1991), the other one is a key protein involved in the control of proper folding of proteins in the endoplasmic reticulum (Martiniuk et al. 1985). Between enzymes from these two families (GH13 and GH31), a remote but significant homology was revealed (Rigden 2002; Janecek et al. 2007).
Presented work is a bioinformatics study of the α-glucosidase multigene family in eukaryotes. We investigated spatial organization of particular α-glucosidase genes, compared amino acid sequences encoded by these genes, analyzed their mutual relationships, and considered evolutionary processes that have shaped this multigene family.
Materials and Methods
Collecting the Sequences
Sequences were obtained using protein BLAST (Altschul et al. 1990) against the default non-redundant database. Characterized α-glucosidase I from Apis mellifera (Huber and Thompson 1973; Kimura et al. 1990) (GenBank accession number: NP_001035326; GeneID: 409889, Hbg1) was used as a query and search was limited to eukaryotes. To assure that a complete set of α-glucosidases (from the family GH13) from the given species was obtained, only sequences from the species with fully sequenced genomes were used. Position of studied genes on a chromosome and intergene distances were inferred from the data obtained from the GenBank (Benson et al. 2012).
Sequence Alignment
The obtained amino acid sequences were aligned using ClustalX (Jeanmougin et al. 1998) and then manually fine tuned with regard to known CSRs known from the literature (Janecek et al. 1997; MacGregor et al. 2001; Janecek 2002; Oslancova and Janecek 2002).
Evolutionary Analyses
MEGA 5.05 package (Tamura et al. 2011) was used to calculate both neighbor-joining (NJ) (Saitou and Nei 1987) and maximum parsimony (MP) (Eck and Dayhoff 1966) phylogenetic trees. For the NJ tree Jones–Taylor–Thornton model (Jones et al. 1992) of amino acid change was used and unequal rates among sites were assumed. Gamma shape parameter (α = 0.95) was estimated using the program PhyML 3.0 (Guindon and Gascuel 2003). Gaps were retained. To infer phylogenetic relationship between eukaryotic and bacterial α-glucosidases, gaps and highly divergent regions were removed and then NJ tree was recalculated. For the MP tree close-neighbor-interchange algorithm with default parameters was used. Reliability of tree topologies was evaluated using the bootstrap test (Felsenstein 1985) with 1,000 replications. WAG+I+G was estimated as the best model (according to AIC) of sequence evolution for analyzed data using the program PROTTEST (Abascal et al. 2005). Maximum likelihood (ML) (Felsenstein 1981) tree was calculated using the PhyML 3.0 algorithm (Guindon and Gascuel 2003) with WAG substitution model and four substitution rate categories. Proportion of invariable sites and gamma shape parameter (α = 0.95) were estimated by program PhyML 3.0 itself (Guindon and Gascuel 2003). The starting tree was calculated using BIONJ, NNI was used for tree improvement; both branch lengths and topology were optimized. Bootstrap test was limited to 100 replications. Characterization of orthology/paralogy relationships of particular genes was based on the phylogenetic reconstruction.
Intron/Exon Composition
The gene structure was estimated by comparison between the amino acid sequence of the respective protein and its nucleotide sequence obtained from GenBank (Benson et al. 2012) using the program GeneWise (Birney et al. 2004).
Results and Discussion
In presented work we have investigated the presence of α-glucosidase genes coding for proteins from the α-amylase family GH13 in eukaryotes where complete genome information was available. We have found unambiguous α-glucosidases from this family only in two groups—in fungi and Arthropoda (Table S1). Although it is known that no α-glucosidases from the family GH13 are present in vertebrates (where α-glucosidase from a related family GH31 are used), we found α-glucosidase-like genes in two deuterostome genomes: in Branchiostoma floridae and Strongylocentrotus purpuratus. Since chordates possess heavy chains of HAT proteins with high similarity to GH13 α-glucosidases (Wells and Hediger 1992; Janecek et al. 1997; Gabrisko and Janecek 2009), classification of these two found genes deserves caution (especially when diagnostic 5′ part of the gene from S. purpuratus is missing). Moreover, the gene from B. floridae groups together with human rBAT protein in phylogenetic tree and it is, therefore, likely that it is indeed hcHAT protein and not an enzyme (Fig. S1). We have identified ambiguous genes (hcHAT or enzymes) in basal Metazoa in Nematostella vectensis (Cnidaria), Trichoplax adhaerens (Placozoa), and in Amphimedon queenslandica (Porifera) (Table S1).
Hymenoptera
In the phylum Arthropoda, the family GH13 α-glucosidase genes are organized in gene clusters. Concerning the order Hymenoptera we studied α-glucosidase genes in Apis mellifera, Nasonia vitripennis, and Bombus terrestris (Table S1).
We confirmed that Apis mellifera possesses only three already known α-glucosidase genes: Hbg1 (BAE86926; Gene ID: 409889), Hbg2 (BAE86927; Gene ID: 411257), and Hbg3 (BAE86928; Gene ID: 406131). Genes Hbg1 and Hbg3 are situated next to each other on the chromosome LG6 (Fig. 1) and gene Hbg2 is positioned on the chromosome LG8. Interestingly, based on the Apis mellifera genome data (Honeybee Genome Sequencing Consortium 2006) the genes Hbg1 and Hbg3 are situated in the intron region (but on a complementary strand) of the phosphodiesterase 1c (Pde1c). This is true also for orthologs of these two genes from N. vitripennis and B. terrestris. Of note are also striking differences in intron number between these genes. While genes Hbg1 and Hbg3 are both intron-rich (7–8 introns), gene Hbg2 is intronless (Nishimoto et al. 2007). It is likely that Hbg2 is a processed gene, result of transcription, splicing, and retroposition. Characteristic features of processed genes are the lack of introns, poly-A tail, and small direct flanking repeats 9–14 bp long (Vanin 1985; Brosius 1999). A comparison between cloned and sequenced cDNA of Hbg2 and corresponding genome DNA sequence revealed that this gene is not divided by introns (Nishimoto et al. 2007). We, therefore, tried to find two other processed gene typical traits, poly-A tract and flanking repeats, but we failed to find any. This could be caused by a relatively high age of Hbg2 gene. We found clear intronless orthologs of this gene in other two studied Hymenoptera species: Bombus terrestris and, more importantly, in distantly related Nasonia vitripennis (Fig. 1). It is, therefore, likely that the gene Hbg2 originated before the split between Aculeata (Apis) and Chalcidoidea (Nasonia) lineages. Clearly distinguishable fossils belonging to Aculeata and Chalcidoidea are believed to be 130–140 million years (MY) old (Grimaldi and Engel 2005) so the two lineages are more than 130 MY old. Therefore, the Hgb2 gene is at least that old. In as much as we did not find this intronless ortholog in any other holometabolan lineage, it is likely that it was not around before major holometabolan divergence took place some 300 million years ago (MYA) (Gaunt and Miles 2002). Alternatively, since Hymenoptera was shown to be the first lineage that branched off from other Holometabola (Savard et al. 2006), it is also possible that this gene was present in a common ancestor of Holometabola. It was retained in Hymenoptera but was lost in the common ancestor of other Holometabola lineages. In any case it is very likely that the Hgb2 is an ancient gene more than 130 MY old. After retroposition both flanking repeats and poly-A tail are presumably dispensable and any signs of them were probably erased after 130 MY of neutral evolution. This hypothesis is further corroborated by low overall similarity between Hbg genes (no more than 50 % identity between amino acid sequences). Thus the only remnants of its processed nature are lack of introns and localization on a different chromosome than the other Hbg genes. Two alternative explanations for Hbg2 origin, i.e., horizontal gene transfer (HGT) of intronless prokaryotic gene or gradual loss of introns, are both less likely. The observation that the Hbg2 gene clusters together with other hymenopteran α-glucosidase genes, rules out the HGT hypothesis (Fig. S1). Since both other Hbg genes have retained seven or eight introns, it seems unlikely that Hgb2 would lose all of them in a gradual manner. Therefore, we think that the Hgb2 fall into a rare category of insect processed genes. Moreover, it is also assumed that the Hbg2 is a functional gene. It has a conserved primary structure, all GH13 catalytic residues are present and no long indels or premature stop codons were identified (Fig. S2). Also a presence of a protein product of the Hbg2 gene was immunologically confirmed in the ventriculus and in the hemolymph (Kubota et al. 2004). It was revealed that the Hbg2 gene codes for enzyme α-glucosidase and its substrate specificity was also characterized (Takewaki et al. 1993). The Hbg2 gene thus unintentionally became one of the most intensively studied insect processed functional gene beside the jing-wei, the alcohol dehydrogenase derived gene found in some Drosophila species (Jeffs et al. 1994; Long et al. 1999).

α-Glucosidases from Hymenoptera. Scheme of spatial organization of GH13 α-glucosidase genes from the order Hymenoptera. Three α-glucosidases (Hbg1, Hbg2, and Hbg3) from Apis mellifera and their orthologs from Bombus terrestris and Nasonia vitripennis are depicted as arrows. Orientation of an arrow shows direction of transcription. Above the arrows is GenBank (Benson et al. 2012) gene identification number (GeneID) for particular gene, and its Hymenoptera Genome Database (BeeBase or NasoniaBase) ID is shown below (Munoz-Torres et al. 2011). Those genes situated next to each other on a chromosome are boxed and the chromosome designation is shown in the left corner. Intergenetic distance is shown as number of nucleotides (nt) between the two genes. Assumed duplication in particular species is represented by two lines connecting the co-orthologous genes. The gene Hbg2 is intronless in all three studied species. The four Hbg2 copies of B. terrestris could be either sequencing artifacts (chromosomal possition of two of them is unknown) or a result of multiple duplications
Substrate and tissue specificity of α-glucosidase coded by other Hbg genes (Hbg1 and Hbg3) from A. mellifera was also studied. The Hbg1 gene is expressed in the ventriculus and product of the gene Hbg3 was found in the hypopharyngeal gland and is also released into the honey (Kubota et al. 2004). Considering the substrate specificity, the Hbg2 gene-coded α-glucosidase is able to hydrolyze isomaltose. The other two α-glucosidases coded by genes Hbg1 and Hbg3 are unable to use isomaltose as their substrate, they utilize only maltose (Kimura et al. 1990; Takewaki et al. 1993; Nishimoto et al. 2001). Although well-studied, their precise in vivo function and substrate specificity remain presently unknown.
Although all hymenopteran Hbg genes cluster together (Fig. S1) and orthology relationships of Hbg genes between three studied species are easily identifiable, it is surprisingly difficult to infer phylogenetic relationship between the three paralogs. Depending on which phylogenetic method is used, the Hbg1 alternately clusters with the Hbg3 or the Hbg2, but every time with a low bootstrap support (around 50 %). It is, therefore, hard to decide, whether the Hbg2 was derived from the common ancestor of Hbg1 and Hbg3 genes or from one of these genes after the common ancestor was duplicated.
Other two Hymenoptera species studied here, Bombus terrestris and Nasonia vitripennis, possess more Hbg genes (both have 6 genes) than Apis (Table S1), although it is not clear if these are true duplicates or artifacts of sequencing process. In genome of B. terrestris we have indentified four copies of Hbg2 ortholog. All are intronless and are localized on different chromosomes (Fig. 1). In the genome of N. vitripennis there are three copies of Hbg1 and two copies of Hbg2 orthologs (intronless). The two Hbg2 copies are situated next to each other on the same chromosome and could be a result of recent duplication.
Beside these three enzyme-coding genes, there are three other genes similar to α-glucosidases in the honey bee genome. Two of them are the heavy chains of HAT proteins (Gabrisko and Janecek 2009). The hcHAT1 gene (XP_624758; Gene ID: 552381) is situated on the chromosome LG5 and hcHAT2 (XP_397564; Gene ID: 409452) is found on the chromosome LG6. Enigmatic is the third GH13 α-glucosidase-like gene (XP_624736; Gene ID: 552357); we designated it as AgluL (α-glucosidase-like gene). It is localized on the chromosome LG5 next to the hcHAT1 gene. AgluL lacks both enzyme and HAT features. It does not possess the complete GH13 catalytic triad (catalytic nucleophile is mutated), what makes it enzymatically inactive. But the transmembrane and intracellular domains necessary for the proper functioning of the hcHAT proteins are also missing; thus it is not a hcHAT gene either (Fig. S2). The AgluL possesses no large indels, nor premature stop codons, and except for missing one of the three catalytic residues it has well conserved typical CSRs and other characteristic features of proteins from the α-amylase family GH13, including domain B (Fig. S2). Moreover, based on the Genbank (Benson et al. 2012) data, mRNA of AgluL gene was isolated from ovary tissue and also from larva and, therefore, it is probably a protein with some non-enzymatic function, i.e., not a pseudogene. Since we found AgluL orthologs in all studied Hymenoptera species including N. vitripennis (NV15743; Gene ID: 100116917, on the chromosome 4), it should be at least 130 MY old. Its position next to hcHAT1 implies that AgluL could be derived from this gene after duplication event. Since there are AgluL orthologs only in Hymenoptera, it could be hypothesized that it originated in the hymenopteran common ancestor after duplicated from the hcHAT1 gene. Unfortunately after phylogenetic analysis this simple scenario seems rather unlikely. HcHAT1 from Apis mellifera is more similar to its orthologs from D. melanogaster and even to the hcHAT1 gene from Acyrthosiphon pisum than it is to the AgluL (Fig. S1). HcHAT1 hymenopteran orthologs also share some distinctive traits not present in the AgluL, e.g., large deletion following CSR around the β8 strand, missing catalytic aspartate on the strand β7, and the presence of transmembrane and intracellular segments characteristic for HAT proteins (Fig. S2). Therefore, if AgluL was derived from the hcHAT1 it should have happened much earlier, probably in the common ancestor of Arthropoda and all AgluL orthologs must have been lost in all Arthropoda lineages other than Hymenoptera. Alternative explanation that the AgluL was derived from Hbg genes is even more improbable, since it does not cluster with any of these genes (Fig. S1). Also why it would be transferred from the close proximity of Hbg genes to the position on the other chromosome next to the hcHAT1 gene need to be explained. There are other known examples of enzymatically inactive GHs which evolved a new function. The plant chitinase-like proteins from the family GH18 that function as lectins and enzyme inhibitors have been well studied (Hennig et al. 1995; Hennig et al. 1996; Payan et al. 2004; Payan et al. 2003; Juge et al. 2004; Durand et al. 2005). Studies of GH13 α-amylases, focused mainly on the genus Drosophila (Da Lage et al. 2000; Zhang et al. 2003) yields one enzymatically inactive protein called Amyrel (Da Lage et al. 1998; Maczkowiak and Da Lage 2006). Its function is still unknown. Further eventual enzymatically inactive α-amylase homologs (one or both catalytic residues are substituted) from the family GH57 were described in prokaryotes (mostly in bacterial order Bacteroidales) (Zona et al. 2004; Janecek and Blesak 2011).
Diptera
Brachycera
From order Diptera we studied the family GH13 α-glucosidases from representatives of suborders Brachycera (genus Drosophila) and Nematocera (Anopheles, Aedes, and Culex). Although we focused on the α-glucosidase (maltase) gene cluster from the genus Drosophila in our previous work (Gabrisko and Janecek 2011), newly available data mainly those on tissue specificity and time of expression (i.e., temporal specificity), could shed more light on a function of particular genes from these gene clusters.
Ten α-glucosidases (Table S1) from the genus Drosophila form two clusters (A and B) located on two chromosomes. They were designated as mal_A1-mal_A8 and mal_B1-mal_B2, respectively (Gabrisko and Janecek 2011). Based on the modENCODE Temporal Expression Data (Graveley et al. 2011) (Fig. 2) and FlyAtlas Anatomical Expression Data (Chintapalli et al. 2007) (Fig. 3) available through the Flybase (McQuilton et al. 2012), most of the ten α-glucosidase genes are transcribed in the midgut of the late embryo (after 20 h) in all three larval instars, extensively in the adult male, and with lower intensity in the adult female (after 5 days the level of transcription decreases). Since the midgut is region where most of the processes of digestion take place, expression pattern of most of the mal genes is consistent with the assumption that they are involved in the utilization of sugars obtained from food. The mal_A1 gene (AAF59089; Gene ID: 35824) is the most intensively transcribed. The gene mal_A2 (AAL49188b; Gene ID: 35825) is transcribed not only in the midgut but also in the hindgut, and the gene mal_A6 (CG30360; Gene ID: 246565) is also transcribed in the larval fat body (Figs. 2, 3). The gene mal_A7 (CG11669; Gene ID: 35829) is transcribed only in the adult life stage (Fig. 2). The presence of ten genes coding for α-glucosidases raises an interesting question, namely: why so many α-glucosidase genes are needed? It is possible that particular genes have specialized for different substrates or reaction conditions. Or they could have evolved some completely different function. Atypical expression pattern of three mal genes (mal_A5, mal_B1, and mal_B2) seems to support this assumption (Figs. 2, 3). The gene mal_A5 (CG30359; Gene ID: 35828) is transcribed already in the early embryo (after first 6 h) (Fig. 2) and, interestingly, instead of dominant transcription in the midgut it is transcribed mostly in the fat body of the larvae (Fig. 3). The larval fat body is known to be an energy reservoir used through the non-feeding period but it is also a major secretory organ. Therefore, it is of interest that the gene mal_A5 is most actively transcribed in the puffstage of the third larval instar and in the white prepuae (Fig. 2). In the adult stage, transcription pattern changes. Transcription in the fat body decreases but there occurs a transcription in many other tissues including spermatheca, thoracic-abdominal ganglion, ovary, heart, brain, and eye. Transcription in these organs is of comparable and relatively high intensity (Fig. 3). This transcription pattern hints at some non-enzymatic, perhaps regulatory function. Similar expression profile has the mal_B2 gene (AAN10789; Gene ID: 34598). High level of transcription detected in the fat body of the larvae contrasts to very low transcription level in the midgut (Figs. 2, 3). In the adult stage this gene is extensively transcribed in the fat body and in the spermatheca. Level of transcription in these two organs is even higher than that of the mal_A5. A unique transcription pattern is possessesed by the gene mal_B1 (CG14934; Gene ID: 34597) (Figs. 2, 3). It is almost exclusively transcribed in the salivary glands of adults (same intensity of transcription in female as seen in male). Interestingly, the highest level of transcription was detected in the late pupae stage (Fig. 2). Although it is possible that the above-mentioned genes play some non-enzymatic role (based on their rather atypical expression pattern), all these three genes possess all amino acid residues needed for their proper enzymatic function (the family GH13 catalytic triad and substrate-binding residues) and their primary structure actually do not differ much from the other mal genes (in the coding regions) (Fig. S2). Since we assume that these genes are at least 60 MY old (Gabrisko and Janecek 2011), to retain all the enzyme characteristics, it is likely that they have been protected by purifying selection. Whatever their function is, it thus probably still involves some enzymatic activity.

Temporal specificity of α-glucosidase genes from D. melanogaster. In the left column particular ontogenetic stages of D. melanogaster are listed. Development of the embryo is divided into 12 substages each representing 2 h of development followed by three larval instars stages, where the third stage is further divided into particular puffstages. Level of transcription was also characterized in the white prepupae (at the early stage and then after 12 and 24 h, respectively) and 2, 3, and 4 days after white prepuae (postWPP). Finally level of transcription was measured in 1, 5, and 30 days old adult male and female. Relative amount of transcription is represented by a rectangle length. Transcription activity is shown for four maltase genes. The gene mal_A1 represents a typical gut maltase involved in the saccharide metabolism, the remaining three genes (mal_A5, mal_B1, and mal_ B2) exhibit different transcription profiles, probable indicating a new, probably non-metabolic function. Where no rectangles are shown, no or low transcription was detected, whereas black-filled are those with intensive transcription activity. The depicted transcription profiles are based on the modENCODE Temporal Expression Data (Graveley et al. 2011) available though the Flybase (McQuilton et al. 2012)

Tissue specificity of α-glucosidase genes from D. melanogaster. In the middle column particular tissues of D. melanogaster, where transcription activity was measured, are listed. Relative amount of transcription is represented by a rectangle length. Transcription activity is shown for four maltase genes. Where no rectangles are shown, no or low transcription was detected, whereas black-filled are those with intensive transcription activity. Transcription activity detected in the larva and in the adult stage is shown in the left and right columns, respectively. The gene mal_A1 represents a typical gut maltase involved in the saccharide metabolism, the remaining three genes (mal_A5, mal_B1, and mal_ B2) show different transcription profiles, probably indicating a new, probably non-metabolic function. The depicted transcription profiles are based on the FlyAtlas Anatomical Expression Data (Chintapalli et al. 2007) available through the Flybase (McQuilton et al. 2012)
Nematocera
From the suborder Nematocera we studied α-glucosidase genes in three mosquito species: Aedes aegypti (the yellow fever mosquito), Anopheles gambiae (the malaria mosquito), and C. quinquefasciatus (the common house mosquito, vector of the West Nile virus). We found out that most of these genes are localized in two gene cluster (A and B; each consists of three to five genes) situated on two different chromosomes (Table S1). The orthology relationship of maltase genes between the three mosquito species is depicted in Fig. 4. C. quinquefasciatus seems to possess (unlike other two mosquitoes) two copies of the second cluster, but is not clear if this extra set of genes represents a true duplication or more likely a sequencing artifact, since genes of C. quinquefasciatus have not been placed on the chromosome yet. Although all genes from one cluster are more similar to each other than any of them is to genes from other cluster, what signalizes that there were no interchromosomal gene transfer, we found signs of intrachromosomal gene movement. It would be expected that neighboring genes should be more similar to each other than the more distant ones, but based on our phylogenetic analysis (using ML, MP, and NJ methods) genes Aglu_1 and Aglu_4 seem to be more similar to each other than either is to in between lying genes Aglu_2 and Aglu_3. The same can be said about genes Aglu_6 and Aglu_8 bordering gene Aglu_7, another example is the gene Aglu_10 that is more similar to distant gene Aglu_11 than it is to its close neighboring gene Aglu_ 9 (Fig. 4 and Fig. S1). These discrepancies are either due to gene movement, possibly caused by small scale inversions or the phylogenetic methods used were unable to reveal true relationships between particular paralogs.

α-Glucosidases from mosquitos. Scheme of spatial organization of GH13 α-glucosidase genes (Aglu_1-Aglu_14) on chromosomes from three mosquito species Aedes aegypti, Anopheles gambiae, and C. quinquefasciatus. Particular genes are depicted as an arrow; its orientation shows direction of transcription. The identification number of a particular gene in the VectorBase (Lawson et al. 2009) is shown above the arrows. Genes situated next to each other on a chromosome are boxed and the chromosome designation is shown in the left corner (“Un” stands for unknown). Intergenic distance is shown as number of nucleotides (nt) between the two genes. Thirteen α-glucosidase genes (Aglu_1–Aglu_14) of A. aegypti are connected by dotted lines with their orthologs from two other mosquito species. The gene Aglu_2 although present in A. gambiae and C. quinquefasciatus is missing in A. aegypti. In parentheses there are names of characterized α-glucosidase genes
Forasmuch studied mosquitoes possess comparable number of α-glucosidase genes as drosophilas do, it would be interesting to reveal their substrate and tissue specificity. Presently only few of these genes were characterized. One of them is gene MalI, which produces α-glucosidase in the salivary glands (in female found only in the lateral lobes of this tissue) of adult A. aegypti (James et al. 1989; Marinotti and James 1990). It corresponds to our genes Aglu_12 (EAT48589; Gene ID: 5576332) and Aglu_13 (EAT38606; Gene ID: 5572111) and is localized outside the two main gene clusters, but is still situated on the chromosome 3 as are genes from the cluster B (Fig. 4). It was believed that salivary α-glucosidase is used for digestion of sugars (in crop or midgut) found in nectar on which mosquitoes feed. But since no α-glucosidase activity was found in the crop of Anopheles aquasalis (Souza-Neto et al. 2007) it is more probable that it participates in the sugar solubilization (Eliason 1963) and intracellular metabolism (Dillon and El Kordy 1997). Its ortholog from A. gambiae (EAA00998; Gene ID: 1281368) was reported to be also expressed in the salivary glands (Kalume et al. 2005). Since the analysis of whole proteome of salivary glands did not reveal any other salivary α-glucosidase, mentioned gene is probably the only Aglu gene expressed in this tissue. Also two midgut α-glucosidases were identified (Zheng et al. 1995). The agm2 gene is expressed only in adult. The transcription of agm1 begins earlier in pupal or pharate adult stage and its intensity increases in the adult stage. This gene is probably transcribed (at very low level) not only in the midgut but also in some other tissues as well (Zheng et al. 1995). Both genes are part of cluster B positioned on the chromosome 3L. The agm1 corresponds to our gene Aglu_7 (EAA00181; Gene ID: 3291576) and the agm2 corresponds to Aglu_8 (EAA00179; Gene ID: 1280462) (Fig. 4). The protein product of genes Agm3 from A. gambiae (Opota et al. 2008) and its orthologs: Cpm1 from C. pipiens (Silva-Filha et al. 1999; Darboux et al. 2001), Cqm1 from C. quinquefasciatus (Romão et al. 2006), and Aam1 from A.aegypti (Ferreira et al. 2010) were identified as a GPI membrane-bound α-glucosidase expressed in the midgut of larvae. The gene Agm3 corresponds to our gene Aglu_5 (EAA14808; Gene ID: 1279927; N-terminal part is missing in the genomic sequence) and is localized on the chromosome 3R as part of the cluster A (Fig. 4). It is expressed in the midgut of fourth instar larvae but not in adult male or non-bloodfed females. Its expression increases in the midgut tissue 3 h after the blood meal. Based on primary structure analysis protein product of Agm3 gene resembles α-glucosidase but enzymatic activity has not been detected so far (Opota et al. 2008). Its ortholog (Aglu_5) from C. quinquefasciatus, the gene Cqm1 (EDS38953; Gene ID: 6046508), is also expressed in the midgut of larvae (Romão et al. 2006). Interestingly, when strains where Cqm1 is expressed are compared with those where it is not, the overall α-glucosidase activity is almost the same, but non-functional Cqm1 still has a negative impact on fitness. It is thus possible that Cqm1 could play also some other probably non-enzymatic role. The Aam1 (EAT37485; Gene ID: 5573501), an ortholog of this gene from A. aegypti, is expressed in the midgut of larvae as well as is in the adult stage (female and male) (Ferreira et al. 2010). Interestingly, the gene Aglu_5 not only codes for a functional α-glucosidase, but it also serves as the binary toxin receptor recognized by binary toxin, which is a toxin produced by Bacillus sphaericus used as an anti-mosquito pesticide (Silva-Filha et al. 1999; Darboux et al. 2001; Romão et al. 2006; Opota et al. 2008; Ferreira et al. 2010). Recently it was shown that although A. aegypti possesses GPI-anchored Bin toxin receptor (the gene Aam1), expressed in the larval midgut, it is still unable to bind the binary toxin binding subunit (BinB) what makes A. aegypti resistant to this toxin. Hypothetical minor substitutions in (presently unknown) binding epitope or influence of glycosylation on conformation of this epitope (since Aam1 is glycosylated and Cpm1 is not) were proposed as an explanation why protein product of Cpm1 (C. pipiens) is capable of binding to Bin toxin and its highly similar ortholog Aam1 is not (Ferreira et al. 2010).
If products of the genes Aglu_12 and Aglu_13 are salivary α-glucosidases and those of Aglu_5, Aglu_7, and Aglu_8 (possibly Aglu_6) are midgut α-glucosidases, it should be of interest to know what is coded by other Aglu genes? All Aglu genes possess the sequence features characteristic of the subfamily GH13_7 α-glucosidases (Oslancova and Janecek 2002; Stam et al. 2006) and thus they likely also code for α-glucosidases (Fig. S2). Since none of them contains large indels and/or premature stop codons, they are either new genes and not enough time has passed to accumulate deleterious mutations or are under purifying selection. It is believed that subfamilies Anophelinae and Culicinae have diverged more than 60 MYA and tribes Culicini and Aedini more than 50 MYA (Foley et al. 1998; Krzywinski et al. 2006). Thus if orthologs of particular genes are present in any two of the three studied species, they have to be at least that old. All Aglu genes have orthologs in at least two species indicating that no one is younger than 50 MY. To determine if Aglu genes are actively transcribed we searched for corresponding ESTs in the GenBank database (Benson et al. 2012). Surprisingly no ESTs were found for the genes Aglu_1 and Aglu_2 in any of the three studied species. The gene Aglu_2 is missing in A. aegypti and since it is present in A. gambiae and C. quinquefasciatus, it was likely present also in the common ancestor of the three mosquito species (A. aegypti and C. quinquefasciatus are more closely related) and lost in the A. aegypti. Moreover, one of the three catalytic residue (GH13 catalytic nucleophile—the β4 strand aspartate) is mutated in the Aglu_2 gene of A. gambiae (EDO63381; Gene ID: 5668268) which would make the product of this gene enzymatically inactive (Fig. S2). The Aglu_2 thus could be a pseudogene. Only short ESTs were found for the genes Aglu_10 (missing in A. gambiae) and Aglu_14. ESTs covering the whole sequence were found for genes Aglu_3, Aglu_4, Aglu_6, Aglu_9, and Aglu_11. These currently uncharacterized genes are probably actively transcribed α-glucosidases. We detected the mRNA of the genes Aglu_4 and Aglu_9 in the head region (possibly from the salivary glands) and mRNA of gene Aglu_11 was found in the midgut (Gomez et al. 2005).
Brachycera/Nematocera Relationship
Based on the phylogenetic reconstruction (methods NJ, MP, and ML) (Fig. S1), common ancestor of Diptera already had two Aglu gene clusters positioned on the two chromosomes (ancestral—A_ch1 and A_ch2) (Fig. 5). On the A_ch1 there were probably four genes (A–D). Three of them were lost (B, C, D) in the Brachycera lineage and the remaining one (A) was duplicated to the extant genes mal_B1 and mal_B2. In Nematocera lineage all four ancestral genes were retained and became the extant genes Aglu_1-Aglu_5 and the gene Aglu_14 (Fig. 5). On the second chromosome (A_ch2) there were probably two genes (E, F) and none of them was lost. In the Brachycera lineage one of these genes (F) became the gene mal_A1, while the second one (E) gave birth to all remaining maltase genes of cluster A (mal_A2-mal_A8). In the Nematocera lineage the gene F was duplicated to genes Aglu_7 and Aglu_12, 13, the other one (E) was ancestor of genes Aglu_6, 8, 9, 10, 11 (Fig. 5). Interestingly, like in the Brachycera lineage, we did not observe any duplication that would take place among Aglu genes in the last 50 MY. Of note are also differences in the tissue specificity of proposed orthologs between the Brachycera and the Nematocera lineage (Fig. 6). The gene mal_A1 from D. melanogaster is transcribed in the midgut but its mosquito orthologs Aglu_12 and Aglu_13 are transcribed in the salivary glands. The only salivary α-glucosidase identified in the Drosophila is the mal_B1 (Fig. 6). The primary structure of this gene is most similar to its Drosophila paralog mal_B2 (transcribed in the fat body) and they both cluster together with mosquito (midgut) gene Aglu_1 (Fig. S1). Six genes transcribed in the midgut of D. melanogaster cluster together with other Drosophila gene—the mal_A5, which is transcribed in the fat body (Fig. S1). It is, therefore, likely that extant tissue specificity was established independently after lineage-specific duplications in Brachycera and Nematocera lineages and was not inherited from their common ancestor. This is also the reason why we were unable to determine tissue specificity of ancestral α-glucosidases.

Diptera phylogeny. Schematic representation of order of duplication of genes from the genus Drosophila and Aedes. Particular genes are depicted as a number in a box (e.g., 1 in Aedes lineage stands for the gene Aglu_1 and A1 in Drosophila lineage stands for the gene mal_A1). Arrows pointing from the boxes represent particular duplication events. Six ancestral genes (A–F) localized on two ancestral chromosomes (ch 1 and ch 2) are shown left. These genes were inherited by Aedes and Drosophila lineages and after multiple duplications became extant genes localized on the chromosomes ch 1, ch 3 and ch 2L, ch 2R (right)

Comparison of tissue specificities of α-glucosidases between Brachycera and Nematocera. Schematic representation of tissue specificities of α-glucosidases from suborders Brachycera and Nematocera. On the left side genes from Brachycera are depicted; and on the opposite side those from Nematocera are depicted. The genes transcribed in the same organ are clustered together in a circle. Assumed orthologs are connected with a dotted line
Other metazoan Groups
Considering other Arthropoda groups (where genome data are available) we have found only two α-glucosidase genes in the Coleoptera representative Tribolium castaneum (Table S1). This small number contrasts with 14 α-glucosidase genes identified in the genome of phloem sap sucking Hemiptera Acyrthosiphon pisum (Table S1). One of these genes (the gene Acypi04 in our classification; ABB55878; Gene ID: 100144774) was characterized as the gut sucrase APS1 (Price et al. 2007). APS1 is probably dominant or perhaps sole gut sucrase in A. pisum because no other family GH13 enzyme was found in the gut-specific EST library; the APS1 being the only PCR product amplified from the gut cDNA using GH13 family specific degenerate primers and no other protein was identified by mass spectrometry in the sucrose enriched gut membrane fractions (Price et al. 2007). It was shown that the gut α-glucosidase is not only involved in the sugar digestion but is also essential for osmoregulation, since sucrose cannot be transported across the gut wall and causes the high osmotic pressure of the phloem sap (Downing 1978; Fisher et al. 1984; Wilkinson et al. 1997). After the sucrose is hydrolyzed to glucose and fructose, glucose is incorporated into glucose oligosaccharides 20 hexose units long via its transglucosidase activity (Walters and Mullin 1988; Rhodes et al. 1997; Ashford et al. 2000; Cristofoletti et al. 2003; Price et al. 2007). Although enzymatic activity of this protein resembles that of other GH13 protein, bacterial amylosucrase from Neisseria polysaccharea (Hehre et al. 1949; Skov et al. 2001), the sucrase from A. pisum clusters together with other arthropodan α-glucosidases. Based on our phylogenic analysis these two proteins thus do not share recent common ancestor and the enzymatic activity similarities are more likely result of a convergent evolution.
Although we have identified 13 other putative α-glucosidase genes coding for the proteins from the family GH13 and found (at least partial) transcripts (searching the GenBank database), for ten of them (genes 1–4, 6, 8, 10, 12–14) we were unable to determine their tissue specificity. Forasmuch genes of A. pisum have not been mapped on the chromosome so far, their precise position is unknown. However, genes 10, 6, 4, 3 are localized next to each other on the same contig and genes 1 and 2 are together on another one. It is thus probable that α-glucosidase genes in A. pisum form gene clusters much like they do in the other studied insect groups.
Interestingly no α-glucosidase genes were identified in the other studied Paraneoptera species—Pediculus humanus corporis. The striking differences in α-glucosidase gene number between these related species could be the result of their different life styles, since A. pisum feed on the phloem sap, whereas P. humanus corporis is highly specialized blood-sucking parasite. This hypothesis is further corroborated by the absence of α-glucosidase genes in Ixodes scapularis, unrelated species (Chelicerata) with similar blood-sucking life style. Two α-glucosidase genes were identified in Daphnia pulex (Crustacea) (Table S1).
Based on phylogenetic reconstruction using all the three methods (NJ, MP, and ML) α-glucosidases from Diptera, Hymenoptera, and Paraneoptera form three separate clusters (Fig. S1). It is thus probable that their common ancestor had only limited number of α-glucosidase genes that went through process of lineage-specific gene expansion after those groups were separated.
Fungi
We have analyzed 103 putative α-glucosidase genes from 37 fungal genomes. Most species contain from one to six genes. Based on the phylogenetic analysis (NJ, MP, and ML; Fig. S1) we assume that the common ancestor of Saccharomyceta possessed two α-glucosidase genes. In the subphylum Saccharomycotina, one of these genes (gene A) was lost and the other one (the gene B) went through process of lineage-specific gene duplication. In the subphylum Pezizomycotina, in the common ancestor of Leotiomyceta, both genes were retained and one of them (the gene B) was duplicated. Different evolutionary histories observed in the two subphyla are of interest. Pezizomycotina experienced two ancient gene duplications already in their common ancestor and orthology/paralogy relationship of these two genes in different species can easily be determined. In contrast, in the subphylum Saccharomycotina we observed relatively recent species-specific duplications. We have detected three putative cases of HGT among fungi, where position of these genes in the phylogenetic trees is in conflict with taxonomic classification. One of two genes (CAG87533; Gene ID: 2902646) from Debaryomyces hansenii (Saccharomycotina) clusters together with Sordariomycetes (Pezizomycotina) although with low bootstrap value (Fig. S1). One α-glucosidase gene from Cryptococcus neoformans (EAL20081; Gene ID: 4936959) and its ortholog from Cryptococcus gattii (ADV23114; GI: 10190495) are found outside of the Fungi group (Fig. S1). Both species possess second α-glucosidase gene that clusters together with other fungal sequences.
Considering enzyme substrate specificity of α-glucosidases, fungi are known to possess both maltases (Winge and Roberts 1950; Barnett 1976; Cohen et al. 1984; Charron et al. 1986; Novak et al. 2004) and isomaltases (Teste et al. 2010). To discern maltase from isomaltase we have looked for valin localized in CSR II on the β4 strand (Fig. S2) that is typical for isomaltases (Yamamoto et al. 2004). In Pezizomycotina all isomaltases are monophyletic, coded by ancestral gene A (Fig. S1). In subphylum Saccharomycotina gene A was lost and isomaltase specificity evolved repeatedly and independently in Saccharomyces, Candida, and Meyerozyma lineage.
Origin of the Eukaryotic GH13 α-Glucosidases
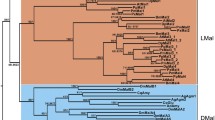
Eukaryotic α-glucosidases from the family GH13 are found mostly in two groups i.e., in Arthropoda and Fungi. Although present in prokaryotes, they are absent in many eukaryotic lineages. We were not able to find GH13 α-glucosidases in most protozoa, plants, nematodes, or vertebrates. Thus if ancestral to eukaryotes (inherited from some prokaryotic ancestor) they would have been lost multiple times in different eukaryotic lineages. Alternatively, the GH13 α-glucosidases could have come from bacteria through HGT. If they were transferred into a common ancestor of Unikonts, they should be present in at least some amebas. We were able to find only two unambiguous enzymes from oligo-1,6-glucosidase subfamily of family GH13 in two members of Amoebozoa. One was protein EHI_130690 from Entamoeba histolytica HM-1:IMSS (GenBank accession number: EAL43779; Gene ID: 3403457) (Tab. S1) the other one was DICPUDRAFT_55969 protein from Dictyostelium purpureum (GenBank accession number: XP_003289239; Gene ID: 10500235) (Tab. S1). The latter one is almost certainly not an α-glucosidase, but more likely trehalose synthase that cluster together with its homolog from Pimelobacter sp. (Fig. 7). Other studied species of Amoebozoa lack GH13 α-glucosidase (Tab. S1).

Phylogenetic tree of α-glucosidases and related proteins from the α-amylase family. The phylogenetic tree was calculated with NJ method using amino acid sequences of eukaryotic α-glucosidases. Bacterial representatives of most closely related enzyme specificities were also included in the analysis: α-glucosidase (AGL), dextran glucosidase (DGL), oligo-1,6-glucosidase (OGL), isomaltulose synthase (ISY), trehalose synthase (TSY), trehalose-6-phosphate hydrolase (T6P), and putative GH13 α-glucosidase representatives of main bacterial and archaeal phyla. Non-arthropod metazoan sequences could be either enzymes or heavy chains of HAT proteins. Bootstrap support is shown as a number (0–1,000) near every node
Considering Opisthokonts, GH13 α-glucosidases are found in Metazoa, Fungi and we identified one also in the genome of Salpingoeca sp. which belongs to order Choanoflagellida. Surprisingly, no GH13 α-glucosidases were found in other choanoflagellate species, Monosiga brevicollis MX1. If GH13 α-glucosidases were ancestral to Opisthokonts, it would be expected that those from Fungi and Arthropoda would cluster together and apart from their bacterial homologs, but they do not (Fig. 7 and Fig. S1). Although with low bootstrap values, fungal α-glucosidases are always rooted deeply in the prokaryotic group, in all phylogenetic reconstructions, using all the three methods (NJ, MP, and ML). Low bootstrap support could signalize that phylogenetic methods used were unable to infer correct evolutionary relationships between α-glucosidases from distantly related taxa. But there could be other explanations of this observation. Either fungal α-glucosidases have retained ancestral similarity with bacterial α-glucosidases while those from Arthropoda have experienced period of accelerated evolution or fungal α-glucosidases were obtained from Bacteria through HGT. This hypothesis is further corroborated by a lack of shared intron position (not shown) between these two groups. Multiple horizontal transfer events were also proposed to explain similarity between α-amylases (related enzyme specificity from the family GH13) of unrelated taxonomic groups (Da Lage et al. 2004; Da Lage et al. 2007). If HGT did occur, it was very likely an ancient event because we were able to identify GH13 α-glucosidase in every fungal genome studied (Tab. S1) and fungal GH13 α-glucosidases seem to be monophyletic.
Although presently GH13 α-glucosidases are found mostly in Arthropoda, there is a good reason to believe that they are ancestral to all Metazoa. Beside enzymes, there are hcHATs—non-enzymatic proteins, universally present in most metazoan lineages, which were shown to be homologous to GH13 α-glucosidases (Wells and Hediger 1992; Janecek et al. 1997; Chillaron et al. 2001; Broer and Wagner 2002; Gabrisko and Janecek 2009). If we assume that ancestral GH13 α-glucosidase was changed in early stages of evolution of Metazoa into the hcHAT protein and its enzymatic nature was retained in Arthropoda, there would be no need for multiple lost in various metazoan lineages. But Arthropoda possess also two types of hcHAT proteins beside their α-glucosidases. Therefore, it is more likely that ancient α-glucosidase gene was duplicated. One of these genes became ancestor of all the hcHATs and the second one, which remained enzymatically active, was retained in Arthropoda and lost multiple times in other lineages after all. Alternatively arthropodan α-glucosidases could have been independently obtained from bacteria through HGT. Since arthropodan α-glucosidases cluster together with hcHAT proteins our phylogenetic analysis did not provide much support for this hypothesis (Fig. 7).
There is one more intriguing possibility able to circumvent the multiple lost problem that need to be evaluated. Ancestral hcHAT protein could have been duplicated in Arthropoda and α-glucosidase enzymatic activity could have been then after restored in one of the two copies. At first glance this scenario seems unlikely because many features responsible for enzyme function, including one or more catalytic residues, are lost in present-day arthropodan hcHAT proteins. On the other hand, complete catalytic triad and most binding residues are present in hcHAT proteins of vertebrates. It is thus probable that the common ancestor of Arthropoda and Vertebrata possessed hcHAT with dormant enzymatic potential and could have been reverted to functional enzyme. Our phylogenetic analysis does lend some support to this interesting scenario. Arthropodan hcHAT proteins cluster together with arthropodan α-glucosidases and not with their orthologs from vertebrates (Fig. 7; Fig. S1).
Conclusions
The α-glucosidases from the α-amylase family GH13 are found mostly in Fungi and in Arthropoda, where they form large multigene families in some species (e.g., Acyrthosiphon or mosquitos) and are missing in parasitic ones (Ixodes and Pediculus). Most studied genes possess all catalytic and binding residues and are thus likely active α-glucosidase enzymes involved in the saccharide metabolism. Although particular paralogs likely have retained their enzymatic activity they differ in tissue and temporal specificity. Some α-glucosidase genes (e.g., mal_A5, mal_B1, and mal_B2) have probably evolved new function different from their original role in the saccharide metabolism. With increased phylogenetic distance between species the amount of detectable orthology among particular α-glucosidase genes decreases. When considering distantly related groups all α-glucosidase genes from one species are co-orthologs of α-glucosidases from the other species. The studied genes thus likely evolve according to birth-and-death model (Nei et al. 1997; Nei and Rooney 2005). We also revealed quite a complex evolutionary history of the eukaryotic α-glucosidases probably involving multiple losses of genes or HGT from bacteria.
Abbreviations
- CSR:
-
Conserved sequence region
- GH:
-
Glycoside hydrolase
- GPI:
-
Glycosylphosphatidylinositol
- ML:
-
Maximum likelihood
- MP:
-
Maximum parsimony
- MY:
-
Million years
- MYA:
-
Million years ago
- NJ:
-
Neighbor-joining
References
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105
Alam MS, Nakashima S, Deyashiki Y, Banno Y, Hara A, Nozawa Y (1996) Molecular cloning of a gene encoding acid alpha-glucosidase from Tetrahymena pyriformis. J Eukaryot Microbiol 43:295–303
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Ashford DA, Smith WA, Douglas AE (2000) Living on a high sugar diet: the fate of sucrose ingested by a phloem-feeding insect, the pea aphid Acyrthosiphon pisum. J Insect Physiol 46:335–341
Barnett JA (1976) The utilization of sugars by yeasts. Adv Carbohydr Chem Biochem 32:125–234
Benson DA, Karsch-Mizrachi I, Clark K, Lipman DJ, Ostell J, Sayers EW (2012) GenBank. Nucleic Acids Res 40(Database issue):D48–D53
Birney E, Clamp M, Durbin R (2004) Genewise and genomewise. Genome Res 14:988–995
Broer S, Wagner CA (2002) Structure-function relationships of heterodimeric amino acid transporters. Cell Biophys 36:155–168
Brosius J (1999) RNAs from all categories generate retrosequences that may be exapted as novel genes or regulatory elements. Gene 238:115–134
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37(Database issue):D233–D238
Charron MJ, Dubin RA, Michels CA (1986) Structural and functional analysis of the MAL1 locus of Saccharomyces cerevisiae. Mol Cell Biol 6:3891–3899
Chillaron J, Roca R, Valencia A, Zorzano A, Palacin M (2001) Heteromeric amino acid transporters: biochemistry, genetics, and physiology. Am J Physiol Renal Physiol 281:995–1018
Chintapalli VR, Wang J, Dow JAT (2007) Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet 39:715–720
Cohen JD, Goldenthal MJ, Buchferer B, Marmur J (1984) Mutational analysis of the MAL1 locus of Saccharomyces: identification and functional characterization of three genes. Mol Gen Genet 196:208–216
Cristofoletti PT, Ribeiro AF, Deraison C, Rahbé Y, Terra WR (2003) Midgut adaptation and digestive enzyme distribution in a phloem feeding insect, the pea aphid Acyrthosiphon pisum. J Insect Physiol 49:11–24
Da Lage JL, Renard E, Chartois F, Lemeunier F, Cariou ML (1998) Amyrel, a paralogous gene of the amylase gene family in Drosophila melanogaster and the Sophophora subgenus. Proc Natl Acad Sci USA 95:6848–6853
Da Lage JL, Maczkowiak F, Cariou ML (2000) Molecular characterization and evolution of the amylase multigene family of Drosophila ananassae. J Mol Evol 51:391–403
Da Lage JL, Feller G, Janecek S (2004) Horizontal gene transfer from Eukarya to bacteria and domain shuffling: the alpha-amylase model. Cell Mol Life Sci 61:97–109
Da Lage JL, Danchin EG, Casane D (2007) Where do animal α-amylases come from? An interkingdom trip. FEBS Lett 581:3927–3935
Darboux I, Nielsen-LeRoux C, Charles JF, Pauron D (2001) The receptor of Bacillus sphaericus binary toxin in Culex pipiens (Diptera: Culicidae) midgut: molecular cloning and expression. Insect Biochem Mol Biol 31:981–990
Dennis JA, Moran C, Healy PJ (2000) The bovine alpha-glucosidase gene: coding region, genomic structure, and mutations that cause bovine generalized glycogenosis. Mammalian Genome 11:206–212
Dillon RJ, El Kordy E (1997) Carbohydrate digestion in sandflies: α-glucosidase activity in the midgut of Phlebotomus langeroni. Comp Biochem Physiol B Biochem Mol Biol 116:35–40
Downing N (1978) Measurements of the osmotic concentrations of stylet sap, haemolymph and honeydew from an aphid under osmotic stress. J Exp Biol 77:247–250
Durand A, Hughes R, Roussel A, Flatman R, Henrissat B, Juge N (2005) Emergence of a subfamily of xylanase inhibitors within glycoside hydrolase family 18. FEBS J 272:1745–1755
Eck RV, Dayhoff MO (1966) Atlas of protein sequence and structure. National Biomedical Research Foundation, Silver Springs
Eliason DA (1963) Feeding adult mosquitoes in solid sugars. Nature 200:289
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Ferreira LM, Romão TP, de Melo-Neto OP, Silva-Filha MH (2010) The orthologue to the Cpm1/Cqm1 receptor in Aedes aegypti is expressed as a midgut GPI-anchored α-glucosidase, which does not bind to the insecticidal binary toxin. Insect Biochem Mol Biol 40:604–610
Fisher DB, Wright JP, Mittler TE (1984) Osmoregulation by the aphid Myzus persicae: a physiological role for honeydew oligosaccharides. J Insect Physiol 30:387–393
Foley DH, Bryan JH, Yeates D, Saul A (1998) Evolution and systematics of Anopheles: insights from a molecular phylogeny of Australasian mosquitoes. Mol Phylogenet Evol 9:262–275
Gabrisko M, Janecek S (2009) Looking for the ancestry of the heavy-chain subunits of heteromeric amino acid transporters rBAT and 4F2hc within the GH13 α-amylase family. FEBS J 276:7265–7278
Gabrisko M, Janecek S (2011) Characterization of maltase clusters in the genus Drosophila. J Mol Evol 72:104–118
Gaunt MW, Miles MA (2002) An insect molecular clock dates the origin of the insects and accords with palaeontological and biogeographic landmarks. Mol Biol Evol 19:748–761
Geber A, Williamson PR, Rex JH, Sweeney EC, Bennett JE (1992) Cloning and characterization of a Candida albicans maltase gene involved in sucrose utilization. J Bacteriol 174:6992–6996
Gomez SM, Eiglmeier K, Segurens B, Dehoux P, Couloux A, Scarpelli C, Wincker P, Weissenbach J, Brey PT, Roth CW (2005) Pilot Anopheles gambiae full-length cDNA study: sequencing and initial characterization of 35,575 clones. Genome Biol 6:R39
Graveley BR, Brooks A, Carlson JW et al (2011) The developmental transcriptome of Drosophila melanogaster. Nature 471:473–479
Grimaldi D, Engel MS (2005) Evolution of the insects. Cambridge University Press, Cambridge
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Han EK, Cotty F, Sottas C, Jiang H, Michels CA (1995) Characterization of AGT1 encoding a general α-glucoside transporter from Saccharomyces. Mol Microbiol 17:1093–1107
Hehre EJ, Hamilton DM, Carlson AS (1949) Synthesis of a polysaccharide of the starch-glycogen class from sucrose by a cell-free, bacterial enzyme system (amylosucrase). J Biol Chem 177:267–279
Henikoff S, Wallace JC (1988) Detection of protein similarities using nucleotide sequence databases. Nucleic Acids Res 16:6191–6204
Hennig M, Pfeffer-Hennig S, Dauter Z, Wilson KS, Schlesier B, Nong VH (1995) Crystal structure of narbonin at 1.8 Å resolution. Acta Crystallogr D Biol Crystallogr 51:177–189
Hennig M, Jansonius JN, Terwisscha van Scheltinga AC, Dijkstra BW, Schlesier B (1996) Crystal structure of concanavalin B at 1.65 Å resolution. An “inactivated” chitinase from seeds of Canavalia ensiformis. J Mol Biol 254:237–246
Hermans MM, Kroos MA, van Beeumen J, Oostra BA, Reuser AJ (1991) Human lysosomal alpha-glucosidase. Characterization of the catalytic site. J Biol Chem 266:13507–13512
Hoefsloot LH, Hoogeveen-Westerveld M, Reuser AJ, Oostra BA (1991) Characterization of the human lysosomal alpha-glucosidase gene. Biochem J 272:493–497
Honeybee Genome Sequencing Consortium (2006) Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443:931–949
Huber RE, Thompson DJ (1973) Studies on a honey bee sucrase exhibiting unusual kinetics and transglucolytic activity. Biochemistry 12:4011–4020
James AA, Blackmer K, Racioppi JV (1989) A salivary gland-specific, maltase-like gene of the vector mosquito, Aedes aegypti. Gene 75:73–83
Janecek S (1995) Close evolutionary relatedness among functionally distantly related members of the (α/β)8-barrel glycosyl hydrolases suggested by the similarity of their fifth conserved sequence region. FEBS Lett 377:6–8
Janecek S (2002) How many conserved sequence regions are there in the α-amylase family? Biologia 57(Suppl. 11):29–41
Janecek S, Blesak K (2011) Sequence-structural features and evolutionary relationships of family GH57 α-amylases and their putative α-amylase-like homologues. Protein J 30:429–435
Janecek S, Svensson B, Henrissat B (1997) Domain evolution in the α-amylase family. J Mol Evol 45:322–331
Janecek S, Svensson B, MacGregor EA (2007) A remote but significant sequence homology between glycoside hydrolase clan GH-H and family GH31. FEBS Lett 581:1261–1268
Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ (1998) Multiple sequence alignment with Clustal X. Trends Biochem Sci 23:403–405
Jeffs PS, Holmes EC, Ashburner M (1994) The molecular evolution of the alcohol dehydrogenase and alcohol dehydrogenase-related genes in the Drosophila melanogaster species subgroup. Mol Biol Evol 11:287–304
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Jones DC, Mehlert A, Güther ML, Ferguson MA (2005) Deletion of the glucosidase II gene in Trypanosoma brucei reveals novel N-glycosylation mechanisms in the biosynthesis of variant surface glycoprotein. J Biol Chem 280:35929–35942
Juge N, Payan F, Williamson G (2004) XIP-I, a xylanase inhibitor protein from wheat: a novel protein function. Biochim Biophys Acta 1696:203–211
Kalume DE, Okulate M, Zhong J, Reddy R, Suresh S, Deshpande N, Kumar N, Pandey A (2005) A proteomic analysis of salivary glands of female Anopheles gambiae mosquito. Proteomics 5:3765–3777
Kimura A, Takewaki S, Matsui H, Kubota M, Chiba S (1990) Allosteric properties, substrate specificity, and subsite affinities of honeybee α-glucosidase I. J Biochem 107:762–768
Krzywinski J, Grushko OG, Besansky NJ (2006) Analysis of the complete mitochondrial DNA from Anopheles funestus: an improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution. Mol Phylogenet Evol 39:417–423
Kubota M, Tsuji M, Nishimoto M et al (2004) Localization of α-glucosidases I, II and III in organs of European honeybee, Apis mellifera L., and origin of α-glucosidase in honey. Biosci Biotechnol Biochem 68:2346–2352
Kunita R, Nakabayashi O, Wu JY, Hagiwara Y, Mizutani M, Pennybacker M, Chen YT, Kikuchi T (1997) Molecular cloning of acid alpha-glucosidase cDNA of Japanese quail (Coturnix coturnix japonica) and the lack of its mRNA in acid maltase deficient quails. Biochim Biophys Acta 1362:269–278
Kuriki T, Imanaka T (1999) The concept of the α-amylase family: structural similarity and common catalytic mechanism. J Biosci Bioeng 87:557–565
Lawson D, Arensburger P, Atkinson P et al (2009) VectorBase: a data resource for invertebrate vector genomics. Nucleic Acids Res 37:D583–D587
Lodge JA, Maier T, Liebl W, Hoffmann V, Sträter N (2003) Crystal structure of Thermotoga maritima α-glucosidase AglA defines a new clan of NAD+-dependent glycosidases. J Biol Chem 278:19151–19158
Long M, Wang W, Zhang J (1999) Origin of new genes and source for N-terminal domain of the chimerical gene, jing-wei, in Drosophila. Gene 238:135–141
MacGregor EA, Janecek S, Svensson B (2001) Relationship of sequence and structure to specificity in the α-amylase family of enzymes. Biochim Biophys Acta 1546:1–20
Maczkowiak F, Da Lage JL (2006) Origin and evolution of the Amyrel gene in the α-amylase multigene family of Diptera. Genetica 128:145–158
Marinotti O, James AA (1990) An α-glucosidase in the salivary glands of the vector mosquito, Aedes aegypti. Insect Biochem 20:619–623
Marinotti O, de Brito M, Moreira CK (1996) Apyrase and α-glucosidase in the salivary glands of Aedes albopictus. Comp Biochem Physiol B Biochem Mol Biol 113:675–679
Martiniuk F, Ellenbogen A, Hirschhorn R (1985) Identity of neutral alpha-glucosidase AB and the glycoprotein processing enzyme glucosidase II. Biochemical and genetic studies. J Biol Chem 260:1238–1242
Matsui H, Iwanami S, Ito H, Mori H, Honma M, Chiba S (1997) Cloning and sequencing of a cDNA encoding alpha-glucosidase from sugar beet. Biosci Biotechnol Biochem 61:875–880
Matsuura Y, Kusunoki M, Harada W, Kakudo M (1984) Structure and possible catalytic residues of Taka-amylase A. J Biochem 95:697–702
McQuilton P, St Pierre SE, Thurmond J, FlyBase Consortium (2012) FlyBase 101—the basics of navigating FlyBase. Nucleic Acids Res 40(Database issue):D706–D714
Munoz-Torres MC, Reese JT, Childers CP, Bennett AK, Sundaram JP, Childs KL, Anzola JM, Milshina N, Elsik CG (2011) Hymenoptera Genome Database: integrated community resources for insect species of the order Hymenoptera. Nucleic Acids Res 39(Database issue):D658–D662
Mury FB, da Silva JR, Ferreira LS et al (2009) α-Glucosidase promotes hemozoin formation in a blood-sucking bug: an evolutionary history. PLoS One 4:e6966
Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39:121–152
Nei M, Gu X, Sitnikova T (1997) Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc Natl Acad Sci USA 94:7799–7806
Nishimoto M, Kubota M, Tsuji M, Mori H, Kimura A, Matsui H, Chiba S (2001) Purification and substrate specificity of honeybee, Apis mellifera L., α-glucosidase III. Biosci Biotechnol Biochem 65:1610–1616
Nishimoto M, Mori H, Moteki T et al (2007) Molecular cloning of cDNAs and genes for three α-glucosidases from European honeybees, Apis mellifera L., and heterologous production of recombinant enzymes in Pichia pastoris. Biosci Biotechnol Biochem 71:1703–1716
Novak S, Zechner-Krpan V, Marie V (2004) Regulation of maltose transport and metabolism in Saccharomyces cerevisiae. Food Technol Biotechnol 42:213–218
Opota O, Charles JF, Warot S, Pauron D, Darboux I (2008) Identification and characterization of the receptor for the Bacillus sphaericus binary toxin in the malaria vector mosquito, Anopheles gambiae. Comp Biochem Physiol B Biochem Mol Biol 149:419–427
Oslancova A, Janecek S (2002) Oligo-1,6-glucosidase and neopullulanase enzyme subfamilies from the α-amylase family defined by the fifth conserved sequence region. Cell Mol Life Sci 59:1945–1959
Payan F, Flatman R, Porciero S, Williamson G, Juge N, Roussel A (2003) Structural analysis of xylanase inhibitor protein I (XIP-I), a proteinaceous xylanase inhibitor from wheat (Triticum aestivum, var. Soisson). Biochem J 372:399–405
Payan F, Leone P, Porciero S et al (2004) The dual nature of the wheat xylanase protein inhibitor XIP-I: structural basis for the inhibition of family 10 and family 11 xylanases. J. Biol. Chem 279:36029–36037
Price DR, Karley AJ, Ashford DA, Isaacs HV, Pownall ME, Wilkinson HS, Gatehouse JA, Douglas AE (2007) Molecular characterisation of a candidate gut sucrase in the pea aphid, Acyrthosiphon pisum. Insect Biochem Mol Biol 37:307–317
Rhodes JD, Croghan PC, Dixon AFG (1997) Dietary sucrose and oligosaccharide synthesis in relation to osmoregulation in the pea aphid, Acyrthosiphon pisum. Physiol Entomol 22:373–379
Rigden DJ (2002) Iterative database searches demonstrate that glycoside hydrolase families 27, 31, 36 and 66 share a common evolutionary origin with family 13. FEBS Lett 523:17–22
Romão TP, de Melo Chalegre KD, Key S, Ayres CF, Fontes de Oliveira CM, de Melo-Neto OP, Silva-Filha MH (2006) A second independent resistance mechanism to Bacillus sphaericus binary toxin targets its α-glucosidase receptor in Culex quinquefasciatus. FEBS J 273:1556–1568
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Savard J, Tautz D, Richards S, Weinstock GM, Gibbs RA, Werren JH, Tettelin H, Lercher MJ (2006) Phylogenomic analysis reveals bees and wasps (Hymenoptera) at the base of the radiation of Holometabolous insects. Genome Res 16:1334–1338
Sikora J, Urinovská J, Majer F, Poupetová H, Hlavatá J, Kostrouchová M, Ledvinová J, Hrebícek M (2010) Bioinformatic and biochemical studies point to AAGR-1 as the ortholog of human acid alpha-glucosidase in Caenorhabditis elegans. Mol Cell Biochem 341:51–63
Silva-Filha MH, Nielsen-LeRoux C, Charles JF (1999) Identification of the receptor for Bacillus sphaericus crystal toxin in the brush border membrane of the mosquito Culex pipiens (Diptera: Culicidae). Insect Biochem Mol Biol 29:711–721
Skov LK, Mirza O, Henriksen A, De Montalk GP, Remaud-Simeon M, Sarçabal P, Willemot RM, Monsan P, Gajhede M (2001) Amylosucrase, a glucan-synthesizing enzyme from the alpha-amylase family. J Biol Chem 276:25273–25278
Snyder M, Davidson N (1983) Two gene families clustered in a small region of the Drosophila genome. J Mol Biol 166:101–118
Souza-Neto JA, Machado FP, Lima JB, Valle D, Ribolla PE (2007) Sugar digestion in mosquitoes: identification and characterization of three midgut α-glucosidases of the neo-tropical malaria vector Anopheles aquasalis (Diptera: Culicidae). Comp Biochem Physiol A Mol Integr Physiol 147:993–1000
Stam MR, Danchin EG, Rancurel C, Coutinho PM, Henrissat B (2006) Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of α-amylase-related proteins. Protein Eng Des Sel 19:555–562
Takewaki S, Chiba S, Kimura A, Matsui H, Koike Y (1980) Purification and properties of α-glucosidases of the honey bee Apis mellifera L. Agric Biol Chem 44:731–740
Takewaki S, Kimura A, Kubota M, Chiba S (1993) Substrate specificity and subsite affinities of honeybee α-glucosidase II. Biosci Biotechnol Biochem 57:1508–1513
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Teste MA, François JM, Parrou JL (2010) Characterization of a new multigene family encoding isomaltases in the yeast Saccharomyces cerevisiae, the IMA family. J Biol Chem 285:26815–26824
Tibbot BK, Skadsen RW (1996) Molecular cloning and characterization of a gibberellin-inducible, putative alpha-glucosidase gene from barley. Plant Mol Biol 30:229–241
Van der Kaaij RM, Janecek S, van der Maarel MJ, Dijkhuizen L (2007) Phylogenetic and biochemical characterization of a novel cluster of intracellular fungal alpha-amylase enzymes. Microbiology 153:4003–4015
Vanin EF (1985) Processed pseudogenes: characteristics and evolution. Annu Rev Genet 19:253–272
Vieira CP, Vieira J, Hartl DL (1997) The evolution of small gene clusters: evidence for an independent origin of the maltase gene cluster in Drosophila virilis and Drosophila melanogaster. Mol Biol Evol 14:985–993
Viigand K, Tammus K, Alamäe T (2005) Clustering of MAL genes in Hansenula polymorpha: cloning of the maltose permease gene and expression from the divergent intergenic region between the maltose permease and maltase genes. FEMS Yeast Res 5:1019–1028
Vongsangnak W, Salazar M, Hansen K, Nielsen J (2009) Genome-wide analysis of maltose utilization and regulation in aspergilli. Microbiology 155:3893–3902
Walters FS, Mullin CA (1988) Sucrose-dependent increase in the oligosaccharide production and associated glycosidase activities in the potato aphid Macrosiphum euphorbiae (Thomas). Arch Insect Biochem Physiol 9:35–46
Wells RG, Hediger MA (1992) Cloning of a rat kidney cDNA that stimulates dibasic and neutral amino acid transport and has sequence similarity to glucosidases. Proc Natl Acad Sci USA 89:5596–5600
Wilkinson TL, Ashford DA, Pritchard J, Douglas AE (1997) Honeydew sugars and osmoregulation in the pea aphid Acyrthosiphon pisum. J Exp Biol 200:2137–2143
Winge O, Roberts C (1950) Identification of the gene for maltose fermentation in Saccharomyces italicus. Nature 166:1114
Yamamoto K, Nakayama A, Yamamoto Y, Tabata S (2004) Val216 decides the substrate specificity of α-glucosidase in Saccharomyces cerevisiae. Eur J Biochem 271:3414–3420
Yuan XL, van der Kaaij RM, van den Hondel C, Punt PJ, van der Maarel M, Dijkhuizen L, Ram AFJ (2008) Aspergillus niger genome-wide analysis reveals a large number of novel α-glucan acting enzymes with unexpected expression profiles. Mol Genet Genomics 279:545–561
Zhang Z, Inomata N, Yamazaki T, Kishino H (2003) Evolutionary history and mode of the amylase multigene family in Drosophila. J Mol Evol 57:702–709
Zheng L, Whang LH, Kumar V, Kafatos FC (1995) Two genes encoding midgut-specific maltase-like polypeptides from Anopheles gambiae. Exp Parasitol 81:272–283
Zona R, Chang-Pi-Hin F, O’Donohue MJ, Janecek S (2004) Bioinformatics of the glycoside hydrolase family 57 and identification of catalytic residues in amylopullulanase from Thermococcus hydrothermalis. Eur J Biochem 271:2863–2872
Acknowledgments
I would like to thank Dr. Stefan Janecek, my supervisor, for all his support and encouragement. This work was supported by the Grant No. 2/0148/11 from the Slovak Grant Agency VEGA.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
239_2013_9545_MOESM2_ESM.pdf
Fig. S1 Phylogenetic trees of α-glucosidases and related proteins from the α-amylase family (pdf file)Supplementary material 2 (PDF 398 kb)
Rights and permissions
About this article
Cite this article
Gabriško, M. Evolutionary History of Eukaryotic α-Glucosidases from the α-Amylase Family. J Mol Evol 76, 129–145 (2013). https://doi.org/10.1007/s00239-013-9545-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-013-9545-4