
Abstract
Diversification patterns and demography of montane species are affected by Pleistocene climate fluctuations. Empirical cases from the Qinling Mountains (QM) region, which is a major biogeographic divider of East Asia, are few. We used DNA sequence data of the complete mitochondrial ND2 gene to detect effects of the Pleistocene glaciations on phylogeographic profiles of a frog species, Feirana taihangnica, which is endemic to the QM. Four distinct lineages consisting of seven sublineages were revealed. The strongest signal of biogeographical structure (F ct = 0.971, P < 0.01) was found when populations were grouped according to these seven sublineages. One narrow secondary contact zone was detected in the middle QM between the lineage from middle QM and the lineage from eastern QM. Coalescent simulations indicated that this species colonized the QM region by a stepping-stone model. Divergences among lineages had likely been influenced by the uplift of the Tibetan Plateau during the late Miocene-to-late Pleistocene, as well as by the Pleistocene climatic cycles. Coalescent simulations also suggested that F. taihangnica populations have persisted through the Pleistocene glacial periods in multiple refugia across the QM region. Demographic analyses indicated that all lineages, except the lineage in the Funiu Mountains, have been experienced postglacial expansion of population size and distribution range. In conclusion, Pleistocene climate fluctuations and tectonic changes during the late Miocene-late Pleistocene have profoundly influenced the phylogeography and historical demography of F. taihangnica.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diversification patterns and genetic architectures of montane species are remarkably affected by climatic changes associated with the Pleistocene glacial cycles (Hewitt 1996, 2000, 2004). Substantial genetic heterogeneity and ongoing gene flow among populations in different Mountains are caused by Pleistocene glacial isolations and interglacial connectivity, respectively. Subsequently, many populations have experienced contractions or expansions of population size and distribution range (Avise and Walker 1998; Knowles 2001; Maddison and McMahon 2000). Furthermore, the levels of genetic differentiation as well as other biological features may influence the lineage sorting, and determine whether different populations would merge into one gene pool within their secondary contact zone (Knowles 2000, 2001; Smith and Farrell 2005; Richards et al. 2007).
Spatial distribution patterns and genetic profiles of species in different regions may reflect the multiple effects of the Pleistocene climate changes on them. Many species in the temperate zone of Europe and North America experienced contractions with southward glacier advances and expansions with northward glacier shrinkages. Consequently, the present genetic diversity-index of them increased with decreasing latitude (Avise et al. 1998; Avise and Walker 1998; Avise 2000; Hewitt 2004). Similar to their temperate counterparts, species in tropical habitats also expanded southward during the Holocene (Hillesheim et al. 2005). East Asia, however, has unique Quaternary Period eco-environments due to the ongoing uplift of the Tibetan Plateau, which has altered the topography of East Asia and driven the formation of the East Asian monsoon. Both changes have profoundly influenced the genetic diversity and dynamic history of organisms in this region (Zhang 1999, 2004; Zhou 2000; Pinot et al. 1999; Qian and Ricklefs 2000; Ju et al. 2007). Several genetic surveys discovered that bird and mammal species took refuge in the central and southeastern China rather than being limited to lower latitudes during the Pleistocene ice ages (Song et al. 2009; Li et al. 2009; You et al. 2010). On the other hand, herpetological species retreated into multiple separated refugia in the southern Korean Peninsula and southeastern China during the Pleistocene glacial periods (Zhang et al. 2008; Ding et al. 2011). Most of these species have experienced expansion of population size and/or distribution range before the last glacial maximum (LGM; Song et al. 2009; Li et al. 2009; Zhang et al. 2008; Ding et al. 2011).
The Qinling Mountains (QM) region, located in the central China, is an important biogeographic divider of East Asia. Historically, drastic tectonic events, such as the significant uplifts of the Tibetan Plateau from the late Miocene, Kun–Huang diastrophisms, and Gonghe movements during the early Pleistocene, led to the development of the Mountains and valleys in the QM region (An et al. 2001; Zhang et al. 2006; Dong et al. 2011). In addition, some high Mountains (e.g., Taibai mountain) greater than 3,000 m above sea level (a.s.l.) in the QM region were likely covered by ice caps during the Pleistocene ice ages (Rost 1994; Shi 2002, 2007; Li et al. 2004). A complex tectonic history and quaternary glaciations may have contributed to genetic-diversity patterns and evolutionary process of organisms in this region. In particular, populations in or near cold areas might shrink into low-elevation refugia during the Pleistocene ice ages and then expand during the postglacial periods. The QM region extends from west to east including Western Qinling Mountains (WQM), Middle Qinling Mountains (MQM), Eastern Qinling Mountains (EQM), Northeastern Qinling Mountains (NEQM), Zhongtiao-southern Taihang Mountains (ZTM), and Funiu Mountains (FNM). Several major rivers, including Jialing River, Yellow River, Hei River, Luo River and Dan River, separate these Mountains (Fig. 1). These topographic features are expected to help maintain significant genetic structure within species. However, limited empirical data (e.g., Song et al. 2009; Li et al. 2009; Wang et al. 2012) restrict our understanding of spatial distribution, lineage sorting, and demography associated with Pleistocene climate changes and tectonic events in this important region.

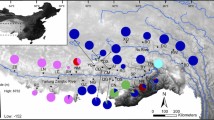
Geographic locations of the 17 sampled populations of Feirana taihangnica. The thinner dashed line mainly encircles the distribution range of F. taihangnica. Populations 1–17 were given in Table 1. The dashed lines with double-headed arrows (I, II, III) standard for the barriers resulted from BARRIER analyses. Lineages and sublineages A–D, B1, B2, and C1–C3 were given in Fig. 3. Size of each circle is proportional to the number of haplotypes. WQM western Qinling Mountains, MQM middle Qinling Mountains, EQM eastern Qinling Mountains, NEQM northeastern Qinling Mountains, ZTM Zhongtiao–southern Taihang Mountains, FNM Funiu Mountains
The frog species Feirana taihangnica (Chen and Jiang 2002), belongs to the family Dicroglossidae and lives in stream habitats across most of the QM region (Wang et al. 2007, 2009; Fei et al. 2009, 2010; Fig. 1). Poor dispersal ability and strict habitat requirements (Yang 2007) likely make this species vulnerable to climatic fluctuations. Thus, F. taihangnica is an excellent model species to test hypotheses concerning the effects of the past climatic cycles on the genetic architecture and demography of species in the QM region.
In this study, we examined phylogeographic patterns and demographic history of F. taihangnica using DNA sequence data of complete mitochondrial ND2 gene. With intensive sampling, we mainly focus on two scientific questions: (1) were all populations derived from one glacial refugium or multiple refugia? (2) Has this species undergone glacial bottlenecks and postglacial expansions?
Materials and Methods
Sampling and Molecular Data
We sampled 192 individuals from 17 sites that cover most of the distribution range of Feirana taihangnica (Table 1; Fig. 1). Muscle tissue samples were preserved in 95 % ethanol. Voucher specimens were deposited in the Herpetology Collection of the Chengdu Institute of Biology of the Chinese Academy of Sciences (CIB, CAS) and the College of Life Sciences of Henan Normal University (HNNU).
Total genomic DNA was extracted using a standard phenol–chloroform extraction procedure (Hillis et al. 1996). The data for 15 individuals (GeneBank Accession Nos.: GQ225989–GQ226003) were first published in Wang et al. (2009). The complete ND2 gene sequence of each sample was amplified and sequenced according to the procedures of Wang et al. (2009). All sequences were deposited in GenBank (accession numbers KC510464–KC510640). Based on Jiang et al. (2005), Che et al. (2009, 2010), and Wang et al. (2009), F. quadranus and Gyndrapaa yunnanensis were used as outgroups. The data for these species were also downloaded from GenBank (GQ225933–34, GQ225954–55, GQ225958, and GQ225877).
Phylogenetic Analyses
All sequences were aligned using Clustal x v1.83 (Thompson et al. 1997) with the default settings, and unique haplotypes were identified by DnaSP v5 (Librado and Rozas 2009). The gene genealogy was reconstructed using maximum parsimony (MP), maximum likelihood (ML), and Bayesian inference (BI), as implemented in PAUP v4.0b10 (Swofford 2002), PhyML v3 (Guindon and Gascuel 2003), and MrBayes v3.0b4 (Ronquist and Huelsenbeck 2003), respectively. Both the Corrected Akaike Information Criterion (AICc; Hurvich and Tsai 1989) and the Bayesian information criterion (BIC; Schwarz 1978) selected the TrN+G model as the best-fit model, using jModeltest v0.1.1 (Posada 2008; Guindon and Gascuel 2003). Under this model, the proportion of invariant sites (I) and the shape of the Gamma distribution (G) were set as 0 and 0.191, respectively. The base frequency and ratio of transitions/transversions were optimized by the maximum likelihood criterion in PHYML. The default tree search parameters were used to estimate tree topologies, including the simultaneous Nearest Neighbor Interchange (NNI) method and BioNJ tree as the starting tree. In the MP analysis, we used heuristic searches of 1,000 random-addition replicates using tree bisection-reconnection (TBR) branch swapping, with characters unordered and equally weighted, and gaps treated as missing data. The nodal confidence of MP and ML trees was assessed using 10,000 non-parametric bootstrap resampling replicates (Felsenstein 1985; Felsenstein and Kishino 1993; Hedges 1992; Huelsenbeck and Hillis 1993). For the BI analyses, we initiated two dependent runs each with four simultaneous Markov chain Monte Carlo chains (MCMC) for 50 million generations with sampling every 1,000 generations and the first 10 % of generations as “burn-in”. The convergence of chains was confirmed when the split-standard deviation was smaller than 0.001, and then the posterior probabilities were achieved using the sampled trees from the remaining generations. A 95 % of bipartition’s posterior probability or greater was considered to be significant support (Leaché and Reeder 2002; Parra-Olea et al. 2004).
Population Genetic Structure
We used TCS v1.21 (Clement et al. 2000) to construct a haplotype network for ingroup samples with a statistical parsimony method (Templeton et al. 1992; Clement et al. 2000). The cutoff probability value was set at 95 %. To test for population genetic structure and geographical divisions, the hierarchical partitioning of genetic variation was investigated using analysis of molecular variance (AMOVA; Excoffier et al. 1992) implemented in Arlequin v3.11 (Excoffier et al. 2005). AMOVA was conducted based on two grouping schemes; one grouped populations according to the lineages recovered in the gene tree reconstructions, and the other grouped populations based on their geographic arrangements (Fig. 1). The fixation indices Fct, Fsc, and Fst were estimated and the most probable geographical subdivision was inferred with the highest value of F ct (Excoffier et al. 2005). The significance of these statistics was determined by 10,000 permutation replicates. Pairwise F st between groups as well as between populations were calculated in Arlequin. Nucleotide site polymorphism, nucleotide diversity (π), and haplotype diversity (Hd) were estimated in DnaSP.
To test whether the genetic variation among populations fit the isolation-by-distance (IBD) model, a Mantel test (10,000 randomizations) was conducted, which was implemented in the web based program IBDWS v3.14 (Jensen et al. 2005). Furthermore, to highlight geographical features that correspond to pronounced genetic structure and gene flow discontinuity, Monmonier’s maximum difference algorithm (Manni et al. 2004) was also carried out using the program BARRIER v2.2 (Manni et al. 2004). Localities with their geographical coordinates were connected by Delauney triangulation using a pairwise Tamura-Nei 93 genetic distance matrix, and topographic boundaries that restrict gene flow were identified across the landscapes using Monmonier’s maximum difference algorithm (Manni et al. 2004).
Divergence Dating
Under a “relaxed molecular clock” assumption (Drummond et al. 2006), divergence times among lineages were estimated using BEAST v1.6.1 (Drummond and Rambaut 2007). Genealogy was reconstructed under the TrN+G model with an uncorrelated lognormal tree prior, an exponential growth prior, and a lognormal mean substitution rate of 0.65 % change per lineage per million years (see below; stdev = 0.7 and offset = 0.0018). We ran 50 million generations in two-independent iterations, sampled every 1,000 generations, and discarded the first 10 % as “burn-in”. Convergence of the chains to the stationary distribution was checked by visual inspection of plotted posterior estimates using the program Tracer v1.5 (Rambaut and Drummond 2007). The effective sample size (ESS) for each parameter mostly exceeded 200 (Drummond and Rambaut 2007). The final tree including divergence time with 95 % highest posterior densities (HPD) was estimated in TREEANNOTATOR v1.4.5.
The mutation rate of the ND2 gene has been found to be fairly constant at 0.57–0.96 % change per lineage per million years across a wide range of vertebrate groups, including fishes, hynobiid salamanders, Laudakia and Teratoscincus lizards, Bufo, Ranid frogs (e.g., Rana boylii), and Eleutherodactylus toads (Bermingham et al. 1997; Weisrock et al. 2001; Macey et al. 1998a, b, 1999, 2001; Crawford 2003). Feirana taihangnica is a close relative of Ranid frogs (Jiang et al. 2005), which have a universal substitution rate of 0.65 % change per lineage per million years for the ND2 gene (Macey et al. 2001). Thus, we used this mean rate to estimate divergence time among lineages within F. taihangnica. There was no fossil record of this species that could be used for calibrations.
Historical Biogeography
We used coalescent simulations to test the fit of the observed gene genealogy to the hypotheses on the evolutionary patterns of Feirana taihangnica (Knowles and Maddison 2002; Knowles 2001; Carstens and Richards 2007). Three hypotheses were tested. (1) A single-refugium or fragmentation model, in which all current populations were derived from a single refugium at the end of the LGM (~50 ka; Dali glaciation in China; Cui and Zhang 2003; Fig. 2a). (2) A two-refugia or stage fragmentation model, in which the species persisted in two refugia during the LGM. The WQM population and the lineage including all other populations are descended from two ancestral populations, which began divergence at approximately 7.7 Ma, and coalescence occurred at 50 ka (Fig. 2b). (3) A multiple-refugia or colonization model, in which the species has experienced a series of dispersals from one mountain to another followed by isolation and divergence. Lineages occurring in different regions have persisted through the LGM in multiple refugia across the QM region (Fig. 2c).

Three hypotheses concerning refugia during the last glacial maximum (LGM) tested using coalescent simulations for Feirana taihangnica. a The single refugium model hypothesized that all populations derived from one refugium after the LGM, T = 7.7 Ma and T1 = 50 ka. b The two refugium model posited that all populations derived from two refugia in the western Qinling Mountains and other part of the Qinling Mountains, T = 7.7 Ma and T1 = 50 ka. c The multiple-refugia model suggested that all lineages have persisted through the LGM in four glacial refugia, T = 7.7, T2 = 1.5, and T1 = 1.4 Ma. Branch lengths are time in generations based on a 2.5-year generation time in F. taihangnica. Branch widths (effective female population size, N ef) were scaled for each mountain based on the proportion of the total N ef that each mountain comprised (listed below mountain name). Internal branches on the second hypothesis were scaled such that all branch widths summed to the total N ef at any definite point in time
For coalescent simulations, the grouping arrangement of populations was based on their geographical origins (Fig. 1). The female effective population size (N ef) for these groups from different mountainous regions was calculated using the equation θ = N ef * μ for mitochondrial DNA, in which the θ values were estimated in the program Migrate-N v3.3 (Beerli 2008) and μ was set as 1.65 × 10−8 substitution per site per year (estimated from the value of 0.65 % change per lineages per million years). We estimated the θ values using maximum-likelihood estimates (MLE) with the following parameters: ten short chains for 200,000 steps and three long chains for 20 million generations with no heating and chains being sampled every 100 generations following a burn-in of 2 million generations. The process was repeated three times to insure convergence upon similar values for θ. We simulated the coalescent times of the current trees under the assumption ‘without migration’ model using Mesquite v2.75 (Maddison and Maddison 2011) because of the negligible gene flow indicated by high pairwise F st values among assigned groups. Total N ef was obtained by summing all estimated N ef of all given groups. The 95 % CI’s of N ef were calculated based on the 95 % CI’s of θ values (Edwards and Beerli 2000). In the hypothetical population trees, branch widths were scaled proportional to total N ef (Carstens et al. 2004).
We used two sets of coalescent simulations to evaluate the fit of the observed gene tree to the different hypotheses (Knowles 2001; Knowles and Maddison 2002) in Mesquite. In the first set of simulations, we simulated 1,000 coalescent genealogies under the hypotheses based on the observed data set and recorded the distribution of S (Slatkin and Maddison 1989). Then, we tested the model fit by comparing the S value of our ML genealogy with the S values of the simulated genealogies. In the second set of simulations, we simulated 100 gene matrices in Mesquite under the substitution model of the observed data set selected by jModelTest. Then, we reconstructed trees based on these simulated gene matrices in PAUP and recorded the distribution of S. Finally, we compared the S value of the empirical ML genealogy with S values of the simulated genealogies to test whether the observed genealogies fit the given scenarios. In all simulations, both the point estimate of N ef and the 95 % CI’s of N ef were used as model parameters and absolute time (years) was converted to coalescent time (generations) using a generation time of 2.5 years for F. taihangnica (Yang 2007).
Historical geographical origin for each lineage was estimated through reconstructing ancestral states using maximum-likelihood estimations in Mesquite. Each individual was assigned to its mountain of origin and the ancestral areas were estimated using a ML model on the tree. Because no prior hypothesis for migration rate existed, we selected the Markov k-state 1 parameter (Mk1) model with an equal likelihood for the rate of change among different Mountains for estimating ancestral area. We determined the ancestral area for each lineage according to the proportional likelihoods (PL) of each mountainous region.
Historical Demography
Several approaches were used to assess historical demographic dynamics of lineages. First, under the assumption of neutrality, deviations in Tajima’s D (Tajima 1989) and Fu’s F s (Fu 1997) were used to examine recent population expansion or bottlenecks in each lineage (Hartl and Clark 2007). Negative values indicate population growth and/or positive selection, whereas positive Tajima’ D indicates bottlenecks and/or balancing selection (Tajima 1989; Fu 1997). Second, mismatch distributions comparing observed versus expected distributions of pairwise nucleotide differences were examined to detect the historical demographic events and test hypotheses regarding recent population growth. A unimodal model indicates a recent range expansion and bimodal or multimodal patterns indicate probable declining population sizes, structured size, migration, subdivided and/or historical contraction; whereas a ragged distribution indicates that the lineage was probably widespread (Excoffier et al. 1992; Rogers and Harpending 1992; Rogers et al. 1996; Excoffier and Schneider 1999; Marjoram and Donnelly 1994; Bertorelle and Slatkin 1995; Ray et al. 2003). The fit of the observed to the expected mismatch was compared using the sum of squares deviations (SSD) between observed and expected data, while the Harpending’s raggedness index (HRI; Harpending 1994) and P values were estimated to quantify the smoothness of the observed mismatch distribution. The mismatch distribution parameter tau (time since the recent expansion, in units of mutational time) was also estimated for each lineage and the time since the recent expansion was calculated from tau value using the equation tau = 2μkt, where μ is the mutation rate, k is the sequence length, and t is the time since the recent expansion. These demographic analyses were implemented in Arlequin and the significance of deviation of values was tested with 10,000 permutations.
Nonetheless, neutrality tests and mismatch distribution probably rely excessively on segregating sites and haplotype patterns and perhaps cannot fully extract historical information (Fitzpatrick et al. 2009). Thus, we used a coalescent-based method Bayesian Skyline Plot (BSP; Drummond et al. 2005) implemented in the software Beast to retrieve historical demographic information; this method does not require a pre-specified demographic model to examine population size fluctuations over time. The HKY nucleotide substitution model was selected as the appropriate model for each lineage using jModeltest. The starting tree was randomly generated. We applied 3–10-grouped coalescent intervals (m) for different groups according to the number of their included samples and the piecewise-constant model was selected as a prior skyline model. Two MCMC runs were conducted for 20 million iterations and the first 10 % were discarded as “burn-in”. The mean rate of 0.65 %/Ma suggested above was used to scale the time axis on BSPs and uncorrelated lognormal model was used as a prior to account for rate variation among lineages (standard deviation of mean rate = 0.7 and offset = 0.0018). Convergence of the chains to the stationary distribution was checked by visual inspection of plotted posterior estimates using the program Tracer, and the ESS for each parameter sampled was almost always found to exceed 200 (Drummond et al. 2005). Demographic plots for each lineage were visualized in Tracer.
Results
Phylogenetic Trees
An alignment of DNA sequences of complete ND2 gene of length 1,035 bps was obtained for 192 individuals of Feirana taihangnica and 40 haplotypes were identified (Table 1). There are 126 variable sites including 111 parsimony informative sites, and the nucleotide composition is T = 29.2 %, C = 31.4 %, A = 29.0 %, and G = 10.4 %, with the ratio of T i/T v (Transition/Transversion) being 10.7.
In the MP analysis, 264 maximum parsimonious trees were produced, with tree length of 291 steps, a consistency index (CI) of 0.85, and a retention index (RI) of 0.80. The ML analysis produced 25 trees with -lnL score of 2791.52. Phylogenetic trees produced by MP, ML, and BI analyses were consistent in topologies, except for a few poorly supported tip nodes (Fig. 3). All haplotypes of F. taihangnica comprised a monophyletic group apart from outgroups (all supports of MP/ML/BI > 95 %). Within F. taihangnica, four lineages, consisting of seven sublineages structured across different Mountains, were revealed (Figs. 1, 3). The first diverged lineage (lineage A; all supports = 100 %) included only one population (population 1) in the WQM. The second diverged lineage (lineage B; supports of MP/ML/BI = 57/59/0.75), from the MQM, included sublineages B1 and B2, of which the sublineage B1 included seven samples of population 2 and sublineage B2 included all samples of population 4. The lineage C (all supports >95 %) was comprised of three sublineages with the relationships as (C3, (C1, C2)), of which, the sublineage C1 contained all samples of populations 10 and 11 from the NEQM, C2 included all samples of the three populations (population 12–14) from the ZTM and C3 contained two samples of population 2 from the MQM and all samples of populations (population 3 and 5–9) from the EQM. Lineage D (all supports >95 %) included all samples of the three populations (populations 15–17) from the FNM.

Maximum likelihood tree (a) and haplotype network (b) for the 40 sampled haplotypes of Feirana taihangnica. Bootstrap values calculated by ML and MP analyses and Bayesian posterior probabilities from Bayesian analyses are presented above the main branches; the mean divergence time and 95 % CI’s are presented below the main branches. Seven major nodes (1–7) are encircled on the tree. The size of each circle in the network is proportional to haplotype frequencies (n), and the black dots represent missing haplotypes. Haplotypes H1–H40 were given in Table 1
Population Genetic Structure
Network analysis grouped haplotypes into four clusters with 95 % probability (connect limit = 14 steps), which was compatible with the phylogenetic tree topology (Fig. 3). There was no haplotype shared by populations from different mountainous regions (Fig. 1), except for population 2 from the MQM and population 3 from the EQM which shared the haplotype H12.
We tested the partitioning of genetic variation across three kinds of population grouping arrangements according to geographical features and phylogenetic lineage arrays: grouping populations by four lineages, by seven sublineages, and by six regions (Table 2; Figs. 1, 3). Hierarchical analyses rejected the null hypothesis that suggests no genetic structure within this species and revealed significant genetic variation (P < 0.001) when all grouping arrangements were tested (Table 2). The percentage of genetic differentiations among groups (F ct) accounted for over 90 % of the variance in each hypothesis, but the F ct (= 0.971) was the highest for the hypothesis assuming that all populations were assigned to seven sublineages (Table 2).
High Hd and π (0.921 and 0.04176, respectively) were found for all populations as a whole. The sequence variation of each lineage/sublineage differed in estimated Hd and π (Table 3). Low Hd and extremely low π (0.569 and 0.00066) were found in the lineage A, while the other three lineages (B–D) had relatively high Hd and π (Table 3). Sublineages B1, B2, and C1 possessed higher Hd (0.672–0.831) than C2 and C3, although the latter two sublineages had a similar quantity of samples with B2 and C1 (Table 3). Nevertheless, sublineages B1 and C1 possessed higher π than that of B2, C2, and C3 (Table 3). Only 22 out of a total 289 pairs of populations shared haplotypes (Table S1). Most pairwise F st values between populations (272 out of 289 pairs) were found to be significant (P < 0.05; Table S2). Similarly, all pairwise F st values in various grouping arrangements were found to be significant and high (Table S3).
IBD analysis revealed that the correlation between genetic distance (F st/1−F st) and geological distance was not significant (P = 0.975), indicating that the genetic differentiation within F. taihangnica did not fit the IBD model. The barrier analysis highlighted geographical barriers restricting gene flow among major lineages of F. taihangnica. The predicted barriers corresponded well with major valleys (barriers I–Ш: Jialing River, Hei River, Luo River-Dan River; Fig. 1). Noticeably, the barrier П partially separated the lineages B and C (Fig. 1).
Divergence Time, Ancestral Area Reconstructions and Hypotheses Testing
Although the divergence dating in this study must be taken with extreme caution because there is no available internal calibration, this dating does provide an approximate time frame. The estimated divergence time of the sister species (F. taihangnica and F. quadranus) is comparable with that of Che et al. (2010) using secondary correction methods. Feirana taihangnica diverged from the closely related species F. quadranus c. 16.9 Ma (95 % CI’s: 2.8–53.7 Ma). The divergence time among lineages or sublineages of F. taihangnica was from the late Miocene c. 7.7 Ma (95 % CI’s: 0.9–15.2 Ma) to the late Pleistocene c. 0.56 Ma (95 % CI’s: 0.05–1.33 Ma) in a broad timescale (Fig. 3a).
The overall θ value was estimated as 0.0152 (95 % CI’s: 0.0103–0.0246) and the total N ef was calculated as 779,487 (95 % CI’s: 528,205–1,261,538). We calculated S = 5 for our ML genealogy. In the first set of simulations, regardless of using upper or lower bound of N ef, the single-refugium/fragmentation and two-refugia/stage fragmentation hypotheses were always rejected (95 % CI’s of S-values of them >11; P < 0.01), but the multiple-refugia/colonization model was not rejected (95 % CI’s of S values for this model is 3–6; P > 0.05). Similarly, the second set of simulations also rejected the single-refugium/fragmentation model (95 % CI’s of S values >13; P < 0.01) and the two-refugia/stage fragmentation model (95 % CI’s of S values >14; P < 0.01) but did not reject the multiple-refugia/colonization model (95 % CI’s of S values: 3–6; P > 0.05).
Reconstruction of the ancestral area for F. taihangnica assigned the highest probability (PL = 1.000) to the WQM (Table 4). The ancestor of lineages B, C, and D likely originated from the MQM (PL = 0.979) and the ancestral area of the lineages C and D was probably the MQM (PL = 0.423) although the PL of the FNM was similarly high (0.309). The ancestral area of the lineage C was probably a wide region (EQM–NEQM) because the PL of these two regions was similar (0.453 for NEQM and 0.431 for EQM). These results indicated that the evolutionary patterns of F. taihangnica fit the colonization model, which was also suggested by coalescent simulations.
Demographic History
We conducted neutral tests and mismatch distribution analyses for lineages as well as sublineages (Table 3; Fig. 4). Among four lineages, only lineage A yielded a significantly negative Fu’s F s value (P < 0.05), and the unimodal mismatch distribution fit the null distribution of sudden expansion, although this lineage yielded a non-significant negative Tajima’ D value. Lineages C yielded non-significant negative values from neutral tests and multimodal mismatch distribution, whereas lineages B and D achieved non-significant positive values from neutral tests and bimodal/multimodal mismatch distributions that indicated these lineages probably have experienced bottlenecks or subdivisions. Among all sublineages, sublineages B2, C2, and C3 yielded significantly negative Fu’s F s values (P < 0.05) and non-significant negative Tajima’s D values, while their mismatch distributions were unimodal, and closely fitted to the distribution under the sudden expansion model. Although the mismatch distributions of sublineage C1 fitted the unimodal curve, the values of Fu’s F s and Tajima’s D tests were positive but not significant. The values from neutral tests for sublineage B1 were non-significantly positive and the mismatch distribution was multimodal, suggesting a bottleneck effect. From the estimates of tau values, the beginning time for expansions of lineage A and sublineages B2, C1, C2, and C3 were measured as c. 24, 27, 38, 14, and 64 ka, respectively (Table 3).

Mismatch distributions for lineages and sublineages of Feirana taihangnica. The histograms represent the observed frequencies of pairwise differences among haplotypes and the line shows the curve simulated for a population that has expanded. Lineages and sublineages were given in Fig. 3
Although we ran 5 × 108 generations with 10 % as burn-in for each of sublineages B1, B2, C1, C2, and C3, Bayesian MCMC chains for these sublineages did not convergence, probably because they contained a small number of populations and scarce haplotypes that were not sufficiently informative about the coalescence time. Thus, we obtained BSPs only for the four lineages of F. taihangnica (Fig. 5). Lineage A showed population size expansion from the beginning of Holocene c. 10 ka and lineage B has experienced population size growth over c. 25 ka, while lineage C has experienced the sharpest population size expansion from c. 55 ka corresponding to the LGM. Furthermore, lineage B maintained a stable population size during the mid-to-late Pleistocene (c. 0.025–0.3 Ma) and lineage C maintained a stable population size during c. 0.07–0.14 Ma. Lineage D has experienced a slight decline in population size from c. 0.14 Ma.

Demographic patterns of each lineage and a combined pattern as determined from Bayesin Skyline Plots. The central solid line represents the median value of the log of the population size (N ef * τ) and the area between two thinner lines represents the 95 % higher posterior density. All figures show the population size fluctuation after the lower limit of 95 % CI’s of the estimated time of the most recent common ancestor. Phylogenetic lineages were given in Fig. 3
Discussion
Prolonged Divergence and Multiple Glacial Refugia
Substantial genetic heterogeneity commonly exists between intraspecific populations from different Mountains that are separated by low-elevation areas with disparate environmental conditions (Knowles 2000; DeChaine and Martin 2006; Smith and Farrell 2005; Wiens and Graham 2005; Kozak and Wiens 2006). In the QM region, the gene flow among populations of Feirana taihangnica that live in stream habitats may be restricted by warmer or more xeric valleys. Consistent with this hypothesis, we found that F. taihangnica contains four divergent lineages structured across major Mountains in the QM region (Table 2; Table S2; Figs. 1, 3), and several major valleys (Jialing River, Hei River, Luo River, and Dan River) were predicted to be the barriers that restricted gene flow among these lineages (Fig. 1). Notably, lineage C spans the predicted barrier Hei River for a short distance (Fig. 1). This may be due to recent expansion and migration of lineage C and secondary contact between lineages B and C. This hypothesis was supported by coalescent simulations, ancestral area reconstructions, and demographic analyses. Coalescent simulations and ancestral area reconstructions suggested that lineage C originated from the east side of Hei River after the LGM (Table 4; Fig. 2c), and demographic analyses indicated that this lineage experienced postglacial expansion in population size and distribution range (Table 3; Figs. 4, 5).
The remarkable spatial genetic differentiations among F. taihangnica lineages likely reflect the effects of historical isolations induced by tectonic changes and Pleistocene climatic shifts. The geomorphology and paleoecology of the QM were affected by various orogenic and climatic fluctuations (Zhou 2000; Dong et al. 2011). During the late Miocene, the rapid uplift of the Tibetan Plateau has profoundly changed the geographical features of its adjacent regions, including the WQM (Zhang et al. 2006), and driven the beginning of the Asia monsoons (An et al. 2001; Molnar 2005). Numerous phylogeographic studies have proposed that species diversifications were in response to this tectonic event (e.g., Qi et al. 2007; Jin et al. 2008; Qu et al. 2010; Guo et al. 2011). In F. taihangnica, coalescent simulations indicated that the split between lineage A (from the WQM) and the ancestor of other lineages occurred during the late Miocene at c. 7.7 Ma. Thus, tectonic and climatic changes triggered by the uplift of the Tibetan Plateau during the late Miocene may have driven the initial divergence within this species. Climate fluctuations and tectonic movements during Pleistocene may have driven the divergence among the three eastern lineages (Figs. 1, 2c, 3). During the early-to-mid Pleistocene, the world experienced significant climatic transitions associated with the glacial cycles (Huybers 2009; Lisiecki and Raymo 2007), and in North China, temperatures dropped dramatically (Cheng 1997; Zhou 2000) and the Asian monsoon was strengthened (Zheng and Rutter 1998). Some local tectonic movements also occurred during this period, such as the Kun–Huang diastrophisms, drastically changing the drainages (e.g., Yellow River) in the north QM (Shi et al. 1998, 1999; Zhou 2000). A rapid large-magnitude uplift of the QM also occurred, especially with respect to the features of Taibai Mountain in the MQM (Liu et al. 2010). In summary, historical isolation derived from the Miocene–Pleistocene orogenesis and climatic changes were likely responsible for the remarkable genetic heterogeneity among lineages of F. taihangnica.
Such a pattern of prolonged divergence among lineages of F. taihangnica is in agreement with the scenario that they were derived from multiple refugia during the LGM. The multiple-refugia model is supported by coalescent simulations (Fig. 2c) and the spatial distribution pattern of ND2 haplotypes that are endemic to different regions, as lineages that survived in the refugia had private haplotypes and substantial genetic diversity (Fig. 3; Table 3; Maggs et al. 2008). Our analysis is the first to provide substantial evidence for multiple refugia for a vertebrate species endemic to the QM region. Previous studies did not analyze the QM populations alone or sampled very limited populations from this region (Li et al. 2009; Song et al. 2009). Some refugia proposed by this study (Fig. 2c) are corroborated by other case studies; for example, the WQM, MQM, and FNM were considered as refugia for the tree peony (Paeonia rockii) (Yuan et al. 2012), the WQM was a refugium for the Chinese Hwamei (Leucodioptron canorum canorum) (Li et al. 2009), and the WQM, MQM, and EQM were refugia for the Gray-cheeked Fulvetta (Alcippe morrisonia) during the Pleistocene ice ages (Song et al. 2009). Certainly, we need more cases to make generalizations about the genetic and demographic legacies of the glacial oscillations on organisms in this region.
Coalescent simulations and ancestral area reconstructions indicated that the diversifications in this species occurred in a colonization fashion involving a series of dispersals from one mountain to another, followed closely by vicariances (stepping-stone model; Fig. 2c; Table 4; Knowles and Maddison 2002). This model is comparable to the hypothesis proposed by Wang et al. (2009). The west-east tendency of the QM region (Fig. 1) provides topographic conditions for the historical eastward dispersing of F. taihangnica, though the precise dispersal routes are probably obscured by the coalescence times, accumulation of genetic differentiations or secondary contact.
Distribution patterns of lineages in F. taihangnica may also have been influenced by competitive interactions with the closely related species, F. quadranus. Wang et al. (2012) proposed that F. quadranus experienced sharp postglacial expansions into the WQM, MQM, and EQM. Now this species is broadly sympatric with F. taihangnica and is more dominant than F. taihangnica in many streams in these areas (Yang 2011). This indicates that F. quadranus likely has higher fitness than F. taihangnica in the QM region in the present warm climate. The putative competition from F. quadranus probably leads to the exclusion of the weaker F. taihangnica from some regions (Hardin 1960; Monney 1996) and contributes to the allopatric divergence among lineages of F. taihangnica.
Historical Demography
Lineages B and C have likely experienced postglacial expansions from their own refugia, respectively, and then were subdivided into present sublineages. Recent expansions of lineages B and C were supported by the BSPs but not by the neutral tests and mismatch distributions (Table 3; Figs. 4, 5). The discordance may indicate that these lineages had undergone recent expansion but were recently subdivided (Ray et al. 2003; Castoe et al. 2007; Bertorelle and Slatkin 1995). In accordance with this hypothesis, coalescent simulations suggested that each lineage derived from one refugium after the LGM, and the divergences in each lineage occurred during the contemporary interstadial (Fig. 2c).
Population size fluctuations associated with eco-environmental changes during Pleistocene are expected to be reflected by the levels of genetic variation and coalescence times (Hewitt 1996, 2000, 2004). High Hd and low π of sublineage C3 indicate that it has recently expanded from a large stable population, and low Hd and π of lineage A and sublineages B2, C1, and C2 (Table 3) indicate that they had probably been suppressed and suffered from bottlenecks during the LGM (Hedrick 1999; Fernando et al. 2000). The low genetic diversity may be derived from the recent rapid expansion that brought about a slight accumulation of mutation and loss of genetic variation (Hewitt 2004). However, it was also probably due to the limitation of the single-gene data and limited sampling. Demographic analyses, even the coalescent-based method BSP, which incorporates information from the genealogy and may achieve better estimates of the demographic history, could only detect recent expansions (Table 4; Figs. 4, 5) and are unable to uncover the bottlenecks in the earlier coalescent time (Jesus et al. 2006; Pybus et al. 2001; Heled and Drummond 2010). Thus, we cautiously make demographic inferences and are striving to develop an extension of this study using multiple loci and adequate sampling to reconstruct a credible genealogy (Heled and Drummond 2010).
Populations occurring in different regions or belonging to different species probably have various demographic responses to Pleistocene climate fluctuations (Hewitt 2004; Carstens and Richards 2007). Postglacial expansions were revealed for most populations of F taihangnica (Figs. 4, 5). This pattern was also found for the QM lineage of F. quadranus (Wang et al. 2012), as well as several other temperate species in North America and Europe (e.g., Milá et al. 2006, 2007; Kvist et al. 2004; Burg et al. 2005; Fontanella et al. 2008; Guiher and Burbrink 2008), but differed from that of other vertebrate species with wider distribution in East Asia that experienced population growth before the LGM (Li et al. 2009; Song et al. 2009; Ding et al. 2011). Nonetheless, like other species (Li et al. 2009; Song et al. 2009; Ding et al. 2011; Wang et al. 2012), most lineages of F. taihangnica might have sustained stable effective population size during the mid-late Pleistocene (Fig. 5). Population subdivision or rare migration events among sub-populations probably inflated the total N ef, though a population would lose its habitat during the cold periods (Wright 1943; Nei and Takahata 1993; Wakeley 2000; De and Durrett 2007). In addition, mild environments during the recent glacial periods proposed by palynological and palaeoclimatic studies in the QM (Knutti et al. 2004; Ju et al. 2007; Yu et al. 2007; Kelly et al. 2006) might also play a role in maintaining N ef of the widespread lineages.
Conclusion
Overall, our results suggest that Pleistocene climate fluctuations, as well as tectonic configurations from the late Miocene to the late Pleistocene most likely promoted the remarkable divergence among lineages of Feirana taihangnica. The diversification process in F. taihangnica fits a stepping-stone model. Once differentiated, F. taihangnica likely persisted through Pleistocene glacial periods in multiple refugia across the QM region. After the LGM, most lineages of F. taihangnica have experienced expansions of population size and distribution range. The findings of this study provide further evidence that Pleistocene climatic changes profoundly impacted the diversification patterns and historical demography of vertebrate species living in stream habitats in the QM region. Integration of data from multiple loci and larger sample sizes might provide greater support for the above hypotheses, or make it possible to extend them further. This would be a worthy goal for future research.
References
An ZS, Kutzbach JE, Prell WL, Porter SC (2001) Evolution of Asian monsoons and phased uplift of the Himalaya–Tibetan plateau since Late Miocene times. Nature 411:62–66
Avise JC (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge
Avise JC, Walker D (1998) Pleistocene phylogeographic effects on avian populations and the speciation process. Proceedings of the Royal Society B: Biological Sciences 265(1395):457–463
Avise JC, Walker D, Johns GC (1998) Speciation durations and Pleistocene effects on vertebrate phylogeography. Proceedings of the Royal Society B: Biological Sciences 265(1407):1707–1712
Beerli P (2008) MIGRATE version 2.4.1. http://popgen.csit.fsu.edu/Migrate-n.html
Bermingham E, McCafferty SS, Martin AP (1997) Fish biogeography and molecular clocks: perspectives from the Panamanian Isthmus. In: Kocher TD, Stepien CA (eds) Molecular systematics of fishes. Academic Press, San Diego, pp 113–118
Bertorelle G, Slatkin M (1995) The number of segregating sites in expanding human populations, with implications for estimates of demographic parameters. Mol Biol Evol 12:887–892
Burg T, Gaston AJ, Winker K, Friesen VL (2005) Rapid divergence and postglacial colonization in western North American Steller’s jays (Cyanocitta stelleri). Mol Ecol 14:3745–3755
Carstens BC, Richards CL (2007) Integrating coalescent and ecological niche modelling in comparative phylogeography. Evolution 61:1439–1454
Carstens BC, Sullivan J, Davalos LM, Larsen PA, Pedersen SC (2004) Exploring population genetic structure in three species of Lesser Antillean bats. Mol Ecol 13:2557–2566
Castoe TA, Spencer CL, Parkinson CL (2007) Phylogeographic structure and historical demography of the western diamondback rattlesnake (Crotalus atrox): a perspective on North American desert biogeography. Mol Phylogenet Evol 42:193–212
Che J, Hu JS, Zhou WW, Murphy RW, Papenfuss TJ, Chen MY, Rao DQ, Li PP, Zhang YP (2009) Phylogeny of the Asian spiny frog tribe Paini (Family Dicroglossidae) sensu Dubois. Mol Phylogenet Evol 50:59–73
Che J, Zhou WW, Hua JS et al (2010) Spiny frogs (Paini) illuminate the history of the Himalayan region and Southeast Asia. Proc Natl Acad Sci USA 107(31):13765–13770
Chen XH, Jiang JP (2002) A new species of the genus Paa from China. Herpetol China 9:231
Cheng J (1997) The mammalian faunas showing climatic fluctuation: as an example of the Early Pleistocene mammalian faunas from Zhoukoudian. Earth Sci Front 4:275–279
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9(10):1657–1659
Crawford AJ (2003) Relative rates of nucleotide substitution in frogs. J Mol Evol 57:636–641
Cui ZJ, Zhang W (2003) Discussion about the glacier extent and advance/retreat asynchrony during the last glaciation. J Glaciol Geocryol 25:510–516
De A, Durrett R (2007) Stepping-stone spatial structure causes slow decay of linkage disequilibrium and shifts the site frequency spectrum. Genetics 176:969–981
DeChaine EG, Martin AP (2006) Using coalescent simulations to test the impact of quaternary climate cycles on divergence in an alpine plant–insect association. Evolution 60:1004–1013
Ding L, He SP, Gan XN, Zhao EM (2011) A phylogeographic, demographic and historical analysis of the short-tailed pit viper (Gloydius brevicaudus): evidence for early divergence and late expansion during the Pleistocene. Mol Ecol 20:1905–1922
Dong YP, Zhang GW, Neubauer F, Liu XM, Genser J, Hauzenberger C (2011) Tectonic evolution of the Qinling orogen, China: review and synthesis. J Asian Earth Sci 41:213–237
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Drummond AJ, Rambaut A, Shapiro B, Pybus OG (2005) Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 22(5):1185–1192
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4:699–710
Edwards SV, Beerli P (2000) Gene divergence, population divergence, and the variance in coalescence time in phylogeographic studies. Evolution 54:1839–1854
Excoffier L, Schneider S (1999) Why hunter–gatherer populations do not show signs of Pleistocene demographic expansions. Proc Natl Acad Sci USA 96:10597–10602
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: applications to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin version 3.0: an integrated software package for population genetics data analysis. Evol Bioinf Online 1:47–50
Fei L, Ye CY, Jiang JP (2010) Colored atlas of Chinese amphibians. Sichuan Publishing House of Science and Technology, Chengdu
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Felsenstein J, Kishino H (1993) Is there something wrong with the bootstrap on phylogeny? A reply to Hillis and Bull. Syst Zool Syst Zool 42:193–200
Fernando P, Pfrender ME, Encalada SE, Landa R (2000) Mitochondrial DNA variation, phylogeography and population structure of the Asian elephant. Heredity 84:362–372
Fitzpatrick SW, Brasileiro CA, Haddad CF, Zamudio KR (2009) Geographical variation in genetic structure of an Atlantic coastal forest frog reveals regional differences in habitat stability. Mol Ecol 18:2877–2896
Fontanella FM, Feldman CR, Siddall ME (2008) Phylogeography of Diadophis punctatus: extensive lineage diversity and repeated patterns of historical demography in a transcontinental snake. Mol Phylogenet Evol 46:1049–1070
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Guiher TJ, Burbrink FT (2008) Demographic and phylogeographic histories of two venomous North American snakes of the genus Agkistrodon. Mol Phylogenet Evol 48:543–553
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. BMC Syst Biol 52:696–704
Guo P, Liu Q, Li C, Cheng X, Jiang K, Wang YZ, Malhotra A (2011) Molecular phylogeography of Jerdon’s pitviper (Protobothrops jerdonii): importance of the uplift of the Tibetan plateau. J Biogeogr 38:2326–2336
Hardin G (1960) The competitive exclusion principle. Science 131(3409):1292–1297
Harpending RC (1994) Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol 66:591–600
Hartl DL, Clark AG (2007) Principles of population genetics, 4th edn. Sinauer Associates, Sunderland
Hedges SB (1992) The number of replications needed for accurate estimation of the bootstrap P-value in phylogenetic studies. Mol Biol Evol 9:366–369
Hedrick PW (1999) Perspective: highly variable loci and their interpretation in evolution and conservation. Evolution 53:313–318
Heled J, Drummond AJ (2010) Bayesian inference of species trees from multilocus data. Mol Biol Evol 27:570–580
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276
Hewitt GM (2000) The genetic legacy of the quaternary ice ages. Nature 405(6789):907–913
Hewitt GM (2004) Genetic consequences of climatic oscillations in the quaternary. Philos Trans R Soc B 359(1442):183–195
Hillesheim MB, Hodell DA, Leyden BW, Brenner M, Curtis JH, Anselmetti FS, Ariztegui D, Buck DG, Guilderson TP, Rosenmeier MF, Schnurrenberger DW (2005) Climate change in lowland Central America during the late deglacial and early Holocene. J Quat Sci 20:363–376
Hillis DM, Mable BK, Larson A, Davis SK, Zimmer EA (1996) Nucleic acids IV: sequencing and cloning. In: Hillis D, Moritz C, Mable B (eds) Molecular systematics. Sinauer Associates, Sunderland, pp 321–378
Huelsenbeck JP, Hillis DM (1993) Success of phylogenetic methods in the four-taxon case. Syst Biol 42:247–264
Hurvich CM, Tsai CL (1989) Regression and time series model selection in small samples. Biometrika 76:297–307
Huybers P (2009) Pleistocene glacial variability as a chaotic response to obliquity forcing. Clim Past 5:481–488
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service v. 3.14. BMC Genet 6:13
Jesus FF, Wilkins JF, Solferini VN, Wakeley J (2006) Expected coalescent times and segregating sites in a model of glacial cycles. Genet Mol Res 5:466–474
Jiang JP, Dubois A, Ohler A, Tillier A, Chen XH, Xie F, Stöck M (2005) Phylogenetic relationships of the tribe Paini (Amphibia, Anura, Ranidae) based on partial sequences of mitochondrial 12s and 16s rRNA genes. Zool Sci 22:353–362
Jin YT, Brown RP, Liu NF (2008) Cladogenesis and phylogeography of the lizard Phrynocephalus vlangalii (Agamidae) on the Tibetan plateau. Mol Ecol 17:1971–1982
Ju L, Wang H, Jiang D (2007) Simulation of the last glacial maximum climate over East Asia with a regional climate model nested in a general circulation model. Palaeogeogr Palaeoclimatol Palaeoecol 248:376–390
Kelly MJ, Edwards RL, Cheng H et al (2006) High resolution characterization of the Asian Monsoon between 146 146,000 and 99 99,000 years B.P. from Dongge Cave, China and global correlation of events surrounding Termination II. Palaeogeogr Palaeoclimatol Palaeoecol 236:20–38
Knowles LL (2000) Tests of Pleistocene speciation in montane grasshoppers (genus Melanoplus) from the sky islands of western North America. Evolution 54:1337–1348
Knowles LL (2001) Did the Pleistocene glaciations promote divergence? Tests of explicit refugial models in montane grasshoppers. Mol Ecol 10:691–701
Knowles LL, Maddison WP (2002) Statistical phylogeography. Mol Ecol 11:2623–2635
Knutti R, Flückiger J, Stocker TF, Timmermann A (2004) Strong hemispheric coupling of glacial climate through freshwater discharge and ocean circulation. Nature 430:851–856
Kozak KH, Wiens JJ (2006) Does niche conservatism promote speciation? A case study in North American salamanders. Evolution 60:2604–2621
Kvist L, Viiri K, Dias PC, Rytkonen S, Orell M (2004) Glacial history and colonization of Europe by the blue tit Parus caeruleus. J Avian Biol 35:352–359
Leaché AD, Reeder TW (2002) Molecular systematics of the eastern fence lizard (Sceloporus undulatus): a comparison of parsimony, likelihood, and Bayesian approaches. Syst Biol 51:44–68
Li JJ, Shu Q, Zhou SZ, Zhao ZJ, Zhang JM (2004) Review and prospects of quaternary glaciation research in China. J Glaciol Geocryol 26(3):235–243
Li SH, Yeung CK, Feinstein J, Han L, Le MH, Wang CX, Ding P (2009) Sailing through the Late Pleistocene: unusual historical demography of an East Asian endemic, the Chinese Hwamei (Leucodioptron canorum canorum), during the last glacial period. Mol Ecol 18(4):622–633
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi:10.1093/bioinformatics/btp187
Lisiecki LE, Raymo ME (2007) Plio–Pleistocene climate evolution: trends and transitions in glacial cycle dynamics. Quatern Sci Rev 26:56–69
Liu JH, Zhang PZ, Zheng DW, Wang JL, Wang WT (2010) The cooling history of Cenozoic exhumation and uplift of Taibai Mountain, Qinling, China: evidence from the apatite fission track (AFT) analysis. Chin J Geophys 53(10):2405–2414
Macey JR, Schulte JA II, Ananjeva NB, Larson A, Rastegar-Pouyani N, Shammakovb SM, Papenfuss TJ (1998a) Phylogenetic relationships among Agamid Lizards of the Laudakia caucasia species group: testing hypotheses of biogeographic fragmentation and an Area Cladogram for the Iranian Plateau. Mol Phylogenet Evol 10:118–131
Macey JR, Schulte JAII, Larson A, Fang Z, Wang Y, Tuniyev BS, Papenfuss TJ (1998b) Phylogenetic relationships of toads in the Bufo bufo species group from the eastern escarpment of the Tibetan Plateau: a case of vicariance and dispersal. Mol Phylogenet Evol 9:80–87
Macey JR, Wang Y, Ananjeva NB, Larson A, Papenfuss TJ (1999) Vicariant patterns of fragmentation among gekkonid lizards of the genus Teratoscincus produced by the Indian Collision: a molecular phylogenetic perspective and an area cladogram for Central Asia. Mol Phylogenet Evol 12:320–332
Macey JR, Strasburg J, Brisson J, Vredenburg V, Jennings M, Larson A (2001) Molecular phylogenetics of western North American frogs of the Rana boylii species group. Mol Phylogenet Evol 19:131–143
Maddison WP, Maddison DR (2011) Mesquite: a modular system for evolutionary analysis. Version 2.75. http://mesquiteproject.org
Maddison WP, McMahon M (2000) Divergence and reticulation among montane populations of a jumping spider (Habronattus pugillis Griswold). Syst Biol 49:400–421
Maggs C, Castilho R, Foltz D, Kelly J, Olsen J, Perez KE, Stam W, Väinölä R, Viard F, Wares J (2008) Evaluating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology 89(11 Suppl):S108–S122
Manni F, Guerard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Hum Biol 76(2):173–190
Marjoram P, Donnelly P (1994) Pairwise comparisons of mitochondrial DNA sequences in subdivided populations and implications for early human evolution. Genetics 136:673–683
Milá B, McCormack JE, Castaneda G, Wayne RK, Smith TB (2007) Recent postglacial range expansion drives the rapid diversification of a songbird lineage in the genus Junco. Proc R Soc B 274:2653–2660
Molnar P (2005) Mio-Pliocene growth of the Tibetan Plateau and evolution of East Asian climate. Palaeontol Elect 8(1):1–23
Monney JC (1996) Biologie comparée de Vipera aspis L. et de Vipera berus L. (Reptilia, Ophidia, Viperidae) dans une station des Préalpes bernoises. Unpublished PhD Thesis, Université de Neuchatel, Neuchatel
Nei M, Takahata N (1993) Effective population size, genetic diversity, and coalescence time in subdivided populations. J Mol Evol 37:240–244
Parra-Olea G, García-París M, Wake DB (2004) Molecular diversification of salamanders of the tropical American genus Bolitoglossa (Caudata: Plethodontidae) and its evolutionary and biogeographical implications. Biol J Linn Soc 81:325–346
Pinot S, Ramstein G, Harrison SP, Prentice IC, Guiot J, Stute M, Joussaume S et al (1999) Tropical paleoclimates at the last glacial maximum: comparison of Paleoclimate Modeling Intercomparison Project (PMIP) simulations and paleodata. Clim Dyn 15:857–874
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25(7):1253–1256
Pybus OG, Charleston MA, Gupta S, Rambaut A, Holmes EC, Harvey PH (2001) The epidemic behavior of the hepatitis C virus. Science 292:2323–2325
Qi D, Guo S, Zhao X, Yang J, Tang W (2007) Genetic diversity and historical population structure of Schizopygopsis pylzovi (Teleostei: Cyprinidae) in the Qinghai–Tibetan Plateau. Freshw Biol 52:1090–1104
Qian H, Ricklefs RE (2000) Large-scale processes and the Asian bias in temperate plant species diversity. Nature 407:180–182
Qu Y, Lei F, Zhang R, Lu X (2010) Comparative phylogeography of five avian species: implications for Pleistocene evolutionary history in the Qinghai–Tibetan Plateau. Mol Ecol 19:338–351
Rambaut A, Drummond AJ (2007) Tracer v1.4. http://beast.bio.ed.ac.uk/Tracer
Ray N, Currat M, Excoffier L (2003) Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol 20:76–86
Richards CL, Carstens BC, Knowles LL (2007) Distribution modelling and statistical phylogeography: an integrative framework for generating and testing alternative biogeographical hypotheses. J Biogeogr 34:1833–1845
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Rogers AR, Fraley AE, Bamshad MJ, Watkins WS, Jorde LB (1996) Mitochondrial mismatch analysis is insensitive to the mutational process. Mol Biol Evol 13:895–902
Ronquist FR, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rost KT (1994) Paleoelimatic field studies in and along the Qinling Shan (Central China). GeoJournal 34(1):107–120
Schwarz GE (1978) Estimating the dimension of a model. Ann Stat 6(2):461–464
Shi YF (2002) Characteristics of late quaternary monsoonal glaciation on the Tibetan Plateau and in East Asia. Quatern Int 97(98):79–91
Shi YF (2007) The Pleistocene glaciation of China and environmental change. Henan Science and Technology Publishing House, Zhengzhou
Shi YF, Li JJ, Li BY (1998) Uplift and environmental changes of Qinghai–Tibetan plateau in the Late Cenozoic. Guangdong Science and Technology Press, Guangzhou
Shi YF, Li JJ, Li BY et al (1999) Uplift of the Qinghai–Xizang (Tibetan) Plateau and East Asia environmental change during late Cenozoic. Acta Geogr Sinica 54:10–20
Slatkin M, Maddison WP (1989) A cladistic measure of gene flow inferred from phylogenies of alleles. Genetics 123:603–613
Smith CI, Farrell BD (2005) Phylogeography of the longhorn cactus beetle Moneilema appressum LeConte (Coleoptera: Cerambycidae): was the differentiation of the Madrean sky islands driven by Pleistocene climate changes? Mol Ecol 14:3049–3065
Song G, Qu YH, Yin ZH, Li SS, Liu NF, Lei FM (2009) Phylogeography of the Alcippe morrisonia (Aves: Timaliidae): long population history beyond late Pleistocene glaciations. BMC Evol Biol 9:143. doi:10.1186/1471-2148-9-143
Swofford D (2002) PAUP*: phylogenetic analysis using parsimony (* and other methods), 4.0th edn. Sinauer Associates, Sunderland
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction site endonuclease mapping and DNA sequence data. (III) Cladogram estimation. Genetics 132:619–633
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The Clustal X windows interface: flexible strategies for multiple sequences alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Wakeley J (2000) The effects of subdivision on the genetic divergence of populations and species. Evolution 54:1092–1101
Wang B, Jiang JP, Chen XH, Xie F, Zheng ZH (2007) Morphometrical study on populations of the genus Feirana (Amphibia, Anura, Ranidae). Acta Zootaxonomic Sinica 3:139–145
Wang B, Jiang JP, Xie F, Chen XH, Dubois A, Liang G, Wagner S (2009) Molecular phylogeny and genetic identification of populations of two species of Feirana frogs (Amphibia: Anura, Ranidae, Dicroglossinae, Paini) endemic to China. Zool Sci 26:500–509
Wang B, Jiang JP, Xie F, Li C (2012) Postglacial colonization of the Qinling Mountains: phylogeography of the Swelled Vent frog (Feirana quadranus). PLoS ONE 7(7):e41579. doi:10.1371/journal.pone.0041579
Weisrock DW, Macey JR, Ugurtas IH, Larson A, Papenfuss TJ (2001) Molecular phylogenetics and historical biogeography among salamandrids of the “true” salamander clade: rapid branching of numerous highly divergent lineages in Mertensiella luschani associated with the rise of Anatolia. Mol Phylogenet Evol 18:434–448
Wiens JJ, Graham CH (2005) Niche conservatism: integrating evolution, ecology, and conservation biology. Annu Rev Ecol Syst 36:519–539
Wright S (1943) Isolation by genetics. Genetics 28:114–138
Yang J (2007) Study on the ecology and embryonic development of Feirana taihangnicus [DM]. Graduate School of Henan Normal University, Xinxiang
Yang X (2011) Speciation and geographic distribution pattern of the genus Feirana [M]. Chengdu Institute of biology, Chinese Academy of Sciences, Sichuan
You Y, Sun KP, Xu LJ, Wang L, Jiang TL, Liu S, Lu GJ, Berquist SW, Feng J (2010) Pleistocene glacial cycle effects on the phylogeography of the Chinese endemic bat species, Myotis davidii. BMC Evol Biol 10:208. http://www.biomedcentral.com/1471-2148/10/208
Yu G, Gui F, Shi Y, Zheng Y (2007) Late marine isotope stage 3 palaeoclimate for East Asia: a data–model comparison. Palaeogeogr Palaeoclimatol Palaeoecol 250:167–183
Yuan JH, Cheng FY, Zhou SL (2012) Genetic structure of the tree Peony (Paeonia rockii) and the Qinling Mountains as a geographic barrier driving the fragmentation of a large population. PLoS ONE 7(4):e34955. doi:10.1371/journal.pone.0034955
Zhang RZ (1999) Zoogeography of China. Science Press, Beijing
Zhang RZ (2004) Relict distribution of land vertebrates and quaternary glaciation in China. Acta Zool Sinica 50:841–851
Zhang PZ, Zheng DW, Yin GM, Yuan DY, Zhang GL, Li CY, Wang ZC (2006) Discussion on late Cenozoic growth and rise of northeastern margin of the Tibetan plateau. Quat Sci 26(1):5–13
Zhang H, Yan J, Zhang GQ, Zhou KY (2008) Phylogeography and demographic history of Chinese black-spotted frog populations (Pelophylax nigromaculata): evidence for independent refugia expansion and secondary contact. BMC Evol Biol 8:21
Zheng B, Rutter N (1998) On the problem of quaternary glaciations, and the extent and patterns of Pleistocene cover in the Qinghai–Xizang (Tibet) plateau. Quat Int 45(46):109–122
Zhou HY (2000) Environmental events and catastrophes from the end of early pleistocene to the early stage of middle pleistocene. J Nat Disasters 9:123–129
Acknowledgments
We are grateful to Gang Liang, Xiaohong Chen, Junhua Hu, Xin Yang, and Rongchuan Xiong for their help in the collection of samples. Also thanks to JinZhong Fu from University of Guelph and Li Ding for polishing this manuscript. This study was supported by the National Natural Sciences Foundation of China (NSFC-31071906 and NSFC-31270568 granted to Jianping Jiang, NSFC-31172055 to Cheng Li, and NSFC-31201702 to Bin Wang), the field front project of the knowledge innovation program of CAS (KSCX2-EW-J-22, Y1C2021200, Y1B3021), and Key Laboratory of Mountain Ecological Restoration and Bioresource Utilization in CIB of CAS and Ecological Restoration and Biodiversity Conservation Key Laboratory of Sichuan Province.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
239_2013_9544_MOESM1_ESM.xls
Table S1 Number of shared haplotypes for each pair of the 17 sampled populations of Feirana taihangnica. Populations 1–17 were given in Table 1. (XLS 26 kb)
239_2013_9544_MOESM2_ESM.xls
Table S2 Fst among the 17 sampled populations of Feirana taihangnica. Populations 1–17 were given in Table 1. * P > 0.05. (XLS 29 kb)
239_2013_9544_MOESM3_ESM.xls
Table S3 Fst among groups in different grouping arrangements according to phylogenetic arrays and geographic occurrences for populations of Feirana taihangnica. All P < 0.01. (XLS 27 kb)
Rights and permissions
About this article
Cite this article
Wang, B., Jiang, J., Xie, F. et al. Phylogeographic Patterns of mtDNA Variation Revealed Multiple Glacial Refugia for the Frog Species Feirana taihangnica Endemic to the Qinling Mountains. J Mol Evol 76, 112–128 (2013). https://doi.org/10.1007/s00239-013-9544-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-013-9544-5