
Abstract
The recent availability of genome sequences of four different Fusarium species offers the opportunity to perform extensive comparative analyses, in particular of repeated sequences. In a recent work, the overall content of such sequences in the genomes of three phylogenetically related Fusarium species, F. graminearum, F. verticillioides, and F. oxysporum f. sp. lycopersici has been estimated. In this study, we present an exhaustive characterization of pogo-like elements, named Fots, in four Fusarium genomes. Overall 10 Fot and two Fot-related miniature inverted-repeat transposable element families were identified, revealing a diversification of multiple lineages of pogo-like elements, some of which accompanied by a gain of introns. This analysis also showed that such elements are present in an unusual high proportion in the genomes of F. oxysporum f. sp. lycopersici and Nectria haematococca (anamorph F. solani f. sp. pisi) in contrast with most other fungal genomes in which retroelements are the most represented. Interestingly, our analysis showed that the most numerous Fot families all contain potentially active or mobilisable copies, thus conferring a mutagenic potential of these transposable elements and consequently a role in strain adaptation and genome evolution. This role is strongly reinforced when examining their genomic distribution which is clearly biased with a high proportion (more than 80%) located on strain- or species-specific regions enriched in genes involved in pathogenicity and/or adaptation. Finally, the different reproductive characteristics of the four Fusarium species allowed us to investigate the impact of the process of repeat-induced point mutations on the expansion and diversification of Fot elements.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, great progress has been made in the characterization, the genomic distribution, dispersion, and maintenance of transposable elements (TEs) in fungal species (for a review, see Daboussi and Capy 2003). Such studies have highlighted interesting features such as the prevalence of pogo-like elements among class II elements (or DNA transposons) in these organisms. An interesting feature of this family in fungal genomes is that it contains active members since several copies have been identified through their transposition in a target gene in F. oxysporum (Migheli et al. 1999; Trouvelot et al. 2002), Botrytis cinerea (Levis et al. 1997), Aspergillus niger (Nyyssönen et al. 1996), and Magnaporte grisea (Kang et al. 2001). Most of these elements share common features with Fot1, the first active pogo-like element discovered in fungi (Daboussi et al. 1992). However, comparative analysis with other members of this family showed that they are diverse, some of them containing introns such as Nht1 of Nectria haematococca (Enkerli et al. 1997), Occan of M. grisea (Kito et al. 2003), and Taf1 from A. fumigatus (Monroy and Sheppard 2005). In the genome of some species exemplified by F. oxysporum, different pogo-like elements coexist. In this genome, in addition to Fot1, four Fot members have already been identified and partially characterized: Fot2 through transposition (Daboussi, unpublished results), Fot3 and Fot4 in nests of transposons (Hua-van et al. 2000) and Fot5 identified in the proximity of the SIX1 gene (Rep et al. 2004).
The recent release of the genome sequences of four distinct Fusarium species F. oxysporum, F. verticillioides, F. graminearum, and F. solani—telomorph Nectria haematococca—either by the Broad Institute (Fusarium Comparative Database: http://www.broad.mit.edu/annotation/genome/fusarium_group/MultiHome.html) or the Joint Genome Institute (http://www.jgi.doe.gov/nectria) offers the opportunity to exhaustively investigate the genomic representation and abundance of TEs. These four Fusaria are quite representative of the diversity of the Fusarium genus. All being plant pathogenic species, they exhibit different host ranges as well as reproductive strategies (Agrios 2005; Samuels et al. 2001). Among the four species, F. oxysporum and F. verticillioides are the most closely related and N. haematococca the more distant one (for an overview, see Fig. 1 in Ma et al. 2010). The corresponding genomes exhibit a variable number of core chromosomes sharing high sequence similarity and extensive synteny (Rep and Kistler 2010; this work, Table 1). In addition to the core genomes, species-specific regions could be found. Interestingly, the genomes of F. oxysporum and to a lesser extent of N. haematococca are particularly enriched in such specific sequences, which are mainly localized on particular chromosomes (or parts of), named lineage-specific regions for F. oxysporum (Ma et al. 2010) and conditionally dispensable (CD) chromosomes for N. haematococca (Coleman et al. 2009). The overall content in repetitive sequences has been recently estimated in these genomes and revealed an extraordinary variation in the TE content (Coleman et al. 2009; Ma et al. 2010, The Fusarium Comparative Database, for an overview, see Table 1). In this study, we focused on pogo-like elements, namely Fots. BLASTN or TBLASTX searches using known Fot elements as queries on the four Fusarium genomes were performed. Overall, 10 Fot families (Fot1 to Fot10) and two Fot-related miniature inverted-repeat transposable element (MITE) families (mFot4 and mFot5) were identified, with a much higher diversification and representation in the genomes of F. oxysporum and N. haematococca which totalize 665 and 84 Fot/mFot copies, respectively. As compared with the recent analysis of overall TE content in these genomes (Ma et al. 2010; Coleman et al. 2009) as well as in other fungal genomes (Galagan et al. 2005a, b; Thon et al. 2006; Espagne et al. 2008; Neafsey et al. 2010; Nowrousian et al. 2010), our study revealed an extraordinary prevalence of pogo-like elements not only among DNA transposons (around 50%), but also among overall TEs (more than 30%, Ma et al. 2010). Indeed, to date, retroelements were by far the most represented elements in most fungal genomes analyzed.
An exhaustive analysis of structural and functional characteristics of all Fot families revealed that the more numerous ones all contain potentially active copies. In the genomes of F. oxysporum and N. haematococca, as already described in Ma et al. (2010) and in Coleman et al. (2009), we found a strong bias of distribution toward peculiar genomic regions, LS regions in F. oxysporum and CD chromosomes in N. haematococca. Interestingly, in the two genomes, these regions are also enriched in pathogenicity/adaptation determinants (Ma et al. 2010; Coleman et al. 2009), which, together with the presence of active TEs, may contribute to fast adaptation of strains to external factors and/or hosts.
In filamentous fungi, repeat-induced point mutation (RIP) has been shown to be involved into genome defense, in particular against mobile genetic elements. This process is acting during the sexual cycle and allows detection of duplicated sequences, >400 bp and sharing more than 80% nucleotide identity. Consequently, the duplicated sequences are mutated through C:G to T:A mutations (Galagan and Selker 2004). Interestingly, among the four Fusarium species studied herein, all different reproductive habits described for filamentous fungi are represented. F. oxysporum is an asexual species while the three other exhibit sexual reproduction: F. graminearum being mostly homothallic, whereas F. verticillioides and N. haematococca are heterothallic. We have exploited this situation to investigate the impact of RIP on the diversification of Fot elements among the four Fusarium species. Altogether, this study allowed us to propose a scheme for the evolutionary history of the Fot elements in the Fusarium genus.
Materials and Methods
Fungal Material
The Fusarium oxysporum strain FOM24 (F. oxysporum f. sp. melonis) was used in RT-PCR experiments to demonstrate the presence of and precisely locate the two intronic sequences in active Fot2 elements. To obtain spores, flasks or 2 ml Eppendorf tube containing 20 or 1 ml of liquid potato dextrose broth (PDB), respectively, were inoculated with a plug from an 8-day-old potato dextrose agar (PDA) plate and incubated with moderate shaking at 26°C for 2–5 days. For long-term storage, a plug of the culture was put on 500 μl PDA poured in a 2 ml Eppendorf tube and after growth for 2–3 days at 26°C, was stored at 4°C.
RT-PCR Analyses
10 ml of liquid PDB or minimal medium were inoculated with 107 spores of F. oxysporum FOM24 strain. Following incubation of 24 h at 26°C, the mycelia were recovered, frozen in liquid nitrogen and kept at −80°C until use. Three replicas were done per medium condition. Total RNA were extracted from the above described mycelia using RNA plus (MP Biomedicals, Illkirch, France) according to the manufacturer’s instructions. First-strand cDNA were synthesized from oligo(dT)12–18 using the Superscript RT(II) First-strand Synthesis System for RT-PCR (Invitrogen, Illkirch, France). 2 μg total RNA were used for each sample. Controls consisting in reaction mixes devoid of reverse-transcriptase were performed for each experimental condition. PCR experiments were performed on 1/10 (2 μl) of each RT reaction using primers Fot2(RT)A (5′-CGTAACTGGGTGATCAAGTTTATCAATCGCGCAGAC-3′) and Fot2(RT)C (5′-CGCGAAGTCGGCCAGTTGCCAGTGACCTCCC-3′). PCR conditions were the following: 5 min at 94°C then 35 cycles of 1 min at 94°C, 1 min at 65°C, and 2 min at 72°C.
PCR products were directly cloned into the pGEM-T Easy vector (Promega) using 3 μl of either the rough or the purified PCR product following the manufacturer’s instructions. Sequencing of PCR products either as double-stranded linear DNA or as plasmids (into the pGEM-T Easy vector) was performed by Genome Express (Meylan, France) using an ABI Big Dye Terminator kit (Perkin Elmer, France) and the universal M13/pUC sequencing primer (−20) (forward) and pR primer (5′-GGAAACAGCTATGACCATG-3′; reverse).
In Silico Extraction of TE Copies
A preliminary BLASTN (Altschul et al. 1997) search was performed on the F. oxysporum sequences available at the Broad Institute website (https://www.broad.mit.edu/annotation/genome/fusarium_group/MultiHome.html), using known sequences of F. oxysporum pogo-like elements (GenBank accession numbers: Fot1, X64799; Fot2, JN624854; Fot3, AF076631; Fot4, AF076632 and Fot5, AJ608703), with the default parameters. To perform the most exhaustive search possible, a new BLASTN search and a TBLASTX search were realized against the four Fusarium genomes available either at the Broad Institute or the DOE Joint Genome Institute (for N. haematococca, http://genome.jgi-psf.org/Necha2/Necha2.home.html). The full-length elements, including a newly identified Fot6 element, were used as batch queries (e-value threshold set to 0.001). An automated procedure was set up to gather contiguous hits separated by <1,000 bp into one copy, in each genome. The BLASTN search served to determine precisely the copy number of each family in each genome. Nucleotide sequences of these different copies were extracted (along with 500 bp of flanking sequences on each side) and batch BLASTed against the six Fot elements. For each sequence, the Fot element giving the highest score was considered as the family to which the sequence belongs. Results were then checked manually to remove false positives or to detect insertions larger than 1,000 bp that could have split copies into two distant parts. All copies that aligned on <400 bp with the Fot reference sequences were arbitrarily excluded from further analysis. The TBLASTX search was intended to identify potentially divergent new elements, and proved to be efficient since it allowed the detection of copies heavily mutated by RIP, not found with the BLASTN procedure, and the identification of two out of the four new families (Fot9 and Fot10). For each family, duplicated copies were identified on the basis of the identity of flanking sequences. For further analyses, independent copies only were considered. The resulting copy number for each family is shown in Table 2.
For the identification of Fot-associated MITE elements, we first used an ad hoc Perl script based on an algorithm already described in Bergemann et al. (2008). The nucleotide sequences of each target terminal inverted repeat (TIR) of a sequence length l were compared with the sequence of all sliding windows of the same length extracted from the F. oxysporum genome. A nucleotide divergence of 25% was tolerated, slightly higher than the maximum value observed for TIRs examined manually. Once a candidate 5′-TIR identified, a potential 3′-TIR was searched in a 500 bp range, in the range of maximum sizes usually reported for MITEs, with the same cutoff value of 75% identity. This recovered 25 MITE copies associated with Fot5. The four more divergent copies were then used in a BLASTN search, and allowed to identify a total of 138 sequences. Queries used were either Fot elements or MITEs identified with the first strategy. With this latter strategy, incomplete MITEs or MITEs with altered TIRs could also be recovered. For other Fusarium genomes, MITEs were detected by analyzing BLASTN hits covering only the TIR part of the query.
Sequence Comparison and Phylogenetic Analysis
For a given family, sequences were aligned using muscle software (Edgar 2004), and refined manually. Distance and phylogenetic analysis was performed using Distmat of the EMBOSS package (Rice et al. 2000), PAUP (Swofford 2002), and PHYML (Guindon and Gascuel 2003). Maximum likelihood and parsimony methods were both used. Bootstrap values were calculated after 100 replicates. For maximum likelihood analyses, the best evolution model was systematically evaluated using Modeltest3.7 (Posada and Crandall 1998) or ProtTest (Abascal et al. 2005). Trees were visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Clustering Analysis
Each cluster was split into 10 kbp non-overlapping windows in each of which the number of Fot elements was counted. Each independent window with more than two elements or adjacent window with more than one element have been further split into 400 bp non-overlapping windows. Each 400 bp window was then coded “0” if the majority of the window is constituted by a Fot element or coded “1” otherwise. Thus, each 10 kbp windows was coded into a 1/0 vector. We further used the MACML model (Zhang and Townsend 2009) to check for elements clustering by using both model selection and model averaging. Finally, each individual element was located on the MACML output.
Results
Identification of Known and Novel pogo-Like Elements, and Associated Elements in Four Fusarium Genomes
To identify members of the five known families previously characterized (Fot1, Fot2, Fot3, Fot4, and Fot5, Daboussi et al. 1992; this work; Hua-Van et al. 2000; Rep et al. 2004), the reference sequences were first used in BLASTN searches against the genome of F. oxysporum strain FOL4287 (F. oxysporum). A preliminary analysis of the BLAST results revealed that a group of copies exhibits homology with small parts of either Fot2 or Fot3. These copies were classified as a sixth family named Fot6. This analysis also permitted to identify full-length Fot4 elements for which only a partial sequence was previously known.
A more detailed family-per-family analysis also uncovered few sequences that were too divergent to be considered as members of the other six families, hence defining two novel minor families, Fot7 and Fot8, related to Fot5 and Fot1, respectively (Fig. 1, Phylogenetic analysis). BLASTN and TBLASTX searches using prototypes of the eight Fot families identified in F. oxysporum was then conducted on the available genome sequence Nectria haematococca (Coleman et al. 2009), F. graminearum (Cuomo et al. 2007), and F. verticillioides. In F. graminearum, five copies of a novel related-family named Fot9 were found. When BLASTed back against F. oxysporum, these sequences provided hits corresponding to 21 new copies that were split into two families, Fot9 and Fot10, based on nucleotide divergence.

Maximum likelihood tree of pogo-Fot transposase DDD domain (232 AA). Fusarium families are in bold. Families with introns are circled, and the position of introns (black triangles) relative to the catalytic DDD triad is displayed. The evolution model was Blosum62+I+G+F. Bootstrap values over 85 (100 replicates) are indicated for internal branches. Bootstrap values for clade 1, clade 2, and non-fungi clade are in bold. Accession numbers for transposase sequences or coordinates of newly identified elements are as indicated. Fusarium oxysporum and F. graminearum Fot elements (FotFo and FotFg): see coordinates on Supplementary Table 1; Nectria haematococca Fot elements (FotNh): Fot2 (Nht1): AAB71335, Fot3 (Nht1-like): AF294788 [2230:3132, 3192:3968], Fot6 (Pot3-like): AF294788 [1915:1923,4253:5534], Fot5: sca_5 2371865:2375474; Aspergillus niger Tan1: U58946; Aspergillus fumigatus Taf1: AAX83011; Magnaporthe grisea Pot2: Z33638, Pot3: U60989, Occan: XP_369247; Botrytis cinerea Flipper: U74294; Cochliobolus carbonum Fcc1: U40479; Phanaerochaete nodorum Molly: CAD32687, Pixie: CAD32689; Drosophila melanogaster pogoR11: S20478; Arabidopsis thaliana Lemi1: AAD14510; Yarrowia lipolytica Fotyl: CAG33729; Candida albicans Cirt1: XP_710204; Homo sapiens Tigger1: AAB61714; Ustilago maydis USMAY: XP_760029; Caenorhabditis elegans Tc4 variant: AAA28148; Sclerotinia sclorotiorum SCSCL: XP_001592252; Paracoccidioides brasiliensis PABRA: ACY56713; Ophiostoma novo-ulmi Ophio1: ABG26269; Chaetomium globosum Cg08137: EAQ86884
A global analysis of pairwise divergence allowed to define a Fot family by an average nucleotide polymorphism below 35%, whereas the divergence between families ranged between 36 and 50%.
Presence of associated MITE families was investigated using different strategies (see “Materials and Methods” section) that, in fine, recovered 138 MITE sequences in F. oxysporum which are all associated with Fot5, based on TIRs similarities and were thus named mFot5. In the other three species, the search for MITE revealed three Fot4-associated copies of a novel element named mFot4, in the genome of F. verticillioides.
Overall, 10 Fot families (Fot1 to Fot10) and two MITE families, mFot4 and mFot5, related to Fot4 and Fot5, respectively, could be identified among the four Fusarium genomes. An overview of the types and copy numbers of elements identified in each of the four Fusarium genomes is presented in Table 2 and coordinates of a representative copy of each type of element or of all copies are shown in Supplementary Table 1 and Supplementary file 1, respectively. The copy number varies greatly between Fot families. When considering the F. oxysporum and N. haematococca genomes in which the greater number of Fot and mFot elements were found, Fot5 represents the largest family (48 and 71% of all Fot elements in F. oxysporum and N. haematococca, respectively) while Fot1, Fot7, Fot8, and Fot10 are present in <10 copies.
By far, the F. oxysporum genome contains the highest number of copies, with a total of 527 and 138 copies of Fot and mFot elements, respectively. This genome carries several segmental duplications (Ma et al. 2010, and our lab, unpublished results). To identify the number of copies corresponding to independent transposition events, flanking regions were analyzed. Only 284 independent Fot- and 75 mFot-insertion events could be detected (Table 2), indicating that genome segmental duplications account for more than 45% of the total Fot and mFot copies. For this genome, the phylogenetic analysis will focus on independent Fot and mFot copies. In N. haematococca, apart from two Fot5 copies, all elements resulted from independent insertion events (Table 2).
Overall, genomic localization of Fot and mFot elements in the F. oxysporum genome fits the biased distribution already described for DNA transposons (Ma et al. 2010). Indeed more than 95% of the elements are located in LS regions defined as four distinct chromosomes (3, 6, 14, and 15), part of chromosomes 1 and 2 and most of the scaffolds not anchored to the optical map (Table 3). A similar distorted distribution could be observed in the N. haematococca genome in which more than 70% of the Fot sequences are located either on CD chromosomes and unmapped scaffolds (Table 3). In F. verticillioides and F. graminearum genomes, in which much fewer copies were identified (see Table 2), elements are dispersed among the chromosomes with no obvious bias.
The potential clustering of Fot and/or mFot elements in the Fol genome was statistically analyzed using the MACML hierarchical clustering method (Zhang and Townsend 2009). Among the overall 665 copies (527 Fots and 138 mFot5), 188 copies were found to be grouped in 78 clusters, the remaining copies (477) being isolated. Concerning Fot elements, the proportion of clusterized elements reflected the representation of each family in the genome. On the contrary, a significantly higher proportion of mFot5 copies (36.7%) was found in these clusters, either close to or inside other Fot elements.
Structural and Functional Analysis of Fots Families
Length of canonical elements ranged from 1,848 (Fot4) to 2,226 bp (Fot6). All full-length elements were inserted into a TA site, which appeared duplicated upon insertion. Except for an enrichment in T, A, or C in 3′ of Fot5 elements and a preference for TA from CTAG tetranucleotides for Fot9 and Fot10 (data not shown), no obvious consensus other than the conserved target TA dinucleotide was found for other Fot families. Two types of TIRs could be observed among the different Fot families, differing in length. Seven families contain short TIRs (Fot1, Fot4, Fot5, Fot7, Fot8, Fot9, and Fot10) ranging from 42 to 46 bp, while three families (Fot2, Fot3, and Fot6) exhibit long TIRs (from 79 to 88 bp if allowing five mismatches). One hallmark of all Fot elements is the presence of short direct repeats within the TIRs, ranging in length from 8 to 17 bp. No obvious correlation between the length of direct repeats and the size of TIRs could be noted.
As reported for Nht1, the canonical Fot2-like element in N. haematococca (Enkerli et al. 1997), the presence of two short introns became apparent in Fot2, Fot3, and Fot6 elements. For Fot2, the sequences of the 5′ and 3′ splice junctions of the introns, located at nucleotide positions 820–884 and 1,225–1,273, respectively, match the consensus defined for fungal genes (Gurr et al. 1987). Splicing sites were confirmed by RT-PCR experiments (data not shown). The nucleotide sequences of two introns of 64 and 58 bp do not match with any known sequence in the databases. The two introns are at the same nucleotide positions in Fot2, Fot3, and Fot6. These three families containing introns also corresponded to the longest elements (ca. 2,200 bp) and to those with long TIRs, thus defining a sublineage with introns.
Considering that a copy needs at least two TIRs with direct repeats to be mobilizable, we estimated the proportion of mobilizable copies in the families exhibiting more than 10 copies, i.e., Fot5 in N. haematococca and Fot2, Fot3, Fot5, Fot6, and Fot9 in F. oxysporum. In N. haematococca, Fot5 contained 88% of mobile copies, and <2% (1 copy) contained a deletion of a direct repeat within TIR (DR deletion) likely affecting the mobility and thus not counted as mobile. In contrast, in F. oxysporum, the percentage of mobile copies was always much lower, ranging between 30% for Fot3 and 56% for Fot2, Fot5, and Fot6 showing intermediate proportions (49 and 45%, respectively) (Table 2). In each family, about 12–15% of full-length copies (with two TIRs) were impaired for transposition because of DR deletion (data not shown). Other non-mobile copies were copies truncated either in 5′ or in 3′ (with no bias for one particular end), sometimes at both ends. In contrast, copies with internal deletion (longer than a couple of nucleotide) were rare (always below 10%), suggesting that this is a minor event. The majority of internal deletion events were unique but in one case for which the same internal deletion was found in four Fot5 copies, suggesting that these copies have amplified after one unique deletion event. Size variations due to the insertion of small TEs was even more seldom (for example, a solo-LTR in Fot4), with no evidence of further transposition. However, we can suspect that in most cases, insertion of large elements will not be detected and the Fot copy considered as truncated.
Intra-family polymorphism was estimated through pairwise divergence. The average divergence is indicated in Table 2 for families with more than 20 copies. It gives a first approximate idea of the age of the families, and suggests that Fot3 and Fot5 seem older than Fot2 and Fot6.
Transpositional activity of Fot1 and Fot2 was previously evidenced in the F. oxysporum FOM24 strain pathogenic to melon and FO47 non-pathogenic strain, through their transposition in the nia gene encoding nitrate-reductase (Daboussi and Langin 1994, MJ Daboussi, unpublished results). For all other families, transposition ability can only be inferred from sequences, as the potential to encode an uninterrupted transposase (Table 2). Elements encoding a potentially active transposase could be detected in the four major Fot families: Fot2, Fot3, and Fot5 in F. oxysporum and N. haematococca and Fot6 in F. oxysporum only. Among the minor families, potentially active copies were observed in Fot1 (F. oxysporum), Fot9 (F. oxysporum and F. graminearum), and Fot4 (N. haematococca). Fot7, Fot8, and Fot10 represent inactive families with very low copy numbers in F. oxysporum. As indicated in Table 2, the proportion of potentially active copies varies greatly between the Fot families with significant lower and higher percentages in the Fot3 and Fot5 families, respectively.
The four species used in this study exhibit different reproductive strategies. In contrast with F. oxysporum which is asexual, the three other Fusarium species can reproduce sexually. F. graminearum (Gibberella zeae) is a homothallic fungus, whereas both F. verticillioides and F. solani f. sp. pisi (Nectria haematococca) are heterothallic. As already mentioned above, the RIP process occurs through meiosis and frequently gives rise to an inactivation of the target sequences. Recently, the occurrence of RIP was demonstrated experimentally using transgene sequences in both F. graminearum (Cuomo et al. 2007) and N. haematococca (Coleman et al. 2009). In F. verticillioides, a severe form of RIP has been reported in a group of class I elements (Cuomo et al. 2007) but no experimental evidence is available.
In order to determine whether RIP could be involved at least in part of the observed nucleotide polymorphism within each Fot family and could also explain the different proportions of potentially active elements (Table 2), an extensive analysis was performed on Fot non-redundant copies in the different Fusarium genomes. Sequences mutated by RIP are typically A+T rich and also exhibit distorted frequencies of dinucleotides due to sequence preference of the process itself. Two parameters are thus commonly used to estimate differences in overall base composition: the global percentage in A+T and the ratio of TpA to ApT dinucleotides. Sequences that have been mutated by RIP should exhibit both a higher A+T percentage than average for a given genome and a high TpA/ApT ratio (usually above 1.0) due to the introduction of C:G to T:A mutations (Margolin et al. 1998; Galagan and Selker 2004). In F. oxysporum, albeit the absence of sexual reproduction, 23 out of 258 (8.9%) Fot copies belonging to the four most numerous families (Fot2, Fot3, Fot5, and Fot6) showed hallmarks of RIP (Table 2, Fig. 2). Among these, 19 are located in the LS regions, a situation which is not surprising since more than 95% of the overall Fot elements are located in these genomic areas (Ma et al. 2010). However, a strong bias is observed within the LS, since 11 of the ripped copies map to a region of chromosome 2 covering <450 kbp. This genomic region also contains seven additional non-ripped copies of Fot elements interspersed with the abovementioned ripped Fot copies. A mixture of ripped and non-ripped copies is also observed for other TEs carried by this locus, belonging either to retroelements or to other DNA transposons superfamilies (data not shown).

Distribution of F. verticillioides (light gray bars), F. graminearum (average gray bars), N. haematococca (dark gray bars), and F. oxysporum (black bars) Fot copies in relation with their RIP index, measured by calculating the TpA/ApT ratios (Margolin et al. 1998). Values indicated on the y axis correspond to the percentage of Fot elements exhibiting a specific RIP index among the overall number of Fot elements. The threshold value of 1.0 for the TpA/ApT ratio, above which sequences are considered to have been mutated by RIP is indicated by the dashed vertical line
In the other three species, Fot copies exhibiting hallmarks of RIP were identified but at different levels depending on the Fot family. In F. graminearum and F. verticillioides, the very few Fot4 and Fot5 copies detected all showed extensive RIP (Table 2, Fig. 2) and were thus highly polymorphic with F. oxysporum copies. All those barely recognizable copies were nevertheless full length. In contrast, among the five Fot9 copies from F. graminearum, only one displayed traces of RIP, two copies out of the remaining four being potentially active. In N. haematococca, 32% (27 out of 84) of Fot copies were found to be ripped whatever the Fot family (Table 2). When considering the distribution of the RIP index in the three sexual species, we observed an irregular pattern in F. verticillioides and F. graminearum, whereas in N. haematococca, it seems to evolve more continuously (Fig. 2). This suggests that RIP, likely occurring at each round of meiosis, is mild allowing the presence of potentially active Fot copies.
Phylogenetic Analysis
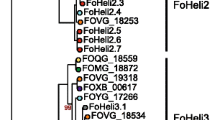
First the diversity of Fusarium Fot families was compared to the diversity of pogo-Fot superfamily through a phylogenetic analysis of a portion of the transposase sequence (the DDD catalytic domain, Yuan and Wessler 2011) (Fig. 1). Fungal pogo-Fot elements form an exclusive and abundant group constituted of two major clades. While structural characteristics of Fot2, Fot3, and Fot6 (introns, TIRs length, and sequences) suggested they form a group divergent to the other Fusarium Fot, the phylogenetic analysis based on amino-acid sequence comparison indicated that they are related to Fot1, Fot4, Fot8, Fot9, and Fot10, all elements gathered within a first clade. Fot5 and Fot7 more distant families form a second supported clade with some other ascomycetous elements.
A phylogenetic analysis of the most numerous families in F. oxysporum (Fot2, Fot3, Fot5, and Fot6) was performed using nucleotide sequences. For each, N. haematococca sequences have been detected and were thus included in the analysis. The trees obtained are presented on Fig. 3.

Phylogenetic relationships between and within intron-containing Fot families. a Maximum likelihood tree derived from CDS alignment of Fot2, Fot3, and Fot6 from F. oxysporum and Nectria haematococca, with GTR+I+G as a substitution model. b, c, d nucleotide Maximum likelihood tree on full independent copies of Fot2, Fot3, and Fot6, respectively, from F. oxysporum and N. haematococca (Nh). Crosses indicate ripped copies, black plain circles, potentially active copies (exhibiting an intact ORF) and open circles, copies located on core chromosome. The copy indicated by an asterisk (*) is duplicated, with one potentially active duplicate in LS region, and one inactive duplicate on a core chromosome. Substitution models used were TrN+G for Fot2 and GTR+G for Fot3 and Fot6. Bars indicate the number of substitutions per site. Bootstrap values above 50 (calculated among 100 replicates) are indicated
The open reading frames (ORFs) of the three intron-containing families, Fot2, Fot3, and Fot6, were first aligned together (Fig. 3a) allowing rooting of the individual family trees. Different levels of structuration were observed. In Fot2 (Fig. 3b), copies clustered in four well-supported clades. Potentially active copies were observed in three of the four clades, however, only one exhibited traces of “recent” transposition events as reflected by short terminal branches. Few copies exhibiting hallmarks of RIP were detected. For Fot3 (Fig. 3c), the copies were dispatched between two deep branches. The few copies with an uninterrupted ORF were dispersed in two clades and each clade contained highly divergent copies, some of them showing traces of RIP (Fig. 3c). The Fot6 tree (Fig. 3d) was the less well-structured and displayed two sets of divergent copies with RIP characteristics, and a bunch of moderately divergent copies. Apart from the highly divergent copies (some resulting from RIP), the three families also contained some closely related copies, indicating that transposition events occurred in a recent past.
When examining the distribution of N. haematococca copies into each family, two contrasted situations were observed. Whereas all N. haematococca sequences grouped into one clade for the Fot2 family, they appeared much more dispersed among F. oxysporum sequences in the Fot3 and Fot6 families suggesting that these three families have different histories within the Fusarium genus.
The large Fot5 family may be divided in several clades and isolated copies. It is much more polymorphic than the other Fot families, which suggests that it is of more ancient origin but also displays traces of recent transposition in N. haematoccoca and F. oxysporum, as evidenced by the presence of closely related or identical copies (Fig. 4). The proportion of potentially active elements is variable depending on the clade (Fig. 4). Even if potentially active and/or mobilisable Fot5 elements are found in LS regions, an unusually high proportion of the Fot5 copies encountered on the core chromosomes are either mobile (at least 11 out of 14) and/or potentially active (at least 7 out of 14). Interestingly, most of these copies are very closely related, some of them being identical although independent, and constitute most of the sequences of a clade reflecting recent transposition events (Fig. 4). The 59 N. haematococca Fot5 elements were grouped in five clusters, interspersed with F. oxysporum sequences. This suggests that Fot5 was present as a large family before speciation with occasional species-specific amplifications. All F. graminearum and F. verticillioides copies grouped together along with some N. haematococca and F. oxysporum copies but this could be due to a composition bias, since all display a high TA content, characteristics of heavily ripped copies (data not shown).

Fot5 phylogenetic tree. Midpoint rooted phylogenetic tree of all Fot5 independent copies, obtained by maximum likelihood with substitution model GTR+I+G. Bootstrap values above 50 are indicated. Species-specific (*) or mixed clades with bootstrap support (100 replicates) are represented by triangles with different gray levels. The horizontal measure of each triangle reflects the length of the longest branch of the corresponding clade to the most recent common ancestor. Pie charts display the proportion of ripped (black), potentially active (gray) and inactive but non-ripped copies (white) within each supported clade
The mFot5 analysis revealed that two subgroups coexist, one with 72 independent sequences and the other represented by three independent sequences only. Between these two subgroups, homology seems limited to the TIRs only. Within the largest group, no clear structuration could be detected, and nucleotide divergence within copies was globally high reflecting that it is probably an old family. A few groups of closely related copies were found, illustrating occasional transposition activity (data not shown). In some cases, TIRs of such recently transposed copies were more conserved with those of some Fot5 elements which could favor the hypothesis of a potential cross-mobilization of mFot5 by the Fot5 transposase.
Discussion
Diversity of Fot and mFot Elements in Fusarium Genus
By exploiting the genome sequences of F. oxysporum and three phylogenetically related species, we identified a total of 10 Fot families (Fot1 to Fot10), belonging to the widespread pogo–Fot–Tigger superfamily. Most of the families could be detected through the BLASTN search but Fot9 and Fot10. These two families being more divergent at the nucleotidic level, they were missed in the first analyses most likely because copies only gave rise to bad short hits. The TBLASTX search is thus a more powerful methodology to exhaustively identify all Fot copies, even from different families, as soon as they still contain part of the coding region. An analysis of excluded reads has provided 687 supplementary hits, representing around 10% of the reads (data not shown). However, no novel element has been identified among these hits. Heavily ripped copies may also have been missed as they exhibit high nucleotide divergence with the original sequence. It is therefore likely that, even if the number of copies in each family has been underestimated, our analysis provides a rather exhaustive identification of the Fot families in the Fusarium genus.
Among the 10 identified Fot families, all are present in the genome of F. oxysporum and are either inactive, represented by few copies, either potentially active with few or more than 50 copies. However, these results should not be generalized to the whole F. oxysporum species as TE copy number has already been reported to be strain dependent. For example, the Fot1 family has originally been identified in more than 100 copies from a F. oxysporum strain originating from melon (Daboussi et al. 1992) and later shown to be poorly represented in other F. oxysporum strains (Daboussi et al. 2002) as is the case for the tomato strain used in this study. In other Fusarium species, both the total copy number and the number of coexisting Fot families are lower, which fits well with the global TE (or repetitive sequences) content estimated for these species (Ma et al. 2010; Coleman et al. 2009). Also, noteworthy is that Fot5 is the only Fot family present in the four species and is also the most abundant one in both F. oxysporum and N. haematococca. Altogether, this suggests that Fot elements are globally old components of Fusarium genomes, but have suffered various fates in the different genomes.
Associated high-copy number MITE families have been detected for some of pogo–Fot elements outside fungi (Casacuberta et al. 1998; Feschotte and Mouches 2000). In Fusarium species, our analysis identified MITEs related to Fot5 and Fot4 only (named mFot5 and mFot4, respectively). mFot5 elements were encountered in the F. oxysporum genome in more than 130 copies. However, compared to other MITE-transposon associations well studied in F. oxysporum (mimp-impala, Bergemann et al. 2008), or in other eukaryotes (Feschotte and Mouches 2000; Santiago et al. 2002), the copy number of mFot5 remains lower than that of its putative master element Fot5. In none of these MITEs, homology with their suspected autonomous partner could be detected outside the TIRs, which does not allow concluding on the origin of these elements: internal deletion of the master element and further degeneration of internal sequences or de novo origin. A high proportion of mFot5 elements were found in clusters together or with Fot5 elements, suggesting that a high concentration of homologous TIR sequences may favor the generation of novel elements, as already suggested for other MITEs (Jiang and Wessler 2000).
Albeit the presence of potentially active Fot5 elements in the N. haematococca genome no mFot5 were found. Such a species-dependent situation is reminiscent of that described for the Emigrant family (Feschotte and Mouches 2000; Santiago et al. 2002). In Arabidopsis, the Emigrant MITE, related to the pogo element Lemi1, amplified whereas a single copy of the master element is present (Feschotte and Mouches 2000; Santiago et al. 2002). A recent study described a contrasted situation in the genome of Medicago truncatula, in which around 30 copies of Lemi1 were identified but no Emigrant members (Guermonprez et al. 2008). The authors suggested that the ability to produce and amplify MITEs may be the consequence of constraints associated with the host genome.
Evolution of pogo Elements in Fungi, and the Gain of Introns
pogo-Fot-Tigger elements have been found in various species from plants, arthropodes, nematodes, or even humans (Smit and Riggs 1996). However, their amplification has been especially successful in fungi (Daboussi and Capy 2003). Indeed, they have been found in most fungal species in which they have been searched for, and constitute by far the most represented Class II superfamily in fungi. Nevertheless, the diversity of Fot families observed in the Fusarium genus is surprisingly high when compared to other fungal species. This discrepancy may be explained in part by the fact that no exhaustive search has yet been done in other fungi in the genomes of which only global TE content was generally estimated. Yet, the situation of F. oxysporum remains an exception, exhibiting both high diversity and copy number of Fot elements when compared not only to other fungi, but also to its close relative Fusarium species.
A phylogenetic tree deduced from the multiple alignment of several pogo-like transposase sequences revealed that the Fusarium Fot families reflect the diversity of fungal pogo-like elements (Fig. 1). The evolutionary history of these elements in fungi is compatible with an ancient diversification of several lineages that can remain together in the same genome for a long time, as exemplified by M. grisea (Thon et al. 2006) and F. oxysporum elements, or that can be lost. Indeed, in F. oxysporum the process of lineage diversification of Fot elements seems to have been particularly intense but is probably as old as ascomycetous diversification since in several cases, sequences from different species seem more closely related than sequences from different Fot families in F. oxysporum. Hence, we could expect that Fot-like elements more closely related to Fusarium Fots do exist in other fungal species but still remain to be identified.
Interestingly, the large majority of fungal pogo-like elements do not contain introns but they are encountered in five transposase genes. Occan from Magnaporthe oryzae (Kito et al. 2003) and Taf1 from Aspergillus fumigatus (Monroy and Sheppard 2005) contain a single intron whereas Fot2, Fot3, and Fot6 genes contain two intronic sequences (this study). Introns in the Fot2, Fot3, and Fot6 elements are located at the same positions but, in Occan and Taf1 elements they not only correspond to different sequences but are located at different positions. Such a patchy distribution of different Fot-like elements containing introns can be best explained by the independent gain of introns in three lineages, as observed for Mutator-like elements in plants (Feschotte and Wessler 2002).
Evolutionary History of Fot and Fot-Associated Elements Across the Fusarium Genus
The phylogenetic analysis conducted on the four most numerous Fot families in the F. oxysporum genome, along with their structural characteristics (proportion of potentially active and mobile, mean pairwise divergence copies) revealed different dynamics/history. Although it presents evidence of recent or present day transposition activity, in both N. haematococca and F. oxysporum, the Fot5 family seems to be a very ancient family. Among the intron-containing families, Fot2 seems to be the less ancient, and in contrast, Fot3 looks like an older family (equivalent to Fot5). Fot6 appears like intermediate. Nevertheless these three last families all contain moderately divergent copies suggesting they have been more or less active in a relatively recent past. However, the absence of identical independent copies suggests that present day transposition is either absent or rare. These families share a common ancestor but it is not clear whether the coexistence of these families within the same genome results from the ancient presence of the ancestor family in F. oxysporum followed by occasional reactivation of divergent copies in combination with a purge of intermediates, or from successive reintroduction within the genome.
When examining the four Fusarium genomes altogether and incorporating N. haematococca sequences in the phylogenetic analyses, two different situations were encountered. Either N. haematococca sequences clearly grouped together in a distinct clade (Fot2) or they were found interspersed with F. oxysporum sequences (Fot3, Fot5, and Fot6). Along with the low structuration level of the trees, this suggests that the expansion of these last three families predated the speciation event. Fot5 could be maintained in F. graminearum and F. verticillioides but not Fot3 and Fot6, which have been lost. A short fragment corresponding to a Fot3 relic has been detected in F. graminearum (data not shown), arguing for this hypothesis. Concerning the Fot2 family, the species-specific clades indicate that amplification occurred after speciation, a situation compatible with two scenarios. Fot2 was present in the common ancestor of the four species but, in contrast with the Fot3, Fot5, and Fot6 families, distinct subfamilies have been maintained in the genomes of N. haematococca and F. oxysporum, hence becoming species-specific, while old copies have been lost. Nevertheless, it cannot be excluded that horizontal transfer of Fot2 followed by amplification has occurred in one or both species after they diverged.
Even if Fot elements were most likely present in the common ancestor, our analyses showed that Fot diversity has only been maintained in F. oxysporum. This situation is likely due to different sexual behavior in the four Fusarium species and subsequent variable efficiency of inactivation processes, such as the meiosis-dependent process RIP (Galagan and Selker 2004). In F. graminearum and F. verticillioides, Fot elements have been subjected to a severe form of RIP and highly diverged which prevents their identification thus explaining their paucity in these two genomes. The Fot9 family is an exception with only one ripped copy out of five. Some copies might escape RIP processes or these elements may have been acquired recently enough not to have been subjected yet to these modifications. In N. haematococca, RIP appears milder than in F. graminearum and F. verticillioides, thus allowing the maintenance of potentially active copies.
In F. oxysporum, albeit the absence of sexual reproduction, a few copies exhibiting hallmarks of RIP could be identified. RIP has already been described in this species for some impala elements (Hua-Van et al. 2001). Intriguingly, for the Fot elements, most of the ripped copies are located in a single region of the genome which, even if part of a core chromosome, belongs to the LS regions. A more detailed examination of all Fot elements in this region revealed the presence of interspersed ripped and non-ripped copies, with a much higher proportion of copies exhibiting traces of RIP. These observations could be explained by a transfer of the whole corresponding chromosomal region from a species in which RIP is active and thus carrying ripped elements, followed by novel insertions of F. oxysporum Fots through transposition, giving rise to the discontinuous pattern of ripped versus non-ripped copies. The occurrence of such horizontal transfer of chromosomal regions has already been hypothesized to account for the simultaneous presence of two TEs, Fot1 and Hop, in both F. oxysporum and distantly related Fusarium species (Daboussi et al. 2002; Chalvet et al. 2003).
This mechanism is not irrelevant when considering the genomic structure of F. oxysporum as well as of N. haematococca. Indeed, both of these genomes have been found to contain regions exhibiting hallmarks of B chromosomes, namely the LS regions for F. oxysporum (Ma et al. 2010) and the CD chromosomes for N. haematococca (Coleman et al. 2009). B chromosomes are defined as extra chromosomes to the standard complement that occur in many organisms and which are usually characterized by a higher content in repetitive sequences as well as in genes involved in adaptation (Camacho 2005). Horizontal transfer of such chromosomes has been frequently hypothesized or evidenced (Camacho 2005), in particular in fungal species (for a review, see Mehrabi et al. 2011). Recently, the transfer of an entire LS chromosome between two strains of F. oxysporum has been experimentally demonstrated and shown to lead to viable individuals (Ma et al. 2010).
This hypothesis provides an explanation to the apparent contradiction of our results with theoretical models (Hickey 1982) that predict that asexual species should contain few elements due to the lack of genetic exchange and the loss of fitness associated to deleterious high amplification bursts. However, in the Fusarium genomes, elements have a better chance to survive in asexual species in which the inactivating RIP process is absent, than in sexual ones. Furthermore, recurrent horizontal transfers may replace genetic exchange by helping propagating elements.
The frequent occurrence of such horizontal transfers of chromosomes during the evolutionary history of the Fusarium genus could thus have been a driving force which, together with strain-specific characteristics such as the existence of more or less efficient RIP, could explain most of the findings on these genomes (Cuomo et al. 2007; Coleman et al. 2009; Ma et al. 2010; this study).
References
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105
Agrios GN (2005) Chapter 11: plant diseases caused by fungi. In: Plant pathology, 5th edn. Academic Press, New York, pp 383–616
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bergemann M, Lespinet O, M’Barek S, Daboussi MJ, Dufresne M (2008) Whole genome analysis of the Fusarium oxysporum mimp superfamily of MITEs and mobilization of a native and an artifical mimps by the impala transposase. J Mol Evol 67:631–642
Camacho JPM (2005) B chromosomes. In: Gregory TR (ed) The evolution of the genome. Elsevier, San Diego, pp 223–286
Casacuberta E, Casacuberta JM, Puigdomènech P, Monfort A (1998) Presence of miniature inverted-repeat transposable elements (MITEs) in the genome of Arabidopsis thaliana: characterisation of the Emigrant family of elements. Nucleic Acids Res 34:5238–5246
Chalvet F, Grimaldi C, Kaper F, Langin T, Daboussi MJ (2003) Hop, an active Mutator-like element in the genome of the fungus Fusarium oxysporum. Mol Biol Evol 20:1362–1375
Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, Wasmann CC, Grimwood J, Schmutz J, Taga M, White GJ, Zhou S et al (2009) The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet 5:e1000618
Covert SF (1998) Supernumerary chromosomes in filamentous fungi. Curr Genet 33:311–319
Cuomo CA, Güldener U, Xu JR, Trail F, Turgeon GB, Di Pietro A, Walton JD et al (2007) The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317:1400–1402
Daboussi MJ, Capy P (2003) Transposable elements in filamentous fungi. Ann Rev Microbiol 57:275–299
Daboussi MJ, Langin T (1994) Transposable elements in the fungal plant pathogen Fusarium oxysporum. Genetica 93:49–59
Daboussi MJ, Langin T, Brygoo Y (1992) Fot1, a new family of fungal transposable elements. Mol Gen Genet 232:12–16
Daboussi MJ, Davière JM, Graziani S, Langin T (2002) Evolution of the Fot1 transposons in the genus Fusarium: discontinuous distribution and epigenetic inactivation. Mol Biol Evol 19:510–520
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 5:113
Enkerli J, Bhatt G, Covert SF (1997) N. haematococca Nht1, a transposable element cloned from a dispensable chromosome in Nectria haematococca. Mol Plant Microb Interact 10:742–749
Espagne E, Lespinet O, Malagnac F, Da Silva C, Jaillon O, Porcel BM, Couloux A, Aury JM, Ségurens B, Poulain J et al (2008) The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol 9:R77
Feschotte C, Mouches C (2000) Evidence that a family of miniature inverted-repeat transposable elements (MITEs) from the Arabidopsis thaliana genome has arisen from a pogo-like DNA transposon. Mol Biol Evol 17:730–737
Feschotte C, Wessler SR (2002) Mariner-like transposases are widespread and diverse in flowering plants. Proc Natl Acad Sci USA 99:280–285
Galagan JE, Selker EU (2004) RIP: the evolutionary cost of genome defense. Trends Genet 20:417–423
Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHugh W, Ma LJ, Smirnov S, Purcell S et al (2005a) The genome sequence of the filamentous fungus Neurospora crassa. Nature 422:859–868
Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, Lee SI, Baştürkmen M, Spevak CC, Clutterbuck J et al (2005b) Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438:1105–1115
Guermonprez H, Loot C, Casacuberta JM (2008) Different strategies to persist: the pogo-like Lemi1 transposon produces miniature inverted-repeat transposable elements or typical defective elements in different plant genomes. Genetics 180:83–92
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Gurr SJ, Unkles SE, Kinghorn JR (1987) The structure and organization of nuclear genes for filamentous fungi. In: Kinghorn JR (ed) Gene structure in eukaryotic microbes. Academic Press, Oxford, pp 93–139
Hickey DA (1982) Selfish DNA: a sexually-transmitted nuclear parasite. Genetics 101:519–531
Hua-Van A, Davière JM, Langin T, Daboussi MJ (2000) Genome organization in Fusarium oxysporum: clusters of class II transposons. Curr Genet 37:339–347
Hua-Van A, Langin T, Daboussi MJ (2001) Evolutionary history of the impala transposon in Fusarium oxysporum. Mol Biol Evol 18:1959–1969
Jiang N, Wessler SR (2000) Insertion preference of maize and rice miniature inverted-repeat transposable elements as revealed by the analysis of nested elements. Plant Cell 13:2553–2564
Kang S, Lebrun MH, Farrall L, Valent B (2001) Gain of virulence caused by insertion of a Pot3 transposon in a Magnaporthe grisea avirulence gene. Mol Plant Microb Interact 14:671–674
Kito H, Takahashi Y, Sato J, Fukiya S, Sone T, Tomita F (2003) Occan, a novel transposon in the Fot1 family, is ubiquitously found in several Magnaporthe grisea isolates. Curr Genet 42:322–331
Levis C, Fortini D, Brygoo Y (1997) Flipper, a mobile Fot1-like transposable element in Botrytis cinerea. Mol Gen Genet 254:674–680
Ma LJ, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B et al (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium oxysporum. Nature 464:367–373
Margolin BS, Garrett-Engele PW, Stevens JN, Fritz DY, Garrett-Engele C, Metzenberg RL, Selker EU (1998) A methylated Neurospora 5S rRNA pseudogene contains a transposable element inactivated by repeat-induced point mutation. Genetics 149:1787–1797
Mehrabi R, Bahkali AH, Abd-Elsalam KA, Moslem M, Ben M’Barek S, Mirzadi Gohari A, Karimi Jashni M, Stergiopoulos I, Kema GHJ, de Wit PJGM (2011) Horizontal gene and chromosome transfer in plant pathogenic fungi affecting host range. FEMS Microbiol Rev 35:542–554
Migheli Q, Laugé R, Davière JM, Gerlinger C, Kaper F, Langin T, Daboussi MJ (1999) Transposition of the autonomous Fot1 element in the filamentous fungus Fusarium oxysporum. Genetics 151:1005–1013
Monroy F, Sheppard DC (2005) Taf1: a class II transposon of Aspergillus fumigatus. Fungal Genet Biol 42:638–645
Neafsey DE, Barker BM, Sharpton TJ, Stajich JE, Park DJ, Whiston E, Hung CY, McMahan C, White J, Sykes S et al (2010) Population genomic sequencing of Coccidioides fungi reveals recent hybridization and transposon control. Genome Res 20:938–946
Nowrousian M, Stajich JE, Chu M, Engh I, Espagne E, Halliday K, Kamerewerd J, Kempken F, Knab B, Kuo HC et al (2010) De novo assembly of a 40 Mb eukaryotic genome from short sequence reads: Sordaria macrospora, a model organism for fungal morphogenesis. PLos Genet 6:e1000891
Nyyssönen E, Amutan M, Enfield L, Stubbs J, Dunn-Coleman NS (1996) The transposable element Tan1 of Aspergillus niger var. awamori, a new member of the Fot1 family. Mol Gen Genet 253:50–56
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Rep M, Kistler HC (2010) The genomic organization of plant pathogenicity in Fusarium species. Curr Opin Plant Biol 13:420–426
Rep M, van der Does HC, Meijer M, van Wijk R, Houterman PM, Dekker HL, de Koster CG, Cornelissen BJ (2004) A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol Microbiol 53:1373–1383
Rice P, Logden I, Bleasby A (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277
Samuels GJ, Nirenberg HI, Seifert KA (2001) Perithecial species of Fusarium. In: Sumerell BA, Leslie JF, Backhouse D, Bryden WL, Burgess LW (eds) Fusarium, Paul E. Nelson memorial symposium. APS Press, pp 1–14
Santiago N, Herraiz C, Goni JR, Messeguer X, Casacuberta JM (2002) Genome-wide analysis of the Emigrant family of MITEs of Arabidopsis thaliana. Mol Biol Evol 19:2285–2293
Smit AF, Riggs AD (1996) Tiggers and DNA transposon fossils in the human genome. Proc Natl Acad Sci USA 93:1443–1448
Swofford DL (2002) PAUP*: phylogenetic analysis using parsimony (* and other methods). Ver. 4.0b10. Sinauer Associates
Thon MR, Pan H, Diener S, Papalas J, Taro A, Mitchell TK, Dean RA (2006) The role of transposable element clusters in genome evolution and loss of synteny in the rice blast fungus Magnaporthe oryzae. Genome Biol 7:R16
Trouvelot S, Olivain C, Recorbet G, Migheli Q, Alabouvette C (2002) Recovery of Fusarium oxysporum Fo47 mutants affected in their biocontrol activity after transposition of the Fot1 element. Phytopathology 92:936–945
Yuan YW, Wessler SR (2011) The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc Natl Acad Sci USA 108:7884–7889
Zhang Z, Townsend JP (2009) Maximum-likelihood model averaging to profile clustering of site types across discrete linear sequences. PLoS Comput Biol 5:e1000421
Acknowledgments
This study was supported by CNRS funding. We are grateful to Eric Espagne for his advices in some of the statistical analyses and to Pierry Capy, Arnaud Le Rouzic and Hadi Quesneville for helpful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Dufresne, M., Lespinet, O., Daboussi, MJ. et al. Genome-Wide Comparative Analysis of pogo-Like Transposable Elements in Different Fusarium Species. J Mol Evol 73, 230–243 (2011). https://doi.org/10.1007/s00239-011-9472-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-011-9472-1