
Abstract
The complete genome of the German isolate AC of Apple stem grooving virus (ASGV) was sequenced. It encodes two overlapping open reading frames (ORFs), similarly to previously described ASGV isolates. Two regions of high variability were detected between the ASGV isolates, variable region 1 (V1, from amino acids (aa) 532 to 570), and variable region 2 (V2, from aa 1,583 to 1,868). The phylogenetic analysis of the V1 and V2 regions suggested that the ASGV diversity was structured by host plant species rather than geographical origin. The dN/dS ratio between nonsynonymous and synonymous nucleotide substitution rates varied greatly along the ASGV genome. Most of ORF1 showed predominant negative selection except for the two regions V1 and V2. V1 showed an elevated dN and an average dS when compared to the ORF1 background but no significant positive selection was detected. The V2 region of ORF1 showed an elevated dN and a low dS when compared to the ORF1 background with an average dN/dS ≈ 3.0 indicative of positive selection. However, the V2 area includes overlapping ORFs, making the dN/dS estimate biased. Joint estimates of the selection intensity in the different ORFs by a recent method indicated that this region of ORF1 was in fact evolving close to neutrality. This was convergent with previous results showing that introduction of stop codons in this region of ORF1 did not impair plant infection. These data suggest that the elimination of a stop codon caused the overprinting of a novel coding region over the ancestral ORF.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The two most important features of RNA viruses are their small genomes and high mutation rates. These two features have been linked by Holmes (2003), arguing that a lack of genomic space means that RNA viruses will be subjected to important evolutionary constraints because specific sequences are required to encode multiple and often conflicting functions.
One frequent attribute of small genomes is gene overlapping, where the same nucleotide sequence simultaneously encodes for two or more proteins in different open reading frames (ORFs). This overlapping gene arrangement is essential to squeeze a maximum amount of information into a limited size genome (Gibbs and Keese 1995; Krakauer 2000; Normark et al. 1983) and increase the amount of protein diversity expressed by a single nucleotide sequence (Holmes 2009). This overlapping can be caused by several mechanisms, including RNA splicing (Pavesi 2007), the use of non AUG start codons (Baril and Barkier-Gingras 2005) and overprinting, where the original sequence undergoes a modification causing the elimination of a stop codon, thus printing a novel coding region over the original ORF (Belshaw et al. 2007; Keese and Gibbs 1992). Recently, Chirico et al. (2010) have found that the physical constraint exerted by the viral capsid size on the genome length has led to gene overlap as a mechanism for producing more proteins from the same genome length.
Overlapping ORFs influence a number of other evolutionary aspects aside from increasing the amount of genetic information stored in a limited size genome. First, synonymous nucleotide substitutions in one ORF are likely to be nonsynonymous in the second one, which complicates evolution analyses (Hein and Støvlbæk 1995). Second, every nucleotide site within the overlap is expected to have a major impact on the fitness and evolutionary flexibility of the virus therefore ORF overlaps can be considered as extreme forms of pleiotropy. This is exemplified by the lower evolution rates observed in overlapping regions compared to non-overlapping regions in Hepatitis B virus (Zhou and Holmes 2007) and by increased rates of deleterious mutations in these regions (Belshaw et al. 2007). Third, because of the reduced urgency to create protein diversity, viruses with larger genomes tend to show less gene overlap than viruses with smaller genomes (Belshaw et al. 2007).
One such small genome exploiting gene overlap is that of Apple stem grooving virus (ASGV). ASGV is the type member of the genus Capillovirus from the family Flexiviridae (Martelli et al. 2007), which also comprises Cherry virus A. The ASGV virus particles are flexuous filaments, 620–680 nm long, depending on the isolate (Clover et al. 2003; Inouye et al. 1979; Lister et al. 1965), and 12 nm in width. Several isolates of ASGV from different hosts have been fully sequenced (Ohira et al. 1995; Shim et al. 2004; Tatineni et al. 2009b, Terauchi et al. 1997; Yoshikawa et al. 1992, also see Table 1). ASGV has a single-stranded positive-sense monopartite genome of approximately 6,495 nucleotides excluding a poly-A tail. The genomic RNA has two overlapping ORFs. ORF1 (bases 37–6341) encodes a 241–242 kDa polyprotein (2,105 amino acids) containing the replicase region with the consensus motifs of methyltransferase, papain-like protease, nucleotide triphosphate-binding helicase, RNA-dependent RNA polymerase, and the coat protein (CP) in the C-terminal region (Fig. 1) (Jelkmann 1995; Yoshikawa et al. 1992). ORF2 (bases 4,788–5,447) encodes a 36-kDa putative movement protein (MP) (Yoshikawa et al. 1992; 1993) and overlaps with both the replicase and CP coding regions. A highly variable region is located between the conserved replicase and CP regions in ORF1, where codons 1,585–1,868 of ORF1 overlap with ORF2. This variable region has no functional motifs, and no ASGV isolates containing a stop codon in this region have been reported (Magome et al. 1997, 1999). The organization of the polyprotein of ASGV is unusual for members of the Flexiviridae, which normally encode the CP in a separate ORF (Martelli et al. 2007). Two additional 3′ co-terminal viral RNA species, in addition to the genomic RNA, were also detected, with sizes suggesting that the CP and the MP genes were expressed through sub-genomic RNAs (Hirata et al. 2003; Tatineni et al. 2009a).

Similarity plot of ASGV-AC with amino acid sequences of other ASGV isolates available on NCBI. The two most variable regions V1 and V2 are indicated with dotted lines. Open boxes represent the open reading frames (ORF), ORF1 encoding the replicase and coat protein (CP), and ORF2 encoding the movement protein (MP). Black boxes represent the proteic domains: Mt methyltransferase, P-pro papain-like protease, Hel nucleotide triphosphate-binding helicase, RdRp RNA-dependent RNA polymerase. The five indicated regions were analysed separately for selection intensity (Table 2)
In this paper, we report the complete sequence of a German isolate of ASGV, its comparison to other available ASGV sequences, and diversity analysis of two variable regions within the ASGV genome in order to get insights into the molecular evolution of the ASGV genomic RNA.
Materials and Methods
Plant Material and Virus Isolates
The ASGV-AC isolate (Liebenberg et al. 2010) was collected in the field from infected Malus domestica nearby Neustadt an der Weinstrasse, Germany, and maintained in vitro on apple plantlets. Additional ASGV isolates from infected M. domestica material were obtained from Australia, Austria, Canada, France, and Germany (Table 1).
RNA Extraction and RT-PCR Amplification
Total RNAs were isolated infected apple plant material using the D-Genos RNA extraction kit (Ivagen, France) and used as template for the One-tube-RT-PCR amplification using the SuperScript III One-Step RT-PCR System with Platinum® Taq DNA Polymerase kit (Invitrogen), according to the manufacturers’ specifications. Degenerate primers were designed from available ASGV and Citrus tatter leaf virus (CTLV) sequences in the GenBank database of the National Centre for Biotechnology Information (NCBI), and used to amplify the first fragments of the ASGV-AC genome, which were cloned and sequenced. Specific primers were designed subsequently from these sequences, to amplify, clone and sequence the remaining gaps on the genome. To amplify the 3′ end of the viral genomic RNA, an oligod(T) primer was used in combination with an ASGV-specific primer. To determine the 5′ end of the genomic RNA, a 5′ RACE kit (Invitrogen) was used, according to the instructions of the supplier. The amplified fragments were ligated directly into the pTPCR vector (Wassenegger et al. 1994) and transformed into INVα chemically competent Escherichia coli cells (Invitrogen). The recombinant plasmids were purified using the Spin Miniprep Kit (Qiagen) and sequenced. Each nucleotide was sequenced at least three times and in both directions, from independent reactions. The primers used to amplify the two variable regions were: A-V1-1142 s (5′-TGAGRGAYGAYTTTGAYGTSTCAGT-3′) nd A-V1-2049as (5′-CATAYTTYCTRTTAGRACTGCCAT-3′) for the amplification of variable region 1 and A-V2-4558 s (5′-GCHAGRATYAARATGATGAGTGA-3′) and A-V2-5866as (5′-ACAGCRGGRAACTGGGTCTT-3′) for the amplification of variable region 2. Additional sets were also used to amplify variable region 1: a-V1-1180 s (5′-GGRATGAGRGAYGACTTTGA-3′) and a-V1-2100as (5′-ACTTCYTCAATCATYTCATG-3′) and for variable region 2: a-V2-4650 s (5′-CTACAGRTTAGGTGAGAGGC-3′) a-V2-5800as (5′-CAYAGAAGAAGGTAAAGCTC-3′). The RT-PCR cycling conditions were: 1 cycle of 30 min at 42 °C, 1 cycle of 2 min at 94 °C, 40 cycles of 20 s at 94 °C, 20 s at 46–50 °C for V1, and 49–56 °C for V2 (different annealing temperatures were used to obtain the desired fragment, depending on the isolate amplified), 20 s at 72 °C, 1 cycle of 7 min at 72 °C.
Building of the ASGV Phylogenetic Trees
BioEdit (Ver. 7.0.4) (Hall 1999) and DNAStar (Ver. 4.0) were used to perform sequence editing and compilation. Sequence comparisons were performed using the BLAST algorithm (Altschul et al. 1990) against the GenBank database of the NCBI (www.ncbi.nlm.nih.gov). The ASGV V1 and V2 nucleotide sequences were aligned with ASGV sequences downloaded from GenBank using the ClustalW (ver. 1.4) algorithm embedded in the BioEdit software. The maximum likelihood method implemented in PhyML version 3.0 (Guindon and Gascuel 2003) and the neighbour joining method implemented in MEGA version 4 software (Tamura et al. 2007) were used to infer the tree topologies. Branch support was calculates with 100 bootstrap replicates. Bootstrap percentages of ≥75 % were considered as well supported, between 75 and 50 % as moderately supported and values below 50 % as weakly supported. Bootstrap percentages below 50 % are not indicated on the phylograms.
Analysis of Selection Intensity in ASGV Genome
Since recombinant sequences could create biases in positive selection analyses (Schierup and Hein 2000), we used the RDP version 2 software (Martin et al. 2005) to detect such sequences in the five genome regions analysed independently for positive selection (see below). RDP2 implements seven different methods of recombination detection. The distribution of synonymous and nonsynonymous substitutions along ORF1 was analyzed first by using the SNAP program (Korber 2000; available at http://hiv-web.lanl.gov).
The mean values of dN and dS deriving from all pairwise comparisons between these regions within ORF1 sequences or between ORF2 sequences were compared with the Student t statistical test. A new method developed for the simultaneous estimation of dN/dS in overlapping genes by Sabath et al. (2008) was applied to the overlapping regions between ORF1 and ORF2. The rates of synonymous (dS) and nonsynonymous (dN) substitutions per site and their standard errors were evaluated by the Nei–Gojobori method (Nei and Gojobori 1986) using the software MEGA version 4.0 (Tamura et al. 2007). dN and dS were analysed separately for the five regions of the ACLSV genome showing different levels of variability after the SNAP analysis and also separately for the two different ORFs (Figs. 1, 3). dN/dS provides an estimate of the selection intensity (Kimura 1983) and allows identifying positive (dN/dS > 1) or negative (dN/dS < 1) selection in coding regions. To identify codon positions with positive selection, i.e. sites showing a dN/dS ratio significantly higher than one, a maximum likelihood approach implemented in the PAML version 4.2 (Yang 1997) package, was performed using the majority-rule consensus tree topology obtained with the neighbour-joining method. Under PAML, the sequence evolution models M0, M1a, M2a, M3, M7, and M8 were compared to assess the significance of positive selection in the sequence alignments and empirical Bayes methods were used to identify codons under positive selection, i.e. codons showing a dN/dS ratio significantly higher than one (Yang et al. 2000, 2005). Each PAML model was run at least three times with different initial values for dN/dS to avoid local maximum likelihood estimates.
Results
Genome Characteristics of ASGV-AC
The complete nucleotide sequence of ASGV-AC was 6496 nucleotides (nt) long, excluding the poly (A) tail. Computer analysis revealed one large open reading frame (ORF) (nt 37–6354), encoding a 2,105 amino acid (aa) polypeptide containing methyltransferase-like, papain-like protease, helicase-like, and RdRp-like domains, and the coat protein (CP) of 27 K (237 aa) located at the carboxy-terminal end of the polyprotein. This ORF was preceded by a 36 nucleotide-long non-coding region at the 5′ end of the genome, and followed by a 142 nucleotide-long non-coding region at the 3′ end. A second smaller ORF was found nested in ORF1 (nt 4788 to 5750), encoding a 320 amino acid polypeptide (M W 36152 = 36 K), with similarities to the 30-kDa superfamily of cell-to-cell movement proteins (Melcher 2000) (Fig. 1). Beside a putative small ORF encoding a 10 kDa protein (nt 3386–3644), for which no conserved domains nor identity to any other protein were found, no significant additional ORFs were found. The genomic organization of ASGV-AC was similar to those of previously described isolates of ASGV (Ohira et al. 1995; Shim et al. 2004; Tatineni et al. 2009b; Yoshikawa et al. 1992). When compared to other isolates of ASGV, the ASGV-AC isolate showed between 86 and 88 % nucleotide identity for ORF1, 94–96 % for ORF2 and between 95 and 98 % for the coat protein. In addition, two regions of high variability were detected in the genome (Fig. 1), variable region 1 (V1, amino acids 532–570) and variable region 2 (V2, amino acids 1,583–1,868) (Tatineni et al. 2009b). Interestingly, variable region 2 corresponded to the overlapping region between ORF1 and ORF2, and most of the variability found in ORF1 was silent in ORF2, as described previously (Magome et al. 1997; Tatineni et al. 2009b).
Phylogenetic Analyses of Variable Genome Regions of ASGV
In order to analyze more thoroughly the genetic variability in the V1 and V2 regions, additional isolates from Germany and other countries were collected (Table 1) and their variable genome regions cloned and sequenced. For a few of the plant samples, however, only one of the variable regions could be amplified (Table 1), even with varying annealing temperatures and additional primer combinations the amplification was unsuccessful. This could be due to insufficient complementarities between the primers and the viral RNAs in this variable region.
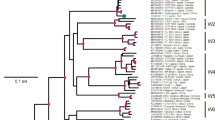
Phylogenetic analyses of the V1 region revealed that the ASGV-AC isolate grouped within one clade of closely related isolates containing European and Canadian isolates (Fig. 2a). Sequences from Asia, the USA, and Australia on the other hand showed much more variability between themselves and with other isolates, and were distributed in several clades, in which clustered also one French (Fr3) and two Canadian isolates (CO4b and CO2).

Neighbour joining phylogenetic trees of the nucleotide sequences of regions V1 (a) and V2 (b) of ASGV. The German AC isolate sequenced is indicated by a box. Bootstrap percentages above 50 % are shown. The scale bar indicates branch lengths in substitutions per nucleotide
Region V2 corresponded to the overlapping region between ORF1 and ORF2. Amino acids in this region were highly variable in ORF1, but conserved in ORF2, which means that the nucleotide changes which induced amino acid substitutions in ORF1 were in most cases silent in ORF2. In addition, no stop codon was found in any of the variable sequences of ORF1, confirming previous results (Magome et al. 1997; Tatineni et al. 2009b). The comparison of the sequences of the V2 regions from the different isolates revealed a similar tree topology as the one observed with V1 and were consistent between the two reconstruction methods used (maximum likelihood and neighbour joining). In these trees, internal branches were associated with low bootstrap values while branches at intermediate levels were particularly long and had high bootstrap values (Fig. 2).
When considering the phylogenetic clustering of the ASGV isolates in relation to their hosts, the three citrus isolates clustered together, as did the two lily isolates. The single pear isolate on the other hand was linked to a separated branch in the tree. Apple isolates of ASGV were grouped into four major clusters. However, this apparent larger diversity could also be due to the higher number of sequences from this host species. By contrast, there is only little evidence of isolate clustering according to their geographical origin. For instance, isolates from Canada and Japan, from Australia and France or from Canada and France clustered together (Fig. 2), which could most likely be explained by vegetative propagation and exchange of infected material between countries.
Evolutionary Patterns Vary Greatly Along ASGV Genome
A sliding-window analysis of dN and dS along ORF1 using the eight full genome sequences of ASGV showed that negative selection predominated (dN < dS) except in the two regions of higher diversity V1 and V2 identified previously, where dN was higher than dS (Fig. 3). Except in these two regions, dN/dS were roughly constant along ORF1. Compared to the background of ORF1, dN increased in region V1 while dS remained constant. By contrast, in the variable region V2, dN increased compared to the background but dS showed a sharp decrease (Fig. 3). This was confirmed by more thorough comparisons of dN and dS in these different regions. For regions 2 and 4 (Fig. 1), no recombination signal was detected with any of the seven methods implemented into RDP2 at the P = 0.05 threshold. For regions 3 and 5, weak (0.001 < P < 0.01) recombination signals were detected by three of the seven RDP2 methods only. For region 1, one sequence (accession number AY646511) was detected as recombinant by a majority of the methods (four of seven; 0.001 < P < 0.01), but the recombinant region was very short (about 100 nucleotide long). As a consequence, no putative recombination events introduced biases in our positive selection analyses of regions 2 and 4 on which we focused in the following, or these events modified only slightly the estimates of dN and dS (regions 1, 3, and 5), which we verified by analyzing datasets where the putative recombinants were withdrawn (data not shown).

Sliding-window analysis of rates of synonymous (dS, in grey) and nonsynonymous (dN, in black) substitutions along ASGV ORF1 using the SNAP program (Korber 2000). The x axis represents the codon positions and the y axis represents the average rates of synonymous or nonsynonymous mutations estimated with a 20 codon window size and a 10 codon step size
As shown in Table 2, the ratio dN/dS between the mean number of nonsynonymous substitutions per site and that of synonymous substitutions was close to 0.05 and remarkably constant in ORF2 and in ORF1 (except in regions V1 and V2 of ORF1). This confirms that ORF1 and ORF2 are overall evolving under strong negative selection. A different feature was observed in regions V1 and V2 of ORF1. In region V1, dN was significantly higher than in the surrounding parts of ORF1 (except V2) but dS was not significantly different (Table 2). This was particularly obvious for the region spanning nucleotides 1629–1748, for which a dN/dS ratio from 0.64 to 0.72 was estimated. This value, close to unity, suggests that some codon positions in this region might undergo positive selection. In region V2, both dN and dS varied significantly from the background of ORF1 (Table 2) and a dN/dS ratio of 2.02 was estimated. Again, this could be indicative of positive selection. It can also be noticed that in the non-overlapping region of the CP-coding region (region 5 in Table 2), both dS and dN were significantly lower than in the background of ORF1, while dN/dS remained unchanged.
Further analyses were performed to confirm or refute previous hypotheses about positive selection. For region V1, analysis of the alignment of the 23 available sequences with PAML did not allow the detection of any codon position under positive selection (data not shown). The best-fit model M3 in PAML indicated that about 9 % of codon positions in this genomic region evolved close to neutrality (dN/dS = 1). In region V2, the use of the dN/ds ratio to estimate selection intensity and to detect positive selection is problematic since the basic assumption is that synonymous substitutions are neutral is violated because of overlapping ORFs. Consequently, we used the method of Sabath et al.’s (2008) for joint estimation of the selection intensity in both ORFs. Since ORF2 overlaps with regions of ORF1 which encode two different proteins (the replicase and the CP) of different functions, we performed these estimations separately for region A where the MP gene overlaps with the replicase gene and for region B where the MP gene overlaps with the CP gene (Table 3). In overlapping region A, the MP gene ORF (ORF2) appears highly constrained with dN/dS = 0.019, while ORF1 (replicase) shows a neutral pattern of evolution (dN/dS = 0.95 ± 0.26). Opposite results were obtained for overlapping region B. The MP gene ORF (ORF2) appears poorly constrained with dN/dS = 0.76 while ORF1 (CP) is highly constrained (dN/dS = 0.18). However, the dN/dS estimate for region B of ORF2 is rather imprecise because of its small length. Largely different estimates of dN/dS would have been obtained, had we ignored the overlap between the two ORFs (Table 3). The SLAC (single-likelihood ancestor counting) maximum-parsimony method implemented in the HyPhy software (Kosakovsky Pond and Frost 2005) available at www.datamonkey.org provided dN/dS estimates highly similar to those obtained with PAML (data not shown). Consequently, the problem of evaluating correctly dN/dS seems to be really due to the overlapping nature of the ORFs rather than to the method used to estimate dN/dS.
Discussion
Two features of interest on the ASGV genome are its high degree of conservation among different isolates, with the exception of two regions, and the fact that it is extremely constrained. About 73 % of the genome (i.e. regions 1, 3, and 5; Fig. 1) show a dN/dS ratio of about 0.05, which is extremely low compared to a value of 1.0 corresponding to neutral evolution. The MP-coding region in the overlapping region 4 shows the same trend with dN/dS = 0.048. Contrasting with this background, two regions (2, 4) (Fig. 1; Table 2), corresponding to variable regions V1 and V2, respectively, show a much higher amino acid diversity. In these two regions, classical dN/dS estimates obtained with the software PAML were close to, or higher than 1.0, suggesting that at least some of the codon positions in these regions could be subjected to positive selection and involved in adaptation of the virus.
Analysis of positive selection in region V2 was more difficult because it consists of overlapping ORFs. “Classical” dN/dS analyses are not suited to analyze selection in overlapping ORFs since they assume that synonymous substitutions are neutral, which is not true in the case of overlapping ORFs because synonymous substitutions in one ORF are frequently nonsynonymous (and consequently frequently not neutral) in the other ORF. In that case, a joint estimation of dN/dS in both ORFs is necessary. Using the dN/dS joint estimation in overlapping ORFs developed by Sabath et al. (2008) showed that evolution of region V2 was very close to neutrality for the replicase ORF (dN/dS = 0.95 ± 0.26). As expected, this estimate contrasted greatly with the “classical” estimate from PAML which predicted positive selection (dN/dS = 2.96).
Taxonomically, capilloviruses are closest to tricho- and citriviruses in the family Beta-Flexiviridae. However, only capilloviruses encode both the replicase and coat protein in a single ORF (Hirata et al. 2010). The introduction of a stop codon in the region V2 of the replicase gene would split the replicase from the CP, as for the tricho- and citriviruses. Interestingly, in a recent study, Hirata et al. (2010) used an ASGV infectious clone to introduce artificial stop codons in the V2 region of ORF1 at the end of the replicase gene, which lead to truncated polyproteins lacking the CP fragment. Four of five constructs were infectious and capable of systemic movement. The only non-infectious construct was mutated at the beginning of region V2 (at position 4795 as compared to position 4825 or beyond in the case of the other four) of the ASGV genome. This demonstrates that the V2 region of the replicase gene downstream of position 4825 is not essential for the replicase activity and systemic movement, confirming the low evolutionary constraint in this region, as attested to by the dN/dS analysis. Our analysis, together with the data of Hirata et al. (2010), fuels the hypothesis of the “compaction theory” (Belshaw et al. 2007; Keese and Gibbs 1992), where the elimination of a stop codon and the overprinting of a novel coding region over the ancestral ORF, increases the amount of genetic data that can be stored in viruses with small genomes.
Evolution analyses of regions V1 and V2 of ORF1 suggest that they correspond to regions with relaxed evolutionary constraints but do not show significant positive selection. They could consequently act as bridges between important functional domains, i.e. between methyltransferase and protease domains of the replicase for region V1 and between the replicase and the CP for region V2 (Fig. 1). The only constraint in regions V1 and V2 is the absence of stop codons that would produce truncated forms of the replicase, lacking important functional domains. As the CP gene is suspected to be produced from a sub-genomic RNA (Hirata et al. 2003; Tatineni et al. 2009a), our results suggest that a CP linked to the C terminal extremity of the replicase could be functionally important for the virus, which would explain the absence of stop codons in region V2.
A clear difference was observed between the selection pressure on regions A and B of ORF2 (Fig. 1; Table 3). Surprisingly, region B of ORF2, which overlaps with the functionally important CP, was much less constrained (and not significantly different from neutral evolution; Table 3) than region A, which overlaps with the low constrained part of the replicase. This suggests that region B of ORF2 might have a more recent origin than region A and could be a new extension of ORF2 with lower selection pressure.
Although the dN/dS-based methods are extremely popular to identify positive selection in protein coding genes, their ability to pinpoint the exact sites under positive selection is limited (Hughes 2007; Kryazhimskiy and Plotkin 2008; Nozawa et al. 2009; Suzuki and Nei 2004; Wyckoff et al. 2005). This is due to positive selection acting on relatively few sites and for a short evolutionary time. This is even more complex for estimating dN/dS in overlapping ORFs (e.g., with the method of Sabath et al. (2008)) because of the inherent constraints of gene overlap (Krakauer 2000). Consequently, there is a strong need for experimental tests of the predictions of these methods in order to exploit their results to understand adaptation mechanisms in species or populations (Moury and Simon 2011; Yokoyama et al. 2008). The convergence between the results obtained in the functional analysis of the V2 region by Hirata et al. (2010) and our analysis of its evolutionary pattern therefore constitutes a first validation of the accuracy of the methods of Sabath et al. (2008)’s method.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Baril M, Barkier-Gingras L (2005) Translation of the F protein of Hepatitis C virus is initiated at a non-AUG codon in a +1 reading frame relative to the polyprotein. Nucleic Acid Res 33:1474–1486
Belshaw R, Pybus OG, Rambaut A (2007) The evolution of genome compression in RNA viruses. Genome Res 17:1496–1504
Chirico N, Vianelli A, Belshaw R (2010) Why genes overlap in viruses. Proc R Soc B 277(1701):3809–3817
Clover GRG, Pearson MN, Elliot DR, Tang Z, Smales TE, Alexander BJR (2003) Characterization of a strain of Apple stem grooving virus in Actinidia chinensis from China. Plant Pathol 52:371–378
Gibbs A, Keese PK (1995) In search of the origin of viral genes. In: Gibbs A, Calisher HC, Garcia-Arenal F (eds) Molecular basis of virus evolution. Cambridge University Press, Cambridge, pp 79–90
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hein J, Støvlbæk J (1995) A maximum-likelihood approach to analyzing nonoverlapping and overlapping reading frames. J Mol Evol 40:181–189
Hirata H, Lu X, Yamaji Y, Kagiwada S, Ugaki M, Namba S (2003) A single silent substitution in the genome of Apple stem grooving virus causes symptom attenuation. J Gen Virol 84:2579–2583
Hirata H, Yamaji H, Komatsu H, Kagiwada S, Oshima K, Okano Y, Takahashi S, Ugaki M, Namba S (2010) Pseudo-polyprotein translated from the full-length ORF1 of capillovirus is important for pathogenicity, but a truncated ORF1 protein without variable and CP regions is sufficient for replication. Virus Res 152:1–9
Holmes EC (2003) Error thresholds and the constraints to RNA virus evolution. Trends Microbiol 11:543–546
Holmes EC (2009) In: Harvey PH, May RM (eds) The evolution and emergence of RNA viruses. Oxford University Press Inc., New York, pp 114–116
Hughes AL (2007) Looking for Darwin in all the wrong places: the misguided quest for positive selection at the nucleotide sequence level. Heredity 99:364–373
Inouye N, Maeda T, Mitsuhata K (1979) Citrus tatter leaf virus isolated from Lily. Ann Phytopathol Soc Jpn 45:712–720
Jelkmann W (1995) Cherry virus A: cDNA cloning of dsRNA, nucleotide sequence analysis and serology reveal a newplant capillovirus in sweet cherry. J Gen Virol 76:2015–2024
Keese PK, Gibbs A (1992) Origins of genes: “big bang” or continuous creation? Proc Natl Acad Sci USA 89:9489–9493
Kimura M (1983) The neutral theory of molecular evolution. Cambridge University Press, Cambridge
Korber B (2000) HIV signature and sequence variation analysis. In: Allen Rodrigo G, Learn GH (eds) Computational analysis of HIV molecular sequences. Kluwer Academic Publishers, Dordrecht, pp 55–72
Kosakovsky Pond SL, Frost SDW (2005) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533
Krakauer DC (2000) Stability and evolution of overlapping genes. Evolution 54:731–739
Kryazhimskiy S, Plotkin JB (2008) The population genetics of dN/dS. PLoS Genet 4(12):e1000304. doi:10.1371/journal.pgen.1000304
Liebenberg A, Wetzel T, Kappis A, Herdemertens M, Krczal G, Jarausch W (2010) Influence of apple stem grooving virus on Malus sieboldii-derived apple proliferation resistant rootstocks. Proceedings of the 21st international conference on virus and other graft transmissible diseases of fruit crops. Julius-Kühn-Archiv 427:186–188
Lister RM, Bancroft JB, Nadakavukaren MJ (1965) Some sap-transmissible viruses from apple. Phytopathology 55:859–870
Magome H, Yoshikawa N, Takahashi T (1999) Single-strand conformation polymorphism analysis of apple stem grooving capillovirus sequence variants. Phytopathology 89:136–140
Magome H, Yoshikawa N, Takahashi T, Ito T, Miyakawa T (1997) Molecular variability of the genomes of capilloviruses from apple, Japanese pear, European pear, and citrus trees. Phytopathology 87:389–396
Martelli GP, Adams MJ, Kreuze JF, Dolja VV (2007) Family Flexiviridae: a case study in virion and genome plasticity. Annu Rev Phytopathol 45:73–100
Martin DP, Williamson C, Posada D (2005) RDP2: recombination detection and analysis from sequence alignments. Bioinformatics 21:260–262
Melcher U (2000) The ‘30 K’ superfamily of viral movement proteins. J Gen Virol 81:257–266
Moury B, Simon V (2011) dN/dS-based methods detect positive selection linked to trade-offs between different fitness traits in the coat protein of Potato virus Y. Mol Biol Evol 28:2707–2717
Nei M, Gojobori T (1986) Simple method for estimating the numbers of synonymous and nonsynonymous substitutions. Mol Biol Evol 3:418–426
Normark S, Bergstrom S, Edlund T, Grundstrom T, Jaurin B, Lindberg FP, Olsson O (1983) Overlapping genes. Annu Rev Genet 17:499–525
Nozawa M, Suzuki Y, Nei M (2009) Reliabilities of identifying positive selection by the branch-site and the site-prediction methods. Proc Natl Acad Sci USA 106:6700–6705
Ohira K, Namba S, Rozanov M, Kusumi T, Tsuchizaki T (1995) Complete sequence of an infectious full-length cDNA clone of citrus tatter leaf capillovirus: comparative sequence analysis of capillovirus genomes. J Gen Virol 76:2305–2309
Pavesi A (2007) Pattern of nucleotide substitution in the overlapping nonstructural genes of influenza A virus and implication for the genetic diversity of the H5N1 subtype. Gene 402:28–34
Sabath N, Landan G, Graur D (2008) A method for the simultaneous estimation of selection intensities in overlapping genes. PLoS ONE 3(12):e3996. doi:10.1371/journal.pone.0003996
Schierup MH, Hein J (2000) Consequences of recombination on traditional phylogenetic analysis. Genetics 156:879–891
Shim H, Min Y, Hong S, Kwon M, Kini D, Kim H, Choi Y, Lee S, Yang J (2004) Nucleotide sequence of a Korean isolate of apple stem grooving virus associated with black necrotic leaf spot disease on pear (Pyrus pyrifolia). Mol Cells 18:192–199
Suzuki Y, Nei M (2004) False-positive selection identified by ML-based methods: examples from the Sig1 gene of the diatom Thalassiosira weissflogii and the tax gene of a human T-cell lymphotropic virus. Mol Biol Evol 21:914–921
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 40. Mol Biol Evol 24:1596–1599
Tatineni S, Afunian MR, Gowda S, Hilf ME, Bar-Joseph M, Dawson WO (2009a) Characterization of the 5- and 3-terminal subgenomic RNAs produced by a capillovirus: evidence for a CP subgenomic RNA. Virology 385:521–528
Tatineni S, Afunian MR, Hilf ME, Gowda S, Dawson WO, Garnsey SM (2009b) Molecular characterization of Citrus tatter leaf virus historically associated with Meyer Lemon trees: complete genome sequence and development of biologically active in vitro transcripts. Phytopathology 99:423–431
Terauchi H, Magome H, Yoshikawa N, Takahashi T, Inouy N (1997) Construction of an infectious cDNA clone of the apple stem grooving capillovirus (isolate Li-23) genome containing a cauliflower mosaic virus 35S RNA promoter. Ann Phytopathol Soc Jpn 63:432–436
Wassenegger M, Heimes S, Sänger HL (1994) An infectious viroid RNA replicon evolved from an in vitro-generated non-infectious viroid deletion mutant via a complementary deletion in vivo. EMBO J 13:6172–6177
Wyckoff GJ, Malcom CM, Vallender EJ, Lahn BT (2005) A highly unexpected strong correlation between fixation probability of nonsynonymous mutations and mutation rate. Tr Genet 21:381–385
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Yang Z, Nielsen R, Goldman N, Petersen AM (2000) Codon substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–444
Yang Z, Wong WSW, Nielsen R (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Yokoyama S, Tada T, Zhang H, Britt L (2008) Elucidation of phenotypic adaptations: molecular analyses of dim-light vision proteins in vertebrates. Proc Natl Acad Sci USA 105:13480–13485
Yoshikawa N, Imaizumi M, Takahashi T, Inouye N (1993) Striking similarities between the nucleotide-sequence and genome organisation of citrus tatter leaf and apple stem grooving capillovirus. J Gen Virol 74:2743–2747
Yoshikawa N, Sasaki E, Kato M, Takahashi T (1992) The nucleotide sequence of apple stem grooving capillovirus genome. Virology 191:98–105
Zhou Y, Holmes EC (2007) Bayesian estimates of the evolutionary rate and age of hepatitis B virus. J Mol Evol 65:197–205
Acknowledgments
Annerie Liebenberg was supported by a grant from the Fonds für die Entwicklung ländlicher Räume in Rheinland-Pfalz (F.E.L.R.). We thank Dr. Fiona Constable, Department of Plant Science, Waite Campus, University of Adelaide, Glen Osmond, South Australia, 5064, Australia, Dr. Margit Laimer, Plant Biotechnology Unit, Institute of Applied Microbiology, University of agricultural sciences, Nussdorffer lande 11, 1190 Vienna, Dr. Mike Bernardy, Pacific Agri-Food Research Centre, Agriculture & Agri-Food Canada, Summerland, British Columbia V0H 1Z0, Dr. Pascal Gentit, Centre Technique Inter professionnel des Fruits et Légumes (CTIFL), 22 rue Bergère, 75009 Paris, France, and Michael Petruschke, Landwirtschaftliches Technologiezentrum Augustenberg—Außenstelle Stuttgart, Rötestrasse 16a, 70197 Stuttgart, Germany, for providing the different isolates of ASGV.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liebenberg, A., Moury, B., Sabath, N. et al. Molecular Evolution of the Genomic RNA of Apple Stem Grooving Capillovirus. J Mol Evol 75, 92–101 (2012). https://doi.org/10.1007/s00239-012-9518-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-012-9518-z