
Abstract
While molecular analyses have provided insight into the phylogeny of ciliates, the few studies assessing intraspecific variation have largely relied on just a single locus [e.g., nuclear small subunit rDNA (nSSU-rDNA) or mitochondrial cytochrome oxidase I]. In this study, we characterize the diversity of several nuclear protein-coding genes plus both nSSU-rDNA and mitochondrial small subunit rDNA (mtSSU-rDNA) of five isolates of the ciliate morphospecies Chilodonella uncinata. Although these isolates have nearly identical nSSU-rDNA sequences, they differ by up to 8.0% in mtSSU-rDNA. Comparative analyses of all loci, including β-tubulin paralogs, indicate a lack of recombination between strains, demonstrating that the morphospecies C. uncinata consists of multiple cryptic species. Further, there is considerable variation in substitution rates among loci as some protein-coding domains are nearly identical between isolates, while others differ by up to 13.2% at the amino acid level. Combining insights on macronuclear variation among isolates, the focus of this study, with published data from the micronucleus of two of these isolates, indicates that C. uncinata lineages are able to maintain both highly divergent and highly conserved genes within a rapidly evolving germline genome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Patterns of molecular evolution among ciliates are known to be shaped by their unusual genome architecture, namely the presence of both germline and somatic nuclei within each cell (McGrath et al. 2006; Zufall et al. 2006). To date, most studies of intraspecific genetic variation in ciliates typically rely on just a single locus, with the notable exception of the model ciliates Paramecium tetraurelia and Tetrahymena thermophila (e.g., Catania et al. 2009; Simon et al. 2008). Yet analyses of several genes from the somatic genome, coupled with data from the germline genome, are needed to assess intraspecific patterns of molecular evolution in ciliates. In light of previous data suggesting both high and low levels of gene conservation in two geographically isolated strains of C. uncinata (Robinson and Katz 2008) and high divergence in the germline genome of the same two strains (Katz and Kovner 2010), we aimed to determine the level of intraspecific variation in protein-coding loci of additional strains. This allowed us to test the hypotheses that the somatic macronucleus is marked by heterogeneous patterns of molecular evolution and that C. uncinata is composed of multiple genetically distinct cryptic species.
The genetics of ciliates must be interpreted in light of the presence of both a germline micronucleus and somatic macronucleus within each cell. The smaller micronucleus, which is not transcribed, resembles a typical eukaryotic genome in that it has few large chromosomes. The transcriptionally active macronucleus contains chromosomes modified from a zygotic nucleus (Juranek and Lipps 2007; Zufall et al. 2006). Macronuclear chromosomes are generated through extensive processing that includes fragmentation, elimination of internal excised sequences, and amplification (McGrath et al. 2006). Extensive processing in some ciliate classes—Spirotrichea, Armophorea, and Phyllopharyngea (including Chilodonella uncinata, the focus of this study)—produces approximately 20,000,000 gene-sized chromosomes (i.e., each macronuclear chromosome contains only a single gene) (Juranek and Lipps 2007; Zufall et al. 2006).
Maintenance of two dimorphic genomes allows ciliates to explore protein space in novel ways compared to other eukaryotes (McGrath et al. 2006; Zufall et al. 2006). For example, ciliates have remarkably divergent histone H4 genes compared to other eukaryotes, with some lineages having multiple paralogs that differ by up to 20% of their amino acids (Katz et al. 2004). Moreover, ciliates with highly processed genomes have greater variation in protein-coding gene families across multiple loci than do ciliates that do not extensively process their somatic genomes (Zufall et al. 2006). These insights are based on comparisons among species and little is known about patterns of protein evolution within ciliate species.
The presence of cryptic species underlying conserved morphospecies—species identified by shared morphology—has been found across the ciliate tree of life. Cryptic species—defined here as morphospecies that contain multiple biological and/or genetic species—were discovered in Tetrahymena and Paramecium as strains within morphospecies that are reproductively isolated from one another (Nanney 1999; Sonneborn 1937, 1957). Some cryptic species have been shown to be ecologically distinct (e.g., differential prey and/or abiotic factors; Weisse and Lettner 2002; Weisse et al. 2008), indicating that elucidating cryptic diversity is important for studies focusing on topics such as food webs and species interactions.
Application of molecular tools has revealed extensive genetic variation underlying morphospecies (Barth et al. 2006; Gentekaki and Lynn 2009; Katz et al. 2005; Nanney et al. 1998; Simon et al. 2008). For example, individuals from multiple populations of both Paramecium caudatum and Paramecium micronucleatum have nearly identical internal transcribed spacer regions of the rDNA locus (i.e., low divergence within species for this locus), but reveal considerably more diversity based on mitochondrial encoded cytochrome oxidase 1 (Barth et al. 2006). In contrast, there are few examples of widely sampled ciliate morphospecies that appear to lack underlying genetic diversity; e.g., Laboea strobila and Pelagostrobilidium neptuni (Doherty et al. 2010; Katz et al. 2005).
Here, we assess genetic data from five strains of the morphospecies C. uncinata: three recently isolated strains are morphologically identical (at least by light microscopy) to two strains, one from the American Type Culture Collection (USA-ATCC) and the other from Poland (POL), that were identified as C. uncinata by ciliate taxonomist Wilhelm Foissner, University of Salzburg (Robinson and Katz 2008). We were motivated by the observation that the strains USA-ATCC and POL have nearly identical nSSU-rDNA, and actin sequences, and both conserved and divergent macronuclear β-tubulin sequences (Robinson and Katz 2008). Moreover, analyses of micronuclear sequences from these two strains reveal that macronuclear-destined sequences (e.g., protein-coding domains) are fragmented (Zufall and Katz 2007) and embedded within rapidly evolving germline-specific sequences (Katz and Kovner 2010). To test whether the patterns of molecular evolution in macronuclear genes extend to other strains and loci, we analyzed macronuclear sequences of mtSSU-rDNA, nSSU-rDNA, actin, α-tubulin, and β-tubulin from all five isolates and assessed patterns of substitution across loci.
Methods
Ciliate Culturing
Three new isolates of C. uncinata are from within the USA: USA-WH was acquired from Woods Hole, MA, and USA-SC1 and USA-SC2 were both collected from Lyman Pond on the Smith College campus in Northampton, MA (Table 1). The two isolates that had been previously characterized are both available at ATCC (USA = ATCC®50194, POL = ATCC®PRA-256), as reported in Robinson and Katz (2008). All isolates were cultured at room temperature in the dark, and maintained in filtered and autoclaved water from a local pond, with a rice grain added to maintain bacterial populations. Clonal lines were generated for each isolate by passing a single cell through three rounds of isolation. Lines were transferred to fresh media every other week. To isolate DNA, cells were pelleted by spinning at 5,000 rpm for 20 min, and DNA was extracted using phenol–chloroform following Riley and Katz (2001).
Characterization of Mitochondrial and Nuclear Genes
All genes were amplified using Phusion Hot Start High Fidelity DNA Polymerase (Finnzymes F 540L). Nuclear SSU-rDNA (nSSU-rDNA), α-tubulin, β-tubulin, and actin were amplified using primers described previously (Robinson and Katz 2008), while mitochondrial SSU-rDNA (mtSSU-rDNA) was amplified using primers described in Dunthorn et al. (2011). Amplified products were cloned using Zero Blunt TOPO kits (Invitrogen 42-0245), and screened using the polymerase TaqGold. Clones were miniprepped using Invitrogen’s 96-well format harvested-cell method, protocol B. Several steps were taken to avoid contamination: only one strain was worked on at any one time; PCRs were set up in a cleaned hood; reported sequences were generally obtained from pooled PCR reactions; and we discarded any reactions for which a negative control turned up positive.
As we aimed to characterize paralogs from PCRs using degenerate primers, we sequenced multiple clones per reaction (~8 initially, with more added, as paralogs were discovered) in just one direction and then fully sequenced at least two representatives of each unique sequence. Sequences were generated using the BigDye terminator RR mix from PE Applied Biosystems (Wellesley, MA, 4303152). Reactions were cleaned using gel filtration columns from Edge Biosystems (Gaithersburg, MD) and analyzed on a PerkinElmer ABI-3100 automated sequencer at the Center for Molecular Biology (Smith College, Northampton, MA). Additional sequencing reactions and sequencing were performed at the Penn State Genomics Core Facility (University Park, PA).
Sequence Analysis
Contigs were assembled in SeqMan (DNAStar), and annotated using Seqbuilder (DNAStar). MacClade (Maddison and Maddison 2005) and MegAlign (DNAStar) were used to create alignments. Genealogies based on nucleotide alignments were estimated using PhyML (Guindon and Gascuel 2003) as implemented in Seaview v. 4.2.4 (Gouy et al. 2010), optimizing the number of invariant sites and using 6 rate classes. We analyzed codon bias by measuring the effective number of codons (Nc) (Wright 1990) using the program CodonW (Peden 2005). For the β-tubulin gene family, we found several sequences that appeared to be PCR recombinants in that they occurred only in single PCR reactions. These recombinants were detected by eye and using GARD (Kosakovsky Pond et al. 2006), and the chimeric sequences were removed from subsequent analyses.
Synonymous and nonsynonymous substitution rates were estimated for the model of Muse and Gaut (1994) using HyPhy (Kosakovsky Pond et al. 2005). Estimates for the conserved paralogs SP1 and SP2 were compared to corresponding estimates for the divergent P3 paralogs across lines.
Results
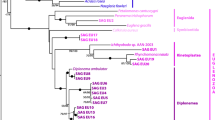
Levels of conservation differ considerably between nSSU-rDNA and mtSSU-rDNA sequences among the five isolates. The isolates share nearly identical nSSU-rDNA sequences: the two haplotypes found (one shared by POL, USA-SC1, and USA-SC2; and the other shared by USA-ATCC and USA-WH) differ by only four single nucleotide polymorphisms (SNPs) across a 1 kb region (Table 3; Fig. 1a). Unlike the nSSU-rDNA, the mtSSU-rDNA gene is highly divergent among the five isolates, with the sequence from USA-SC2 differing by up to 8.0% (nucleotide differences; Table 3; Fig. 1b). POL and USA-SC1, which share an identical nSSU-rDNA locus, also have identical mtSSU-rDNA genes, and USA-ATCC and USA-WH are more similar to each other than either is to USA-SC2 (Fig. 1b).

Actin sequences are highly similar within all five isolates of C. uncinata. We examined between three and six clones in each of the isolates characterized for this study (SC-1, SC-2 and WH) and found that each contained a single sequence differing from one another by <3% at the nucleotide level (Tables 2 and 3). The two lines POL and USA-SC1 share identical sequences and the two lines USA-ATCC and USA-WH are very similar (Fig. 1c). Actins from all five isolates are nearly identical at the amino acid level (Table 3). Again, the USA-SC2 sequence is the most divergent, differing from the other isolates by as much as 2.3% of its nucleotides and by three amino acid substitutions (Table 3).
Alpha-tubulin sequences are also highly conserved across all isolates. In each of USA-SC2, USA-SC1, and USA-WH, we identified only one sequence. These sequences are highly similar at the nucleotide level and identical at the amino acid level (Table 3). In a previous study (Israel et al. 2002), we identified two α-tubulin sequences in USA-ATCC. One of these two paralogs, found less frequently, was highly divergent in some regions from the conserved sequences analyzed in this study. Because we did not find any similar sequences in POL, USA-SC1, USA-SC2, or USA-WH, we did not include this previously published sequence in the analyses here.
The β-tubulin sequences in our three new isolates are similar to those previously characterized by Robinson and Katz (2008). The clonal lines contains a subset of five β-tubulin paralogs: SP1, SP2, P1, P2, and P3 (Table 2; Fig. 2), with the P3 paralogs being most divergent (up to 24% from the conserved SP1/SP2 amino acids sequences). Genealogical analyses indicate that all the haplotypes fall into several major clades and that the topology of each gene family member is consistent with that of the mtSSU-rDNA phylogeny (Fig. 2): USA-SC1 and POL share identical sequences, USA-ATCC and USA-WH are similar, and USA-SC2 is the most divergent at the nucleotide and amino acid levels (Figs. 1 and 2).

Patterns of Substitutions
We calculated the effective number of codons for protein-coding genes using codonW (Wright 1990). Codon bias is variable across protein-coding genes, with more highly conserved haplotypes having a lower effective number of codons (Table 3). Actin, α-tubulin, and two β-tubulin paralogs (SP1 and SP2), use approximately 27–29 effective codons while the β-tubulin P3 paralogs use nearly double that with 52–54 codons.
To assess the patterns of substitutions further, we analyzed the ratios of dN/dS between the conserved SP1/SP2 β-tubulin paralogs and the more divergent P3 sequences. The clade-based estimate of dN/dS for the SP1/SP2 is 0 while that of the P3 clade is 0.050, suggesting a difference in selection intensity between the P3 and SP1/SP2 lineages. We also assessed patterns along individual branches of the β-tubulin topology, though this was not straightforward as not all paralogs were isolated from all lines (for example, we did not find a P3 sequence in USA-SC2). Although variances of the rate estimates are high, estimates of both synonymous and nonsynonymous rates for individual P3 branches are roughly an order of magnitude higher than those for their corresponding SP1 and SP2 branches (Table 4).
Discussion
Analyses of multiple molecular markers isolated from macronuclear and mitochondrial genomes lead to two complementary insights: (1) the morphospecies C. uncinata is composed of multiple genetically distinct cryptic species; and (2) these species maintain both highly conserved and highly divergent loci in the macronucleus, while germline-restricted (micronuclear) sequences for two of the isolates have so many substitutions that they are unalignable (Katz and Kovner 2010).
Cryptic Species of the Morphospecies C. uncinata
Several lines of evidence suggest that there are multiple cryptic species underlying the morphospecies C. uncinata including the high divergence among some loci and the lack of evidence of recombination within or between loci. Four of the five isolates of C. uncinata that we characterized are genetically distinct at mtSSU-rDNA and in protein-coding loci (e.g., the divergent β-tubulin P3 locus; Table 3; Figs. 1, 2). The level of divergence among isolates varies across loci, with the deepest divergence varying from 2.2% for actin to 8.0% for mtSSU to 13.5% for β-tubulin P3 (Table 3). Moreover, we see no evidence of recombination within loci or independent assortment among genes. The lack of recombination indicates that the four genetically distinct C. uncinata—USA-ATCC, USA-SC2, USA-WH and (POL + USA-SC1)—are reproductively isolated. Intriguingly, the most divergent line, USA-SC2, is sympatric with a second isolate, USA-SC1, which in turn is genetically identical to an isolate from Poland. While we took considerable care to eliminate contamination in PCR reactions, we recognize that the identity between the Pol and USA-SC1 lines may still be due to contamination of either molecular data or of the ciliate cultures. Hence, though more collections are needed to assess biogeographical patterns further, our data indicate that the geographic distribution of the cryptic species is complex, involving both sympatry and allopatry.
We propose mtSSU-rDNA as a tool for rapidly identifying cryptic species underlying ciliate morphospecies. Mitochondrial loci, primarily cytochrome c oxidase, have been shown to be effective in assessing intra-morphospecies variation in various ciliate clades (Barth et al. 2008; Catania et al. 2009; Chantangsi et al. 2007; Gentekaki and Lynn 2009; Snoke et al. 2006; Strüder-Kypke and Lynn 2010). To date, mtSSU-rDNA has only been used to assess phylogenetic relationships for deeper nodes in the Colpodea (Dunthorn et al. 2011). Yet, mtSSU-rDNA primers can readily be used to assess the nature of diverse ciliate morphospecies (Dunthorn et al. 2011).
Divergence Among Loci
The cryptic species of C. uncinata maintain both highly conserved and highly divergent macronuclear coding domains. This pattern is particularly striking as micronuclear-specific sequences, at least between USA-ATCC and POL, are evolving at such a rapid rate that they can not be aligned. For example, internal eliminated sequences in the actin gene are more than 40% divergent (Katz and Kovner 2010), while the coding region is ≤2.2% divergent (Table 3; Fig. 1c). The heterogeneity in the levels of divergence among loci is reflected in patterns of codon usage, with more highly conserved genes showing more codon bias as measured by the effective numbers of codons (Wright 1990).
Further evidence for heterogeneity in patterns of evolution is seen in comparing the β-tubulin P3 paralogs to the conserved SP1/SP2 paralogs. These sequences arose by gene duplication within C. uncinata, yet the subsequent patterns of substitutions have varied such that the ratio of nonsynonymous to synonymous changes is higher for P3 sequences compared among strains (dN/dS = 0.05) as compared to the SP1/SP2 sequences (dN/dS = 0). Yet, even the highly divergent P3 sequences (differ by ~24% amino acids from SP1/SP2) remain under some form of constraint, as the dN/dS for this clade is still much less than the value of 1.0 expected for pseudogenes. Moreover, the individual estimates of synonymous rates vary across lineages, with the P3 sequence having at least 10× higher rates of synonymous substitutions (Table 4).
Our data suggest that selection on coding domains in ciliates can be very strong over long evolutionary times. Even though sufficient time has elapsed for substitutions to render germline-specific sequences unalignable (Katz and Kovner 2010), coding domains at some loci (e.g. actin, β-tubulin SP1/SP2) remain conserved even to the level of eliminating most silent site mutations (Figs. 1, 2; Table 2). Within the same genomic context, some protein-coding genes evolve rapidly, such as β-tubulin-P3, and perhaps take on a new function. These observations are consistent with our hypothesis that the genome architecture of ciliates enables these lineages to explore protein space in novel manners (Katz and Kovner 2010; McGrath et al. 2006; Zufall et al. 2006).
Synthesis
The observation of elevated rates of evolution of a mitochondrial locus compared to most nuclear loci (the exception being the highly divergent β-tubulin paralog P3; Table 2) among lineages of the ciliate morphospecies C. uncinata is not surprising given elevated mutation rates in mitochondria in many taxa (Lynch 2007). More unusual, though, is the distinction in patterns of evolution between germline-limited (micronuclear) and somatic (macronuclear) sequences. Here, we document considerable conservation in coding domain, even among the silent sites within codons, while germline-limited sequences between two of the five cryptic species are so divergent that they are unalignable (Katz and Kovner 2010). These observations extend the comparisons of ciliate genome processing (i.e., between germline and somatic genomes) to the chromosomal rearrangements in somatic B and T cells that generate diversity in the adaptive immune systems of some animals (Andersson et al. 2006; Herrick 1994; Kloc and Zagrodzinska 2001; Zufall et al. 2005). In contrast to adaptive immunity, where chromosomal rearrangements are associated with somatic hypermutation (Ginger et al. 2010; Zmasek and Godzik 2011), we observe conservation in coding domains (i.e., somatic sequences) embedded within rapidly evolving germline sequences.
References
Andersson JO, Hirt RP, Foster PG, Roger AJ (2006) Evolution of four gene families with patchy phylogenetic distributions: influx of genes into protist genomes. Bmc Evol Biol 6:18
Barth D, Krenek S, Fokin SI, Berendonk TU (2006) Intraspecific genetic variation in Paramecium revealed by mitochondrial cytochrome c oxidase I sequences. J Eukaryot Microbiol 53:20–25
Barth D, Tischer K, Berger H, Schlegel M, Berendonk TU (2008) High mitochondrial haplotype diversity of Coleps sp. (Ciliophora: Prostomatida). Environ Microbiol 10:626–634
Catania F, Wurmser F, Potekhin AA, Przybos E, Lynch M (2009) Genetic diversity in the Paramecium aurelia species complex. Mol Biol Evol 26:421–431
Chantangsi C, Lynn DH, Brandl MT, Cole JC, Hetrick N, Ikonomi P (2007) Barcoding ciliates: a comprehensive study of 75 isolates of the genus Tetrahymena. Int J Syst Evol Microbiol 57:2412–2425
Doherty M, Tamura M, Costas BA, Ritchie ME, McManus GB, Katz LA (2010) Ciliate diversity and distribution across an environmental and depth gradient in Long Island Sound, USA. Environ Microbiol 12:886–898
Dunthorn M, Foissner W, Katz LA (2011) Expanding character sampling for ciliate phylogenetic inference using mitochondrial SSU-rDNA as a molecular marker. Protist 162:85–99
Gentekaki E, Lynn DH (2009) High-level genetic diversity but no population structure inferred from nuclear and mitochondrial markers of the peritrichous ciliate Carchesium polypinum in the Grand River Basin (North America). Appl Environ Microbiol 75:3187–3195
Ginger ML, Fritz-Laylin LK, Fulton C, Cande WZ, Dawson SC (2010) Intermediary metabolism in protists: a sequence-based view of facultative anaerobic metabolism in evolutionarily diverse eukaryotes. Protist 161:642–671
Gouy M, Guindon S, Gascuel O (2010) SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Herrick G (1994) Germline-soma relationships in ciliates protozoa: the inception and evolution of nuclear dimorphism in one-celled animals. Semin Dev Biol 5:3–12
Israel RL, Kosakovsky Pond SL, Muse SV, Katz LA (2002) Evolution of duplicated alpha-tubulin genes in ciliates. Evolution 56:1110–1122
Juranek SA, Lipps HJ (2007) New insights into the macronuclear development in ciliates. Int Rev Cytol 262:219–251
Katz LA, Kovner AM (2010) Alternative processing of scrambled genes generates protein diversity in the ciliate Chilodonella uncinata. J Exp Zool B 314:480–488
Katz LA, Bornstein J, Lasek-Nesselquist E, Muse SV (2004) Dramatic diversity of ciliate histone H4 genes revealed by comparisons of patterns of substitutions and paralog divergences among eukaryotes. Mol Biol Evol 21:555–562
Katz La, McManus GB, Snoeyenbos-West OLO, Griffin A, Pirog K, Costas B, Foissner W (2005) Reframing the ‘Everything is everywhere’ debate: evidence for high gene flow and diversity in ciliate morphospecies. Aquat Microb Ecol 41:55–65
Kloc M, Zagrodzinska B (2001) Chromatin elimination–an oddity or a common mechanism in differentiation and development? Differentiation 68:84–91
Kosakovsky Pond SL, Frost SDW, Muse SV (2005) HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679
Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SDW (2006) GARD: a genetic algorithm for recombination detection. Bioinformatics 22:3096–3098
Lynch M (2007) Origins of genome architecture. Sinauer Associates, Sunderland
Maddison WP, Maddison DR (2005) MacClade v. 4.0.8. Sinauer Assoc, Sunderland
McGrath C, Zufall RA, Katz LA (2006) Genome evolution in ciliates. In: Katz LA, Bhattacharya D (eds) Genomics and evolution of eukaryotic microbes. Oxford University Press, New york
Muse SV, Gaut BS (1994) A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates with application to the chloroplast genome. Mol Biol Evol 11:715–724
Nanney DL (1999) When is a rose?: the kinds of Tetrahymena. In: Wilson RA (ed) Species: new interdisciplinary essays. The MIT Press, Cambridge, pp 93–118
Nanney DL, Park C, Preparata R, Simon EM (1998) Comparison of sequence differences in a variable 23S rRNA domain among sets of cryptic species of ciliated protozoa. J Euk Microbiol 45:91–100
Peden J (2005) CodonW version 1.4.2. http://codonw.sourceforge.net/culong.html#License
Riley JL, Katz LA (2001) Widespread distribution of extensive genome fragmentation in ciliates. Mol Biol Evol 18:1372–1377
Robinson T, Katz LA (2008) Non-Mendelian inheritance of two cytoskeletal genes in the ciliate Chilodonella uncinata. Mol Biol Evol 24:2495–2503
Simon EM, Nanney DL, Doerder FP (2008) The “Tetrahymena pyformis” complex of cryptic species. Biodiv Conserv 17:365–380
Snoke MS, Berendonk TU, Barth D, Lynch M (2006) Large global effective population sizes in Paramecium. Mol Biol Evol 23:2474–2479
Sonneborn TM (1937) Sex, sex inheritance and sex determination in Paramecium aurelia. Proc Natl Acad Sci USA 23:378–383
Sonneborn TM (1957) Breeding systems, reproductive methods, and species problems in protozoa. In: Mayr E (ed) The species problem. American Association for the Advancement of Science, Washington DC, pp 155–324
Strüder-Kypke MC, Lynn DH (2010) Comparative analysis of the mitochondrial cytochrome c oxidase subunit 1 (CO1) gene in ciliates (Alveolata, Ciliophora) and evaluation of its suitability as a biodiversity marker. Syst Biodivers 8:131–148
Weisse T, Lettner S (2002) The ecological significance of intraspecific variation among freshwater ciliates. Verh Intern Verein Limnol 28:1880–1884
Weisse T, Strüder-Kypke MC, Berger H, Foissner W (2008) Genetic, morphological, and ecological diversity of spatially separated clones of Meseres corlissi Petz & Foissner, 1992 (Ciliophora, Spirotrichea). J Eukaryot Microbiol 55:257–270
Wright F (1990) The ‘effective number of codons’ used in a gene. Gene 87:23–29
Zmasek CM, Godzik A (2011) Strong functional patterns in the evolution of eukaryotic genomes revealed by the reconstruction of ancestral protein domain repertoires. Genome Biol 12:13
Zufall RA, Katz LA (2007) Micronuclear and macronuclear forms of beta-tubulin genes in the ciliate Chilodonella uncinata reveal insights into genome processing and protein evolution. J Eukaryot Microbiol 54:275–282
Zufall RA, Robinson T, Katz LA (2005) Evolution of developmentally regulated genome rearrangements in eukaryotes. J Exp Zool Part B-Mol Dev Evol 304B:448–455
Zufall RA, McGrath C, Muse SV, Katz LA (2006) Genome architecture drives protein evolution in ciliates. Mol Biol Evol 23:1681–1687
Acknowledgments
This work was supported by two grants to L.A.K.: NSF DEB award (DEB 0816828) and an NIH AREA award 1R15GM081865-01.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Katz, L.A., DeBerardinis, J., Hall, M.S. et al. Heterogeneous Rates of Molecular Evolution Among Cryptic Species of the Ciliate Morphospecies Chilodonella uncinata . J Mol Evol 73, 266–272 (2011). https://doi.org/10.1007/s00239-011-9468-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-011-9468-x