
Abstract
Previous studies on organisms with well-differentiated X and Y chromosomes, such as Drosophila and mammals, consistently detected an excess of genes moving out of the X chromosome and gaining testis-biased expression. Several selective evolutionary mechanisms were shown to be associated with this nonrandom gene traffic, which contributed to the evolution of the X chromosome and autosomes. If selection drives gene traffic, such traffic should also exist in species with Z and W chromosomes, where the females are the heterogametic sex. However, no previous studies on gene traffic in species with female heterogamety have found any nonrandom chromosomal gene movement. Here, we report an excess of retrogenes moving out of the Z chromosome in an organism with the ZW sex determination system, Bombyx mori. In addition, we showed that those “out of Z” retrogenes tended to have ovary-biased expression, which is consistent with the pattern of non-retrogene traffic recently reported in birds and symmetrical to the retrogene movement in mammals and fruit flies out of the X chromosome evolving testis functions. These properties of gene traffic in the ZW system suggest a general role for the heterogamety of sex chromosomes in determining the chromosomal locations and the evolution of sex-biased genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Previous studies in species with well-differentiated X and Y chromosomes consistently revealed a significant excess of retrogenes moving out of the X and obtaining male-biased or male-specific expression/functions (Potrzebowski et al. 2008; Emerson et al. 2004; Betran et al. 2002; Dai et al. 2006; Bai et al. 2007; Sturgill et al. 2007). This pattern can be explained by either selection-based evolutionary mechanisms or mutation-based mechanistic processes. The major selection-based evolutionary mechanisms are meiotic sex chromosome inactivation (MSCI, Betran et al. 2002; Vibranovski et al. 2009a) in the male germline, sexual antagonism (Rice 1984; Charlesworth et al. 1987; Wu and Xu 2003), dosage compensation (Bachtrog et al. 2010; Vicoso and Charlesworth 2009), and meiotic drive (Tao et al. 2007a, b).
MSCI could drive the movement of genes out of the X chromosome, which could be favored for being located in the autosomes since they could compensate the silent expression of their X-linked parental genes during male meiosis (Betran et al. 2002; Vibranovski et al. 2009a). In mammals, X-linked parental genes are expressed more in the pre-meiotic stage of spermatogenesis, but are silent from meiosis to post-meiosis, while autosomal-linked retrogenes are often higher expressed in meiotic than in mitotic stages (Potrzebowski et al. 2008). In Drosophila, such a complementary expression pattern is also found for retrogenes and parental genes across male mitosis and meiosis (Vibranovski et al. 2009a). The observations in mammals and fruitflies could be explained by MSCI. However, in mammals, there is an excess of male-biased genes on the X expressed during the pre- and postmeiotic stages of spermatogenesis (Wang et al. 2001; Mueller et al. 2008) and in Drosophila there is an under-representation of somatic male-biased genes on the X chromosome (Parisi et al. 2003). The nonrandom distribution of those sex-biased genes could be explained by sexual antagonism, in which we expect male-beneficial/female-harmful mutations to be more frequently spread on the X or on the autosomes depending on their dominance (Rice 1984; Charlesworth et al. 1987). Moreover, meiotic drive has also been proposed to account for the traffic of genes because autosomal retrogenes may evolve as potential suppressors of X-distorters (Tao et al. 2007a, b). In addition, dosage compensation has been proposed to play a role in retrogene trafficking in Drosophila, as hyper-transcription on the X in males could further limit the up regulation of male-biased genes on the X chromosome and contribute to the movement of male-biased genes out of the X (Bachtrog et al. 2010; Vicoso and Charlesworth 2009).
Recently, a mutation mechanistic process based on the nuclear chromosome dynamics in the spermatogenesis has been proposed to explain the nonrandom genomic distribution of retrogenes in Drosophila (Diaz-Castillo and Ranz 2012). Retrogenes with testis-biased expression would be more frequently inserted in the autosomes than in the X chromosome because the former contain more chromosomal domains occupying accessible positions for genomic insertion during the male germline.
Retrogene movement has been associated with testis-biased retrogene expression in several organisms bearing the X and Y chromosomes: Drosophila, mosquito, humans, and mouse (Betran et al. 2002; Emerson et al. 2004; Vinckenbosch et al. 2006; Zhang et al. 2010a, b; Baker and Russell 2011). If the movement of genes off the X chromosome is advantageous for organisms with well-established heteromorphic sex chromosomes, and if selection-based rather than mutation-based mechanisms drive this movement, these patterns should also appear in other heterogametic species with female heterogamety (ZW organisms such as birds and Lepidoptera) and should be associated with ovary-biased rather than testis-biased retrogene expression.
However, to date, no studies investigating retrogene traffic in ZW species, such as chicken (Hillier et al. 2004) and Populus (Zhu et al. 2009) have found nonrandom patterns of gene movement. There are several possible reasons for this lack of success. First, in chicken, retrotransposition events are rare because the reverse transcriptase of the avian CR1 retrotransposon cannot efficiently recognize polyadenylated mRNA and hence is unable to promote the retrotransposition of mRNAs from other genes (Hillier et al. 2004). Second, Populus Z and W chromosomes show less than 5% divergence, which could indicate that they are too young to show an appreciable number of movements between sex chromosomes and autosomes (Zhu et al. 2009).
Fortunately, the recently available genome annotation (Xia et al. 2004) and expression (Xia et al. 2007) profiles of the domesticated silkworm Bombyx mori, a model organism of lepidopteran insects with ZW chromosomes (Fujii and Shimada 2007; Traut et al. 2007), provide another opportunity to test the conjecture of biased gene trafficking patterns in a species with female heterogamety.
As a species with female heterogamety, B. mori has some peculiarities in its chromosome configurations and genetics compared to species with male heterogamety. First, B. mori females are ZW, whereas males are ZZ (Fig. 1). Second, in B. mori, a genetic factor (Fem) on the W chromosome (although its precise location has not yet been determined) determines the female sex, rather than the ratio of Z chromosomes to autosomes (Traut et al. 2007; Goldsmith and Marec 2010). Third, in B. mori and lepidopterans in general, meiotic recombination does not occur in females, but in males, which are the homogametic sex (Sturtevant 1915). Fourth, although no crossing-over occurs during female meiosis, the W chromosome pairs with the Z chromosome until metaphase I (Goldsmith and Marec 2010). It remains unclear whether MSCI occurs in Lepidoptera, as cytologically observed in female chicken (Schoenmakers et al. 2009) and male mammals (Richler et al. 1992). Fifth, in B. mori, it is controversial whether the Z chromosome is dosage compensated. Initially, it was shown that only 35% of Z-linked genes are hyper-transcribed in females, suggesting incomplete dosage compensation of Z between male and female B. mori (Fujii and Shimada 2007; Xia et al. 2007; Zha et al. 2009). However, a re–analysis of the original data (Xia et al. 2007) showed that the global male:female expression ratio does not differ between the Z and autosomes, which is in agreement with complete dosage compensation model (Walters and Hardcastle 2011). Moreover, the same work suggested that dosage compensation is achieved by inactivation of one of the Zs in males as global Z chromosome expression is significantly reduced relative to autosomes (Walters and Hardcastle 2011).

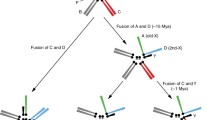
Origin of the ZW chromosomes in Bombyx mori. Bombyx mori is a species in the “Bombycidae” family of the lepidopteran insects. Among the insects, female heterogamety with the Z/ZZ sex determination system evolved in the common ancestor, before Trichoptera and Lepidoptera split more than 190 MYA. The W chromosome was a later acquisition of “advanced” Lepidoptera, Ditrysia and Tischeriina. Hence, W was acquired between 180–190 MYA and 97 MYA. The W chromosome was occasionally and independently lost again in some species of the “advanced” Lepidoptera (Adapted from Figure 12.1 of Grimaldi and Engel 2005 and Fig. 6 of Traut et al. 2007)
Bombyx mori belongs to the Bombycidae family of lepidopteran insects. It split from its nearest wild ancestor, B. mandarina, ca. 7.1 MYA (Goldsmith et al. 2005) and was domesticated ca. 5000 years ago. It is mainly composed of geographic races (e.g., Chinese, Japanese, Korean, European, and tropical varieties) and strains that have been genetically improved (e.g., laboratory mutants; Goldsmith et al. 2005; Xia et al. 2009). The genome sequence used in our study was derived from an inbred domesticated variety, Dazao (posterior silk gland, fifth-instar day 3, from a mix of 1,225 males; Xia et al. 2004). We investigated the distribution of inter-chromosomal retrotransposition events, especially between the Z chromosome and autosomes. In addition, by analyzing gene expression profiles, we explored the role of natural selection underlying retrogene movement.
Materials and Methods
Identification of Inter-Chromosomal Retrogenes in B. mori
We downloaded all the Bombyx mori genome data from http://www.silkdb.org/silkdb/doc/download.html (Duan et al. 2010, see Table S1).
To identify paralog-pair candidates, we aligned all peptide sequences against all peptide sequences with FASTA34 (Pearson 2000) and screened out gene pairs. Here we refer to a gene pair as the two genes present in the FASTA34 alignment result, i.e., the paralog-pair candidate. We set the screen for gene pairs with ≥30% alignment identity and ≥40% alignment coverage (22118 gene pairs, which were composed of 6095 genes). Then we divided the 22118 paralog-pairs into two groups. One group contained the TE-occupied paralog pairs (4834 gene pairs and 873 genes); in each of these, at least one gene had ≥70% CDS occupied by transposable elements (TEs). The other group contained the TE-free paralog pairs (17284 gene pairs and 5642 genes); neither of the two genes in each pair had ≥70% CDS occupied by TEs. We only choose the group of TE-free paralog pairs for further analyses because TE-occupied genes had a higher potential to be pseudogenes, and the TE inserted regions rather than their own CDS might have contributed to the similarity of two genes, which we used as criteria to assign two genes as paralogs. Thus, TE-occupied gene pairs may not be generated by duplication and may not be true paralogs (see demonstration in “Examination of the identified paralog relation of TE-associated gene pairs” in Supplementary Methods). Among the TE-free paralog pairs, we only considered 2304 gene pairs (1886 genes) as potential paralog pairs generated by inter-chromosomal retrotransposition, which contain one single-exon gene (potential retrogene) and one multiple-exon gene (potential parental gene), and of which the two genes were located on different chromosomes. Thus, we ensured the filtering out of paralog pairs that were generated by tandem duplication with/without following inversion. From the 2304 paralog gene pairs, we extracted 270 pairs (492 genes), of which at least one gene was the best hit of the other gene or both genes were reciprocal best hits. After that, we ensured that one retrogene (single-exon gene) had only one parental gene and appeared in only one paralog pair. However, we allowed one parental gene to have several retrogenes, and this parental gene could appear in more than one paralog pair (in our final dataset of 68 retro-paralog pairs, there was only one case where the same parental gene, BGIBMGA011777, generated two retrogenes, BGIBMGA004200 and BGIBMGA004157, both from autosomes to autosomes). Thus, we further screened out 228 gene pairs (449 genes) from the 270 pairs (For details and confirmation of this step, see “Confirmation of the origination relation within each paralog pair” in Supplementary Methods). Then, we increased the paralog alignment criteria to ≥50% coverage and ≥40% identity to define a more conservative paralog set (83 gene pairs and 165 genes). Last, we manually inspected the CDS alignment for each of the 83 paralog pairs (the CDS alignment was generated according to their protein alignments). Specifically, we checked whether the boundaries of at least two exons (usually most of the exons) from each parental gene were aligned inside of the single exon of the offspring retrogene to avoid cases generated by exon-gaining after single-exon gene duplication (see Figure S1 for the comparison of distribution of exon number of parental genes between B. mori and Drosophila, whose genes, in general, had similar composition in terms of exon number). Thus, we finally identified 68 gene pairs (135 genes, File S1) as a set of conservative inter-chromosomal retrotransposition paralog pairs and performed the rest of the analyses based on this set.
Functionality Analysis of Retrogenes
Computing Ka/Ks Ratio
To examine the functional constraints on these new genes, we computed K a/K s ratios (ω) of the identified retrotransposition paralog pairs with PAML (Yang 1997) and estimated whether they were significantly smaller than 0.5 (Betran et al. 2002). Usually, a K a/K s ratio significant lower than unity indicates the genes under functional constraint. A K a/K s ratio under unity but higher than 0.5 suggests the divergence between a functionless new gene and a functional parental gene (Li 1997). Conservatively, we considered a K a/K s ratio significant smaller than 0.5 to indicate functional constraints on both genes (parental gene and retrogene). We aligned the CDSs of each paralog pair according to their protein alignment with MAFFT. Then, we ran Codeml of PAML twice, first fixing ω at 0.5, and then estimating ω freely. Finally, we tested whether the likelihood of the ω free model was significantly smaller than that of the ω fixed at 0.5 model by comparing two times the log likelihood difference, 2L = 2(L0.5 – L0) to a Chi-square distribution with one degree freedom to compute a P value (Yang 1998). Note that L0,5 means log likelihood with ω fixed at 0.5, and L0 means log likelihood with ω estimated freely.
Polymorphism Analysis to Confirm the Functionality of the 68 Retrogenes
We collected SNPs in the CDS regions of the 68 retrogenes based on the SNP data generated from a complete re-sequencing of 29 strains of B. mori (Xia et al. 2009). Next, based on the SNP data, we used the software DNasp to estimate non-synonymous nucleotide diversity, synonymous nucleotide diversity and nucleotide variation in CDS regions of the 68 retrogenes.
EST-Based Expression Analysis
We did an EST-based expression search analysis to support the functionality of the 68 retrogenes. We downloaded the NCBI EST library collection of B. mori from http://www.silkdb.org/silkdb/doc/download.html. The collection contains 184509 ESTs from 79 EST libraries expressed in 27 tissues. We used BLAST (Altschul et al. 1997) to identify the corresponding genes of the ESTs. The criteria to define the corresponding gene of an EST were as follows: 1) the CDS of this gene was the first best hit of the EST; 2) the alignment of the EST and this best hit gene satisfied ≥90% identity, equal/longer than 100 nt, ≤ 1e-20 E value, ≥100 blast score; and 3) the blast score of this first best gene hit was at least 2 points higher than that of the second gene hit of the EST. Thus, the corresponding relation between ESTs and 8215 (56.18%) current annotated genes were constructed.
Retrogene Movement Pattern Analysis
We compared the observed movement patterns to the expected ones under the null hypothesis according to a simplified random model with a Chi-square test (Betran et al. 2002). Briefly, the expectation considered the amount of available transcripts from the source chromosome, the dosage compensation on the source chromosome, the length of the target chromosome, and the chance of a transcript from the source chromosome inserting into a given target chromosome. Further, we conducted Monte Carlo simulation based on a similar random model with a multinomial distribution (Emerson et al. 2004) to estimate the significance of excess/deficit of nonrandom gene movement (for more details, see “Analysis of the movement pattern of retrotransposition events” in Supplementary Methods).
Retrogene Expression Pattern Analysis
We analyzed retrogene expression patterns based on microarray expression profiles of B. mori generated by Xia et al. (2007). We downloaded the raw microarray data from http://www.silkdb.org/microarray/download.html. We analyzed the raw microarray data with PTL within array normalization, “normexp” background correction and “Aquantile” between array normalization (Walters and Hardcastle 2011), which are available from the LIMMA package (Smyth 2004) in the BioConductor software suite of R statistical computing software. We used a maximum likelihood method for discerning expressed probes (personal communication with JR Walters). We generated the expression heat map of retrotransposition paralog pairs with R. We computed the tissue specificity index, τ (Yanai et al. 2005) and the tissue expression components for each gene (see “Statistical analysis of gene expression pattern” in Supplementary Methods for more detail). Then, we analyzed the gonad-biased (namely, testis-biased and ovary-biased) expression pattern of each gene with one-tailed paired t test on ovary and testis gene expression from 6 microarray replicates (The reason why we used one-tail paired t test is because we specifically wanted to know whether a gene has higher expression in testis or ovary). If a gene has significant t test P value, we will define it as ovary-biased or testis-biased gene (see “Analysis of the association of retrogene expression patterns with their movement patterns” in Supplementary Methods for detail). We computed the associated q value for each t test’s P value to adjust for multiple comparisons.
We conducted all the intermediate steps with PERL, R scripts and Excel functions, but the exon boundary alignments of each retrogene and the corresponding parental gene were manually confirmed.
Results
Retrogenes of B. mori
We identified a conservative set of 68 paralog pairs as functional inter-chromosomal retrogenes and corresponding parental genes based on a refined procedure of previous gene traffic studies (Emerson et al. 2004; Betran et al. 2002; Dai et al. 2006; Bai et al. 2007; Vibranovski et al. 2009b; for the list of the 68 paralog pairs, see File S1). Essentially, the procedure is based on protein sequence alignment, where gene pairs with ≥40% identity and ≥50% coverage that are not occupied by transposable elements were selected (for more details, see “Identification of Inter-Chromosomal Retrogenes in B. mori ” section).
Several lines of evidence indicate that these retrogenes are functional. First, among the 68 identified paralog pairs, the K a/K s ratios of 67 pairs were significantly <0.5, which indicate functional constraints on both genes (Betran et al. 2002; Table 1). Only in one case (BGIBMGA000596 vs. BGIBMGA008879), the K a/K s ratio was higher than 0.5 (K a/K s = 0.677, K a = 0.0271, and K s = 0.04). Second, to further investigate the functionality of the 68 retrogenes, we performed a polymorphism analysis. For the 68 retrogenes together, the non-synonymous nucleotide diversity is significantly smaller than the synonymous nucleotide diversity, Wilcoxon rank sum test, P = 2.196e−09 (see Figure S2a, File S8). Similarly, the variation in the CDS region of the retrogenes was significantly lower than the average variation in the CDS region of all the genes of B. mori (see Figure S2b, File S8). Taken together, the low variation present in the retrogenes argues in favor the functionality hypothesis because usually functionless DNA is more variable than the functional DNA (Betran et al. 2002). In addition, we obtained individual supporting evidence for the functionality of the retrogene BGIBMGA000596 using the polymorphism analysis with SNP data. There were no synonymous or non-synonymous mutations in this retrogene in 29 B. mori strains. Moreover, among the 68 paralog pairs, 48 retrogenes (70.59%) and 55 parental genes (80.88%) had EST data available, constituting additional evidence supporting the functionality for most retrogenes (for more details, see “EST-Based Expression Analysis” section). Thus, the EST-based expression data, the population genetic analysis and K a/K s values significantly lower than 0.5 for most genes suggest that almost all the retrogenes identified are subject to strong functional constraints.
Retrogene Movement Patterns of B. mori
Next, we defined the chromosomal movement direction of each of the 68 paralog pairs as from a parental gene to its retrogene (Betran et al. 2002; Emerson et al. 2004; Dai et al. 2006; Bai et al. 2007; Vibranovski et al. 2009b). We grouped the movement directions of the 68 retrotransposed paralog pairs into three categories (Fig. 2): from Z to autosomes, from autosomes to Z, and between autosomes. We compared the observed distribution of movements in the three categories with the expected random distribution (random pattern, Betran et al. 2002) using a Chi-square test (Betran et al. 2002; Table 1). Because some expected cell counts were less than 5, we estimated the significance levels of gene movements with a multinomial Monte Carlo simulation via 106 re-samplings of the genome-wide distribution of retrotransposition events based on a random pattern (Emerson et al. 2004; Table 1). The P values calculated from the Chi-square test and the Monte Carlo simulation indicated that “out of Z” retrotranspositions occurred significantly more frequently than would be expected under a random model (Chi-square test P = 0.0069, df = 2; Monte Carlo simulation test, three directions together P = 0.0063, “out of Z” P = 0.0154 and “into Z” P = 0.0836; Table 1). Thus, this is the first study demonstrating a significant excess of retrogenes “out of” the homogametic sex chromosome in an organism with female heterogamety (ZW species). These nonrandom movement patterns were similar to those in species with male heterogamety (XY species), such as fruitfly (Betran et al. 2002).

The 68 identified inter-chromosomal retrotransposition events in Bombyx mori genome. The circle schematically represents Bombyx mori’s genome, where each arc is one of the chromosomes (scale based on the effective number of genes on each chromosome). The movement direction of each of the 68 identified retrotransposition events is indicated with an arrow pointing from the parental gene-locating chromosome to the retrogene-landing chromosome; the three different colors of the arrows correspond to the three types of movement directions, namely, A → A, white; A → Z, blue/grey; and Z → A, pink/black. Both observed Z → A and A → Z retrotransposition events were significantly higher than neutrally expected patterns (7 vs. 2.72, and 5 vs. 2.50; P values of both Chi-square test and Monte Carlo simulation <0.05, see Table 1). Although retrotransposition events associated with other chromosomes also showed certain degree of excess or deficit, most of them were not statically significant (e.g., excess of chr3→, chr27→, and →chr27; deficit of chr10→; but Chi-square test, df = 1, P > 0.05). The only exception was the excess of →chr19 (Chi-square test, df = 1, P < 0.05), but the expression pattern of the “into chr19” retrogenes was randomly distributed (Fisher test, P > 0.05) (Color figure online)
Moreover, we obtained similar results when we analyzed several subsets of retrogenes under more stringent criteria. For instance, we tested whether the movement pattern would change if we only analyzed paralog pairs with the following characteristics: (i) alignment that satisfied the criteria of ≥70% coverage and ≥50% identity (Table S3a); (ii) at least a 2 intron loss between the parental gene and the retrogene (Table S3b); or (iii) confirmation based on another independent source of gene family annotation of Bombyx mori genome from SilkDB (Table S3c). We found no qualitative difference from our previous conclusion (for more details, see “Test on the excess movement pattern in more restrictedly defined retrotransposition paralog pairs” in Supplementary Methods).
If a retrotransposition event occurred before Drosophila melanogaster and Bombyx mori diverged, when the B. mori Z chromosome had not yet originated, we cannot determine whether retrogene movement is linked to the sex chromosome or to the autosomes. Therefore, we used Blastp to detect the orthologs of the B. mori paralog pairs in D. melanogaster, and, when possible, in Apis mellifera, Anopheles gambiae, Aedes aegypti, Homo sapiens, Gallus gallus, and Takifugu rubripes (see “Dating the age of 68 paralog pairs in B. mori” in Supplementary Methods). We excluded the paralog pairs with retrogenes that might have orthologs in these outgroup species (i.e., those that may have originated before the formation of the Z chromosome). Nevertheless, we still observed the significant excess of out of Z retrogene movement (48 cases, Table 2 and File S1).
To additionally test the pervasiveness of our results, we analyzed the movement pattern of retrogenes selected through a less stringent alignment criterion, namely, protein sequence alignment between gene pairs with ≥30% identity and ≥40% coverage. This less stringent analysis might contain false positives because, by including more distantly related duplicates, noise to the potential retrotransposition distribution signals was likely added. However, we detected a nonrandom retrogene movement pattern. We identified 159 paralog pairs (68 pairs in the conservative set, plus 91 new cases, as a nonconservative set of inter-chromosomal retrogenes and corresponding parental genes) by performing the same refined procedure described for the conservative set. Because all the expected and observed cell counts were larger than 5 in this analysis, we only compared the observed distribution of movement in the three categories (Z → A, A → A, A → Z) with the expected distribution using a Chi-square test (Table S3d). Although the comparison of all three categories in the same test suggests that retrotransposition events were not significantly different from randomly expected events (Chi-square test, P = 0.0630, df = 2; see Table S3d); when we compared A → Z and Z → A with A → A movement separately, we found a significant excess of retrotranspositions from the Z to autosomes (Z → A Chi-square test, P = 0.0210, df = 1; Table S3d), confirming the excess of “out of Z” but not “into Z” retrogene movements in silkworm (A → Z Chi-square test, P = 0.5520, df = 1; Table S3d), which is similar to the observations found in Drosophila (Betran et al. 2002).
Comparison to the Retrogene Analysis from Toups et al. (2011)
A recently published study identified 22 retrotransposition events in B. mori and found no excess movement out of the Z chromosome (Toups et al. 2011). We compared their dataset with ours and found only 13 events with overlap (see “Analysis of Toups et al.’s data” in Supplementary Methods). Curiously, our manual check of all nine remaining events (41% of their sample size) revealed that they were all misidentified as cases of retrotransposition mostly due to sequence similarity produced by recent TE-insertions into unrelated genes. It is known that 43.6% of the silkworm genome is occupied by transposable elements (Duan et al. 2010), a proportion much higher than the Drosophila genome. Thus, it is very likely that the transposable element-derived regions rather than real duplicated gene regions are contributing to the similarity of a gene family selected in their study (see Supplementary File S10). In addition, we noticed that in six cases (their gene families 13, 14, 19, 20, 21, and 22, see Supplementary File S10), the alignment between retrogene and parental gene reveals no signature of intron loss, the hallmark of a retrotransposition event (see Supporting File S10). These problems made it unlikely to identify retrotransposition events with certainty.
Note that all those misclassified cases correspond to A → A movements. Their inclusion in the final dataset inflated the number of events following the random model and therefore misled the authors to conclude that there is no excess of retrogene movement off the Lepidopteran Z chromosome. In the re-analysis of the data confirmed by their analysis and this study independently consisting of the remaining 11 cases of retrotransposition (we excluded two cases where the duplication may have occurred before speciation), we still found the excess of out of Z movement, although not significant due to the small sample size (Table S3e, Monte Carlo simulation P = 0.0622). Therefore, their dataset, after filtering out TE contamination, confirmed the excess of retrogene movement out of the Z in silkworms, which verifies that the excess of retrogene moving out of the Z chromosome in silkworm is unambiguous.
Germline Gene Expression Does Not Affect Retrogene Traffic
The excess of retrogenes deriving from the Z chromosome could be generated if the Z-linked genes produce a higher amount of transcripts than the autosomes in the gonads. In that case, a higher amount of inheritable Z-originated retrogenes would be produced via retrotransposition of those transcripts. To test this hypothesis, the EST number per gene can be a reasonable numeric approximation of transcript abundance. We therefore tested if the germline expression of genes in different chromosomes could affect the frequency of the retrogene traffic. We conducted the comparison of the total number of ESTs on the Z chromosome and autosomes in testes and ovaries, respectively, with the total number of genes found on those chromosomes (see file S2 and Table S7). We found that, in testes, the Z chromosome and autosomes generate an amount of ESTs (transcripts) proportional to the gene number on the chromosome (Fisher test P = 0.1864). However, in ovaries, the Z chromosome produces significantly fewer ESTs (transcripts) (Fisher test P < 2.2e−16). Thus, our results support that the significant excess of out of Z retrogene movement is not generated by a higher amount of transcripts produced from the Z chromosome in gonads.
General Expression Properties of Retrogenes and Parental Genes in B. mori
Three hypotheses, MSCI, sexual antagonism, and dosage compensation, explained the nonrandom gene traffic in species with male heterogamety (Potrzebowski et al. 2008; Emerson et al. 2004; Betran et al. 2002; Bachtrog et al. 2010; Vicoso and Charlesworth 2009). These hypotheses explained chromosomal movement patterns of retrogenes based on their sex-biased expression patterns. To interpret the traffic patterns in the B. mori genome, we analyzed the expression patterns of 68 retrogenes and their parental genes based on microarray expression profiles of B. mori genes (Xia et al. 2007) using Walters and Hardcastle’s (2011) normalization procedure.
We analyzed the patterns of expression of retrogenes and their parental genes, respectively. We calculated the tissue specificity index, τ, of 62 retrogenes and the corresponding parental genes (Yanai et al. 2005). Sixty-two of the 68 pairs had microarray data available for both retrogenes and parental genes (66 retrogenes and 63 parental genes had microarray data available). We conducted a one-tailed paired t test on the τ values of the 62 pairs and find that parental genes are expressed in significantly more tissues than retrogenes (t test, P = 0.0220; Figure S3 and File S4). Although all retrogenes appear to be expressed in relatively few tissues, we found that retrogenes are more highly expressed in testis than any other tissue in 12 out of the 17 pairwise comparisons (one-tailed paired t test; all the P values <0.05, Table S4 and File S5). The trend is maintained even if we repeat the analysis just for retrogenes moving between autosomes (P < 0.05, in 9 out of the 17 pairwise comparisons). Thus, the extensive expression spectrum of parental genes and the higher testis expression level of retrogenes found in B. mori, a species with female heterogamety, is consistent with the testis-expression tendency of retrogenes in species with male heterogamety, whose parental genes also tend to be housekeeping genes (e.g., mammals: Vinckenbosch et al. 2006; Potrzebowski et al. 2008; Marques et al. 2005; and fruit flies: Dai et al. 2006; Bai et al. 2007; Vibranovski et al. 2009a).
“Out of the Z” Retrogenes Tend to Develop Ovary-Biased Expression
Using the conservative set of 68 paralog pairs, we found that a significantly higher proportion of the retrogenes moving from the Z chromosome to autosomes have ovary-biased expression than retrogenes moving between autosomes (Fisher exact test P = 0.0147; Fig. 3; Table 3. For defining the ovary-biased or testis-biased expression, see “Analysis of the association of retrogene expression patterns with their movement patterns” in Supplementary Methods for detail). Mainly, we defined those genes showing higher expression in ovary than in testis as ovary-biased genes. The same pattern can be found for the 48 paralog pairs whose retrogenes do not have orthologs in other species (Fisher exact test P = 0.0196; Table 4).

Heat maps of expression level of 62 retrogenes with their parental genes in 18 tissues of Bombyx mori. The left side of the heat maps shows the expression level of 62 parental genes in 18 tissues (i.g. testis: Gonad.m; ovary: Gonad.f; male head: Head.m; female head: Head.f; male integument: Ig.m; female integument: Ig.f; male mpg_tube: mt.m; female mpg_tube: mt.f; male am_silkgland: asg.m; female am_silkgland: asg.f; male post_silkgland: psg.m; female post_silkgland: psg.f; male Fat_body: Fb.m; female Fat_body: Fb.f; male Midgut: Mg.m; female Midgut: Mg.f; male Hemocyte: Hemo.m; female Hemocyte: Hemo.f), and the right side shows that of the corresponding retrogenes. Both the left and right sides of the heat maps were divided into three sections according to the movement directions of associated retrotransposition events, namely, gene pair 1–7: Z → A; gene pair 8–11: A → Z; and gene pair 12–62: A → A (see File S1)
Interestingly, a significantly higher proportion of the retrogenes moving into the Z chromosomes have testis-biased expression than the retrogenes moving between autosomes for the conserved set of 68 paralog pairs (Fisher exact test, P = 0.0408, Table 3). We confirmed the testis expression trend for into Z retrogene, but not the excess of into Z movement pattern itself for the 48 paralog pairs (for movement pattern, Chi-square test P = 0.4107, Table 2; for expression pattern, Fisher exact test P = 0.0461, Table 4).
Note that all the expression analysis done above used the raw microarray data generated by Xia et al. (2007) with normalization approaches by Walters and Hardcastle (2011). The major findings for the conserved set are not changed if we normalize the data using Xia et al.’s (2007) approach. We still found that a significantly higher proportion of the “out of Z” retrogenes have ovary-biased expression (Fisher exact test P = 0.0425, Table S5).
Discussion
Dosage Compensation in Silkworm
The dosage compensation of the Z chromosome of Bombyx mori is controversial (Zha et al. 2009; Walters and Hardcastle 2011). One study using microarray expression data in different male and female tissues (Xia et al. 2007) observed that the Z chromosome was incompletely dosage compensated by the hyper-transcription of only 35% of Z-linked genes in females (Zha et al. 2009). Recently, Walters and Hardcastle (2011) re-analyzed the same microarray data (Xia et al. 2007) using different approaches for normalization and detection of probe activity to conclude that there is no reason to reject the complete dosage compensation model in silkworm. Their re-analysis suggested that the gene expression was completely compensated by the inactivation of one of the Zs in males instead of by the Z chromosome hyper-transcription in females. In our work, we considered the effects of these two types of dosage compensation on the retrogene movement patterns. Dosage compensation impacts the number of copies of Z-linked and autosomal genes actively expressed in the germline and therefore could potentially affect the expected number of Z → A retrogene movements. The dosage compensation model based on the inactivation of the Z chromosomes in males predicts that the ratio of expression between Z-linked genes and autosomal genes is 0.5, whereas the same ratio is 0.8375 for the incomplete (35%) dosage compensation model by hyper-transcription in females. We constructed the random model (expectation) of retrotransposition for all analyses based on the latter dosage compensation model since it has the highest expected value for “out of Z” movement. Therefore, our results are conservative regarding the “out of Z” pattern. As predicted, we obtained a similar, significant excess of “out of Z” retrogene movement when we calculated the random model of retrotransposition based on dosage compensation by inactivation of one of the Z chromosomes in the males of B. mori (Chi-square test df = 2, P = 3.2744E−05, see File S3, Monte Carlo simulation test for three movement direction together, P = 0.0031). Moreover, even if we consider that the most extreme situation (not reported yet) where the dosage compensation of the Z chromosome is complete through total hyper-transcription of the Z chromosome in females, the observed excess of “out of Z” movement was still significant (Chi-square test df = 2, P = 0.0221, Monte Carlo simulation test for three movement direction together, P = 0.0618, Monte Carlo simulation test for “out of Z”, P = 0.0154, Monte Carlo simulation test for “into Z”, P = 0.0834 see File S3).
Nonrandom Gene Traffic Patterns Are Associated with the Establishment of Heteromorphic Sex Chromosomes
The observed difference in gene traffic patterns between taxa with different sex chromosome system reveals that the biased gene traffic pattern is a feature associated with well-established heteromorphic sex chromosomes. For instance, in the genome of Populus, which has primitively developed Z and W chromosomes (sex chromosome differentiation <5%), retrogene traffic is extensive but no biased patterns were found (Zhu et al. 2009). In the Aedes aegypti mosquito, which has homomorphic sex chromosomes with negligible differentiation of the sex chromosome from autosomes (only a small nonrecombining sex-determining region that spans a few megabases), retrogenes also show no significant biased gene traffic pattern between its X chromosome and autosomes (Toups and Hahn 2010). In contrast, species with biased movement patterns have evolved strikingly differentiated heteromorphic sex chromosomes (Potrzebowski et al. 2008; Emerson et al. 2004; Betran et al. 2002; Bai et al. 2007; Vibranovski et al. 2009b; Toups and Hahn 2010; Baker and Russell 2011). Therefore, the comparison between these two classes of organisms suggests that nonrandom gene traffic patterns might be a general property of species with well-differentiated heteromorphic sex chromosomes regardless of which sex is the heterogametic one (Potrzebowski et al. 2008, 2010).
“Out of” and “into” the Z Chromosome Retrogene Traffic
We found that retrogenes moving out of the Z chromosome tend to develop ovary-biased expression in both the conservative dataset (Table 3) and the 48 gene pairs whose retrogenes do not have orthologs in other species (Table 4). We cautiously prefer not to conclude that silkworm has an excess of retrogenes moving into Z chromosome that tend to be testis-biased expressed, because the evidence supporting those conclusions were only obtained in some subsets of the analyses (Tables 3, 4; Table S3c). Therefore, expression patterns combined with movement patterns of inter-chromosomal retrogenes in B. mori are symmetrical or at least partially symmetrical to those found in species with male heterogamety, e.g., mammals and fruit flies. In mammals, the “out of the X” retrogenes tend to evolve testis-biased expression, whereas the “into the X” retrogenes generally have lower expression levels in testis (Potrzebowski et al. 2010). Drosophila only showed an excess of “out of the X” retrogenes acquiring testis-biased expression, without significant excess of “into X” retrogenes.
Additionally, expression patterns combined with movement patterns of retrogenes in B. mori are consistent with what was recently found in birds (also female heterogamety) (Ellegren 2011). Recently, the non-retrogene movement between Z and autosomes was found to be associated with sex-biased expression in the chicken genome, in which “Out of Z” genes tend to develop ovary-biased expression, and “into Z” genes tend to gain testis-biased expression (Ellegren 2011). Remarkably, the tendency of “out of Z” retrogenes in silkworm and DNA-level duplicated genes in birds to evolve ovary-biased expression suggests that processes intrinsic to testis can not drive biased gene movement pattern (e.g., the “out of the testis” and the “nuclear chromosome dynamics in male germline” hypotheses, Vinckenbosch et al. 2006; Diaz-Castillo and Ranz 2012). More specifically, only the selection-based rather than the mutational-based hypothesis predict the ovary-biased retrogene expression. It is possible that the nuclear position of certain chromosomal domains could generate more retrogene insertions on the autosomes. However, such phenomenon is specifically associated with the retrogene testis-biased expression since the mutational mechanism involved is known to occur in the spermatogenesis but not in the oogenesis (Diaz-Castillo and Ranz 2012). In addition, independent evidence in favor of selection-based processes comes from a population genomic analysis, which found excess of fixed retrogenes originating on the X chromosome in comparison to polymorphic ones (Schrider et al. 2011).
Moreover, in silkworm, the pattern of “Out of Z” retrogenes developing ovary-biased expression is consistent with our current finding that the Z chromosome is depleted of ovary-biased genes (File S9, Figure S4). On the other hand, previous findings showed that testis-specific and testis-biased genes are enriched in the Z chromosome of B. mori (Arunkumar et al. 2009), and it may be a product of higher male-driven mutation rate (Naurin et al. 2009), sexual selection that is usually stronger in males than in females (Bateman 1948), and sexual antagonism which prefers the accumulation of male-beneficial allele on the Z chromosome due to the longer evolution time of the Z spending in male (the Z spends 2/3 of evolution time in male, and 1/3 of evolution time in female).
Evolutionary Forces and Mechanisms that Could Lead to Nonrandom Gene Traffic Patterns
Several selection-based mechanisms (MSCI, sexual antagonism, faster-X/Z effect, meiotic drive hypothesis, and dosage compensation) could explain the nonrandom gene movement we observe in both XY and ZW systems. MSCI has been already shown to be one of the driving forces impacting the retrogene movement in mammals and fruitflies. In mammals, “out of X” retrogenes tend to gain male-biased expression specific to spermatocyte and postmeiotic spermatids which compensates the silenced expression of their respective X-linked parental genes during meiosis (Potrzebowski et al. 2008). Similar complementary expression for retrogenes and parental genes has been also shown to occur in Drosophila mitotic and meiotic phases of the male germline (Vibranovski et al. 2009a) in agreement with the MSCI model. In birds, the MSCI observed in the synapsed ZW chromosomes during chicken oogenesis (Schoenmakers et al. 2009; Namekawa and Lee 2009) could explain the ovary-biased expression pattern found for “out of Z” duplicate genes (Ellegren 2011). In silkworm, it is still unknown whether MSCI exists and if it plays a role on the retrogene traffic between chromosomes.
For sexual antagonism, sex-biased genes could be the evolutionary products of the resolved sexual conflict (Innocenti and Morrow 2010; Connallon and Clark 2011). In mammals, the faster exposure of X-linked recessive male-beneficial alleles under selection could explain the excess of X-linked genes expressed in pre-meiotic and post-meiotic stages of spermatogenesis (Wang et al. 2001; Mueller et al. 2008). In fruitfly, the longer evolutionary time of the X chromosome in females (the X spends 2/3 of its evolution time in female and 1/3 in male) and the faster selection of X-linked female-beneficial dominant alleles could explain the feminization of the X chromosome (Rice 1984). Likewise, the faster spread of male-beneficial dominant alleles on autosomes could explain the movement of male genes off the X chromosome via retrotransposition or/and DNA level duplication/relocation mechanisms (Rice 1984; Wu and Xu 2003; Vibranovski et al. 2009b; Gallach et al. 2010), which is also consistent with the under-representation of somatic male-biased genes on the X of Drosophila (Parisi et al. 2003; Sturgill et al. 2007). In birds, sexual antagonism could explain the excess of Z-linked female-biased genes in somatic ovary cells (Mořkovský et al. 2009), following the expectation that the female-biased alleles are recessive, beneficial to female and harmful to male. In B. mori, sexual antagonism could explain the retrogenes movement out of Z and tendency to gain ovary-biased expression, because dominant antagonistic alleles, female-beneficial and male-detrimental, would be more easily spread in autosomes than in the Z chromosome (Rice 1984).
Regarding the Faster-X effect, Zhang et al. (2010a, b) recently identified an excess of young X-linked male biased genes, which disappear with evolutionary time until autosomes accumulated an excess of male biased genes in fruitfly and mammals. Faster-X effect could explain the emergence of young X-linked male-biased genes, in which recessive male-favored X-linked mutation are more often fixed, and they do not necessarily have an adverse effect on female fitness (Zhang et al. 2010a, b). However, in birds, faster-Z effect was found to be generated by random drift rather than selection and thus cannot affect the distribution of sex-biased genes (Mank et al. 2010). In B. mori, faster-Z effect has not yet been studied and therefore its affects on the gene trafficking patterns is unknown.
Dosage compensation could also cause the nonrandom movement patterns in B. mori. Compared to XY species, dosage compensation is controversial in the ZW species investigated to date (Itoh et al. 2007; Zha et al. 2009, Walters and Hardcastle 2011). Dosage compensation in silkworm may affect gene movement patterns in two aspects. One possibility is that the lack of complete dosage compensation may cause the Z chromosome to mostly carry genes required at higher level in males (e.g., male-biased genes) (Fujii and Shimada 2007; Koike et al. 2003), but disfavor genes highly expressed in females. In this hypothesis, the incomplete dosage compensation could be associated with a process of genome shuffling where the Z chromosome only carries genes whose products are required at higher levels in males. In such a scenario, the organism could still function well even without dosage compensation. This hypothesis could explain the “out of Z” retrogenes tendency to develop ovary-biased expression, but not the general small number of retrotransposition events involving the Z chromosome. The other possibility is that dosage compensation may cause the up-regulation of Z in females (Vicoso and Charlesworth 2009; Bachtrog et al. 2010). This up-regulation might limit further increase of the Z-linked gene expression in females, resulting in the paucity of female-biased genes in the Z chromosome, which may also explain our observation that “out of Z” retrogenes tend to gain ovary-biased expression. However, silkworm might achieve dosage compensation by inactivation of the Z chromosome in males (Walters and Hardcastle 2011). In this case, models that invoke up-regulation limitations are not appropriate to explain gene movement patterns.
Meanwhile, the meiotic drive hypothesis was also proposed to account for the gene traffic out of X or Z driven by MSCI (Tao et al. 2007a, b), suggesting that retrogenes may evolve into X-distorters and autosomal suppressors. In this hypothesis, the meiotic drive is avoided efficiently with excess autosomal suppressors, which account for the preferable fixation of retrogenes in autosomes. Additional studies will be needed to test specifically the contribution of those factors on generating the gene traffic pattern in both XY and ZW species.
The Domestication Process of B. mori Should Not Affect Biased Retrogene Traffic
Based on archeological and genetic evidence, B. mori was domesticated in Asia about five thousand years ago (Goldsmith et al. 2005). The minimum origination rate of retrogene in fruit flies is 0.51 genes per MY per lineage (Bai et al. 2007); therefore, for B. mori, it should be <0.51 genes per MY per lineage because the generation time of B. mori is longer than that of fruit flies. During the ~5000 years of domestication, it is likely that no retrogenes have arisen, and gene traffic patterns of B. mori should not be affected.
Conclusion
In conclusion, we demonstrated the following: first, nonrandom retrogene traffic also exists in organisms with female heterogamety, i.e., ZW species; second, retrogenes are more narrowly expressed than their parental genes and have higher expression levels in testis, which is not specific to species with male or female heterogamety; and third, expression pattern accompanied by traffic pattern, i.e. “out of Z” retrogenes acquiring ovary-biased expression, confirm that selection rather than a mutational-based mechanistic force drives nonrandom gene traffic. Moreover, the ovary-biased expression of “out of Z” retrogenes indicates that male- or testis-biased expression is not an intrinsic and exclusive property associated with nonrandom retrogene movements. In summary, although the underlying evolutionary mechanisms of nonrandom retrogene traffic may differ among species, excess traffic patterns are likely to be a general feature of the evolution of heteromorphic sex chromosomes driven by selection and may consistently appear in organisms with well-established heteromorphic sex chromosomes.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z et al (1997) Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Arunkumar KP, Mita K, Nagaraju J (2009) The silkworm Z chromosome is enriched in testis-specific genes. Genetics 182:493–501
Bachtrog D, Toda NRT, Lockton S (2010) Dosage compensation and demasculinization of X chromosomes in Drosophila. Curr Biol 20(16):1476–1481
Bai YS, Casola C, Feschotte C, Betran E (2007) Comparative genomics reveals a constant rate of origination and convergent acquisition of functional retrogenes in Drosophila. Genome Biol 8:R11
Baker DA, Russell S (2011) The role of testis-specific gene expression in sex chromosome evolution of Anopheles gambiae. Genetics. doi:10.1534/genetics.111.133157
Bateman AJ (1948) Intra-sexual selection in Drosophila. Heredity 2:349–368
Betran E, Thornton K, Long M (2002) Retroposed new genes out of the X in Drosophila. Genome Res 12:1854–1859
Charlesworth B, Coyne JA, Barton NH (1987) The relative rates of evolution of sex chromosomes and autosomes. Am Nat 130:113–146
Connallon T, Clark AG (2011) The resolution of sexual antagonism by gene duplication. Genetics 187(3):919–937
Dai H, Yoshimatsu TF, Long M (2006) Retrogene movement within- and between-chromosomes in the evolution of Drosophila genomes. Gene 385:96–102
Diaz-Castillo C, Ranz JM (2012) Nuclear chromosome dynamics in the Drosophila male germline contribute to the non-random genomic distribution of retrogenes Mol Biol Evol. doi:10.1093/molbev/mss096
Duan J et al (2010) SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res 38:453–456
Ellegren H (2011) Emergence of male-biased genes on the chicken Z-chromosome: contrasts between male and female heterogametic systems. Genome Res. doi:10.1101/gr.119065.110
Emerson JJ, Kaessmann H, Betran E, Long MY (2004) Extensive gene traffic on the mammalian X chromosome. Science 303:537–540
Fujii T, Shimada T (2007) Sex determination in the silkworm, Bombyx mori: a female determinant on the W chromosome and the sex-determining gene cascade. Semin Cell Dev Biol 18:379–388
Gallach M, Chandrasekaran C, Betrán E (2010) Analyses of nuclearly encoded mitochondrial genes suggest gene duplication as a mechanism for resolving intralocus sexually antagonistic conflict in Drosophila. Genome Biol Evol 2:835–850
Goldsmith MR, Marec F (2010) Molecular biology and genetics of the Lepidoptera. CRC Press/Taylor and Francis Group, Boca Raton, FL
Goldsmith MR, Shimada T, Abe H (2005) The genetics and genomics of the silkworm, Bombyx mori. Annu Rev Entomol 50:71–100
Grimaldi DA, Engel MS (2005) Evolution of the insects. Cambridge University Press, Cambridge
Hillier LW, Miller W, Birney E et al (2004) Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432:695–716
Innocenti P, Morrow EH (2010) The sexually antagonistic genes of Drosophila melanogaster. PLoS Biol 8(3):e1000335
Itoh Y, Melamed E, Yang X et al (2007) Dosage compensation is less effective in birds than in mammals. J Biol 6:2
Koike Y, Mita K, Suzuki MG et al (2003) Genomic sequence of a 320-kb segment of the Z chromosome of Bombyx mori containing a kettin ortholog. Mol Gen Genomics 269:137–149
Li WH (1997) Molecular evolution. Sinauer Associates, Sunderland, MA
Mank JE, Nam K, Ellegren H (2010) Faster-Z evolution is predominantly due to genetic drift. Mol Biol Evol 27(3):661–670
Marques AC, Dupanloup I, Vinckenbosch N, Reymond A, Kaessmann H (2005) Emergence of young human genes after a burst of retroposition in primates. PLoS Biol 3:1970–1979
Mořkovský L, Storchová R, Plachý J et al (2009) The chicken Z chromosome is enriched for genes with preferential expression in ovarian somatic cells. J Mol Evol 70:129–136
Mueller JL, Mahadevaiah SK, Park PJ et al (2008) The mouse X chromosome is enriched for multicopy testis genes showing postmeiotic expression. Nat Genet 40:794–799
Namekawa SH, Lee JT (2009) XY and ZW: is meiotic sex chromosome inactivation the rule in evolution? PLoS Genet 5:e1000493
Naurin S, Hansson B, Bensch S, Hasselquist D (2009) Why does dosage compensation differ between XY and ZW taxa? Trends Genet 26:15–20
Parisi M, Nuttall R, Naiman D et al (2003) Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 299:697–700
Pearson WR (2000) Flexible sequence similarity searching with the FASTA3 program package. Methods Mol Biol 132:185–219
Potrzebowski L, Vinckenbosch N, Marques AC et al (2008) Chromosomal gene movements reflect the recent origin and biology of therian sex chromosomes. PLoS Biol 6:e80
Potrzebowski L, Vinckenbosch N, Kaessmann H (2010) The emergence of new genes on the young therian X. Trends Genet 26:1–4
Rice WR (1984) Sex chromosomes and the evolution of sexual dimorphism. Evolution 38:735–742
Richler C, Soreq H, Wahrman J (1992) X inactivation in mammalian testis is correlated with inactive X-specific transcription. Nat Genet 2:192–195
Schoenmakers S, Wassenaar E, Hoogerbrugge JW (2009) Female meiotic sex chromosome inactivation in chicken. PLoS Genet 5:e1000466
Schrider DR, Stevens K, Cardeño CM, Langley CH, Hahn MW (2011) Genome-wide analysis of retrogene polymorphisms in Drosophila melanogaster. Genome Res 21(12):2087–2095
Smyth GK (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3. doi:http://www.bepress.com/sagmb/vol3/issl/art3
Sturgill D, Zhang Y, Parisi M, Oliver B (2007) Demasculinization of X chromosomes in the Drosophila genus. Nature 450:238–241
Sturtevant AH (1915) Linkage in the silkworm moth. Am Nat 48:315–317
Tao Y, Masly JP, Araripe L, Ke Y, Hartl DL (2007a) A sex-ratio system in Drosophila simulans. I: An autosomal suppressor. PLoS Biol 5:e292
Tao Y, Araripe L, Kingan SB, Ke Y, Xiao H, Hartl DL (2007b) A sex-ratio meiotic drive system in Drosophila simulans. II: An X-linked distorter. PLoS Biol 5(11):e293
Toups MA, Hahn MW (2010) Retrogenes reveal the direction of sex-chromosome evolution in mosquitoes. Genetics. doi:10.1534/genetics.110.118794
Toups MA, Pease JB, Hahn MW (2011) No excess gene movement is detected off the avian or lepidopteran Z chromosome. Genome Biol Evol. doi:10.1093/gbe/evr109
Traut W, Sahara K, Marec F (2007) Sex chromosomes and sex determination in Lepidoptera. Sex Dev 1:332–346
Vibranovski MD, Lopes HF, Karr TL, Long M (2009a) Stage-specific expression profiling of drosophila spermatogenesis suggests that meiotic sex chromosome inactivation drives genomic relocation of testis-expressed genes. PLoS Genet 5:e1000731
Vibranovski MD, Zhang Y, Long M (2009b) General gene movement off the X chromosome in the Drosophila genus. Genome Res 19:897–903
Vicoso B, Charlesworth B (2009) The deficit of male-biased genes on the D. melanogaster X chromosome is expression-dependent: a consequence of dosage compensation? J Mol Evol 68:576–583
Vinckenbosch N, Dupanloup I, Kaessmann H (2006) Evolutionary fate of retroposed gene copies in the human genome. Proc Natl Acad Sci USA 103:3220–3225
Walters JR, Hardcastle TJ (2011) Getting a full dose? Reconsidering sex chromosome dosage compensation in the silkworm, Bombyx mori. Genome Biol Evol 3:491–504. doi:10.1093/gbe/evr036
Wang PJ, McCarrey JR, Yang F, Page DC (2001) An abundance of X-linked genes expressed in spermatogonia. Nat Genet 27:422–426
Wu CI, Xu EY (2003) Sexual antagonism and X inactivation—the SAXI hypothesis. Trends Genet 19:243–247
Xia Q, Zhou Z, Lu C et al (2004) A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science 306:1937
Xia Q, Cheng D, Duan J et al (2007) Microarray-based gene expression profiles in multiple tissues of the domesticated silkworm, Bombyx mori. Genome Biol 8:R162
Xia Q, Guo Y, Zhang Z et al (2009) Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx). Science 326:433
Yanai I, Benjamin H, Shmoish M et al (2005) Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21:650–659
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13(5):555–556
Yang Z (1998) Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15(5):568–573
Zha X, Xia Q, Duan J et al (2009) Dosage analysis of Z chromosome genes using microarray in silkworm, Bombyx mori. Insect Biochem Mol Biol 39:315–321
Zhang YE, Vibranovski MD, Krinsky BH, Long M (2010a) Age-dependent chromosomal distribution of male-biased genes in Drosophila. Genome Res. doi:10.1101/gr.107334.110
Zhang YE, Vibranovski MD, Landback P, Marais GAB, Long M (2010b) Chromosomal redistribution of male-biased genes in mammalian evolution with two bursts of gene gain on the X chromosome. PLoS Biol 8(10):e1000494. doi:10.1371/journal.pbio.1000494i
Zhu Z, Zhang Y, Long M (2009) Extensive structural renovation of retrogenes in the evolution of the Populus genome. Plant Physiol 151:1943–1951
Acknowledgments
We are grateful for comments from Dr. Yong Zhang and all other Long Lab members. We thank James Walters, Qingyou Xia and Si Tang for help on gene expression analysis. We thank Nick Vankuren for proof reading our manuscript. Computing was supported by the EEgrid of the University of Chicago. The authors were supported by a National Institutes of Health grant (NIH R0IGM078070-01A1), the NIH ARRA supplement grant (R01 GM078070-03S1) and NSF (MCB1051826).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
239_2012_9499_MOESM1_ESM.doc
Supplementary material 1: Distribution of exon-number of parental genes in silkworm and Drosophila. The distributions of exon-number of parental genes were drawn for retrotransposition in three directions for B. mori and all together for B. mori and D. melanogaster. We found the distribution of exon-number of all the parental genes in B.mori is similar to that in D. melanogaster (Bai et al 2007), supporting the reliability of identification of the 68 retrotransposition paralog pairs in B. mori (DOC 253 kb)
239_2012_9499_MOESM2_ESM.doc
Supplementary material 2: a Distribution of Pi(S) and Pi(N) of 68 retrogenes We estimated non-synonymous nucleotide diversity (Pi(N)) and synonymous nucleotide diversity (Pi(S)) for the 68 retrogenes and draw their distribution in this figure (File S8). It was shown that most of 68 retrogenes have fewer (if any) non-synonymous nucleotide diversity than synonymous nucleotide diversity, suggesting that the 68 retrogenes were under functional constraints. b Histogram of θπ for SNPs at the CDS of 68 retrogenes We estimated the nucleotide diversity, θπ, through collecting SNPs in the CDS regions of all the B. mori genes (File S8). We then compared the histogram of θπ in the CDS regions of the 68 retrogenes to the average θπ (0.0061, Xia Q et al 2009) in the CDS regions of all the genes. We found θπ in the CDS regions of the 68 retrogenes was significantly lower than the average θπ in the CDS region of all the genes, which supported that the 68 retrogenes were under functional constraints (DOC 1376 kb)
239_2012_9499_MOESM3_ESM.doc
Supplementary material 3: Histograms of τ of retrogenes and their parental genes. The tissue specificity index τ was computed for the 62 retrogenes and parental genes, which have microarray data available. We found that the τ values of the 62 retrogenes are significantly higher than those of the 62 parental genes with one tailed paired T test (P=0.0220, File S4), suggesting the 62 parental genes were expressed in more tissues than the 62 retrogenes (DOC 974 kb)
239_2012_9499_MOESM4_ESM.xls
Supplementary material 4: Distribution of gonad-biased genes. This figure draws the proportion of testis-, ovary- and non- biased genes on each chromosome with single-probe microarray data available (File S9), which was normalized by Walters and Hardcastle’s (2011) procedure (XLS 262 kb)
239_2012_9499_MOESM5_ESM.pdf
Supplementary material 5: The neighbor-joining tree of the gene pair 16. This figure shows the neighbor-joining tree generated using the protein sequences of BGIBMGA002559, BGIBMGA010222 and their orthologs in D. melanogaster. Since the single-exon gene BGIBMGA002559 clusters with its single-exon ortholog CG18319, and the multiple-exon gene BGIBMGA010222 clusters with its multi-exon ortholog CG2013, we inferred that the duplication event happened before the split between B. mori and D. melanogaster ancestral lineages (PDF 139 kb)
Rights and permissions
About this article
Cite this article
Wang, J., Long, M. & Vibranovski, M.D. Retrogenes Moved Out of the Z Chromosome in the Silkworm. J Mol Evol 74, 113–126 (2012). https://doi.org/10.1007/s00239-012-9499-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-012-9499-y