
Abstract
We have used three independent phylogenomic approaches (concatenated alignments, single-, and multi-gene supertrees) to reconstruct the fungal tree of life (FTOL) using publicly available fungal genomes. This is the first time multi-gene families have been used in fungal supertree reconstruction and permits us to use up to 66% of the 1,001,217 genes in our fungal database. Our analyses show that different phylogenomic datasets derived from varying clustering criteria and alignment orientation do not have a major effect on phylogenomic supertree reconstruction. Overall the resultant phylogenomic trees are relatively congruent with one another and successfully recover the major fungal phyla, subphyla and classes. We find that where incongruences do occur, the inferences are usually poorly supported. Within the Ascomycota phylum, our phylogenies reconstruct monophyletic Saccharomycotina and Pezizomycotina subphyla clades and infer a sister group relationship between these to the exclusion of the Taphrinomycotina. Within the Pezizomycotina subphylum, all three phylogenies infer a sister group relationship between the Leotiomycetes and Sordariomycetes classes. However, there is conflict regarding the relationships with the Dothideomycetes and Eurotiomycetes classes. Within the Basidiomycota phylum, supertrees derived from single- and multi-gene families infer a sister group relationship between the Pucciniomycotina and Agaricomycotina subphyla while the concatenated phylogeny infers a poorly supported relationship between the Agaricomycotina and Ustilagomycotina. The reconstruction of a robust FTOL is important for future fungal comparative analyses. We illustrate this point by performing a preliminary investigation into the phyletic distribution of yeast prion-like proteins in the fungal kingdom.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The fungal kingdom makes up one of the main domains of the eukaryotic tree of life. The exact number of fungal species is unknown but it is estimated to be 1.5 million (Hawksworth 1991, 2001). The fungal fossil record is poor with the oldest fossils dating back 600 million years (Yuan et al. 2005). However, molecular clock studies estimate the origin of the fungi at approximately 1.5 billion years ago (Heckman et al. 2001).
Until recently evolutionary relationships among fungi were poorly understood (Guarro et al. 1999). This was due to their simple morphology, poor fossil record, and high degree of biological and physiological diversity (Guarro et al. 1999). Traditional studies of fungal evolution relied on morphology, sexual states, cell wall composition, cytological testing, ultrastructure, and metabolism (Guarro et al. 1999). More recently molecular phylogenetic analyses have successfully revealed that there are at least seven distinct phyla within the fungal kingdom (Hibbett et al. 2007; James et al. 2006b) these include the Chytridiomycota, Blastocladiomycota, Glomeromycota, Microsporidia, Neocallimastigomycota, Ascomycota, and Basidiomycota. Taxa traditionally placed in Zygomycota phylum are now distributed among the Glomeromycota and several incertae sedis subphyla, including Mucoromycotina, Entomophthoromycotina, Kickxellomycotina, and Zoopagomycotina (Hibbett et al. 2007).
Saccharomyces cerevisiae was the first eukaryote to have its genome completely sequenced (Goffeau et al. 1996). Because of their relative small genome size, roles as human/crop pathogens and importance in the field of biotechnology, 102 fungal species have been since sequenced to date (Supplementary file 1), accounting for approximately 40% of eukaryotic genomic data currently available. This abundance of data has moved the fungal kingdom to the forefront of eukaryotic genomics. While some of the species sequenced are closely related, others have diverged over 1 billion years ago. This enables us to use fungi to study evolutionary mechanisms associated with eukaryotic genome structure, organization, and content. Furthermore it permits us to undertake comparative analysis into fungal virulence (Butler et al. 2009; Faris et al. 2010), evolution (Fitzpatrick et al. 2008), metabolic capabilities (Fitzpatrick et al. 2010), and fate of genes that have arisen through duplication (Scannell et al. 2006). However to fully understand fungal evolution and associated biological processes it is essential we have a reliable fungal tree of life (FTOL).
Initially, the majority of fungal phylogenies were derived from individual ribosomal genes (Lutzoni et al. 2004). However, phylogenies derived from single-genes (SGs) may not be reliable as they may contain too few sites and therefore fail to resolve deep branches. Furthermore, SGs do not always correlate with vital physiological processes or basic adaptive strategies. Recently phylogenomic approaches such as multi-gene concatenation (supermatrix) and supertree methods have been successful in addressing relationships among diverse fungal species (Fitzpatrick et al. 2006; Kuramae et al. 2006; Liu et al. 2009; Marcet-Houben and Gabaldon 2009; Robbertse et al. 2006).
Supertree methods take a set of phylogenetic trees as input and return one or more phylogenetic trees that represent the input trees. Supertrees have many advantages including the capacity to use single and multi-gene families, the ability to analyze each gene individually using the best fitting substitution model and reduced computation time in the reconstruction of large species phylogenies (Holton and Pisani 2010). Disadvantages include the potential for species phylogenies derived from relatively small alignments resulting in significant statistical errors in the phylogenomic supertree (Holton and Pisani 2010). These effects can be minimized, however, using filtering strategies such as the removal of individual gene families that do not contain strong phylogenetic signal (Holton and Pisani 2010). In a supermatrix analysis, SG families are merged into a large multiple sequence alignment that is then analyzed using an appropriate phylogenetic reconstruction method. Supermatrix approaches have the advantage of resolving nodes, basal branches, and improve phylogenetic accuracy (Barrett et al. 1991; Delsuc et al. 2005). Some problems include errors in phylogeny due to systematic biases (e.g., compositional biases) although novel phylogenetic models appear to be adequate at handling these (Lartillot et al. 2007; Lartillot and Philippe 2004). Finally, supermatrix approaches cannot handle multi-gene families meaning the total number of genes being compared can be quite low and not representative of the entire genome (Dagan and Martin 2006). The most robust phylogenomic analyses take a total evidence approach. These endeavor to use all available data (Eernisse and Kluge 1993; Kluge 1989) and cross reference different methodologies (Fitzpatrick et al. 2006).
In this study, we have used a total evidence approach to reconstruct the FTOL using completely sequenced genomes (Supplementary file 1). As well as traditional supertrees derived from SG families, we have also reconstructed the first FTOL supertree that incorporates information from multi-gene families. Genome data for three (Chytridiomycota, Ascomycota, and Basidiomycota) of the seven fungal phyla are available and were analyzed. Three genomes are also available for the incertae sedis subphylum Mucoromycotina, and these were also included in our analysis. The Chytridiomycota is the only fungal phylum to produce zoospores and requires water for their dispersal. They are an ancient group of organisms and are thought to have changed little from early times of eukaryotic evolution. The Ascomycota is the largest fungal phylum accounting for approximately 65% of all know fungal species and includes important biotechnological species such as S. cerevisiae and the human pathogen Candida albicans. The Basidiomycota accounts for approximately 35% of the known fungal species. Well-known edible Basidiomycota mushrooms include Agaricus bisporus (common mushroom) and Pleurotus ostreatus (oyster mushroom).
It is hoped that the FTOL presented here will help resolve a number of currently debated fungal phylogenetic relationships. For example, there is substantial evidence that within the Ascomycota phylum, the Pezizomycotina and Saccharomycotina subphyla are sister groups (Fitzpatrick et al. 2006; Kuramae et al. 2006; Liu et al. 2009; Philippe et al. 2004; Robbertse et al. 2006). However, there is conflicting evidence to suggest that the Taphrinomycotina and Saccharomycotina are sister clades (Baldauf et al. 2000; Bullerwell et al. 2003; Diezmann et al. 2004). Similarly within the Basidiomycota phylum, a consensus regarding the phylogenetic relationships among the Ustilagomycotina, Pucciniomycotina, and Agaricomycotina subphyla is not yet available (Begerow et al. 2004; Hibbett et al. 2007; James et al. 2006b). A phylogenomic-based FTOL can also help address relationships at the class level, for example, the evolutionary relationships among a number of Aspergilli species are currently debated (Galagan et al. 2005a; Peterson 2008).
To illustrate the usefulness of a coherent fungal phylogeny, we have undertaken a preliminary investigation of the phyletic distribution of yeast prion-like proteins in the fungal kingdom and mapped their presence/absence onto our FTOL. A prion is an infectious protein that has the capability of converting native molecules of the same type into the infectious prion form. Prions have been classified as the causative agent of a class of mammalian neurodegenerative diseases termed Transmissible Spongiform Encephalopathies (TSEs) which includes Creutzfeldt Jakob Disease (CJD) in humans and Bovine Spongiform Encephalopathy (BSE—Mad Cow Disease) in cattle (McKintosh et al. 2003). However, Wickner’s proposal that the S. cerevisiae non-mendelian genetic elements [PSI +] and [URE3] were prion forms of the native protein Sup35 and Ure2, respectively, potentially extended the role of prions beyond only being disease-causing agents (Wickner 1994). Since Wickner’s proposal subsequent work, predominantly on [PSI +], was key to confirming that prions exist in yeast and in proving the prion hypothesis (King and Diaz-Avalos 2004; Tanaka et al. 2004) that was first proposed by Prusiner (1982). After a steady increase in the numbers of S. cerevisiae proteins with potential prion-forming ability (for summary, see Wickner et al. 2010), this number dramatically increased to approximately 30 (Alberti et al. 2009) and has fueled the opinion that the formation of prions in vivo may be a naturally occurring phenomenon and that the prion form of some proteins may have functional significance within the cell. Support for such a proposal already exists in the examples of the well-characterized [Het-s] prion of Podospora anserina (Saupe 2007) and more recently the potential functional prion-forming capacity of the Aplysia californica CEPB protein and its role in long-term memory (Si et al. 2010). Given the apparent importance and potential influence of prion-forming ability on protein function and fungal development, we have assessed the distribution of confirmed and potential prions identified in S. cerevisiae across the fungal kingdom.
Methods
Genome Data
Our fungal protein database consisted of 103 genomes and 1,001,217 individual genes (Supplementary file 1). Where available, data were obtained from the NCBI fungal genome FTP site (ftp://ftp.ncbi.nih.gov/genomes/Fungi). The remaining data were downloaded from the relevant sequencing centres (Supplementary file 2).
Reconstruction of Gene Trees
Homologous families were identified using an all-versus-all BlastP (Altschul et al. 1997) search (cutoff E-value = 10−10) followed by a markov clustering (MCL)-based algorithm (Enright et al. 2002). The MCL algorithm implements a user-defined inflation parameter (Enright et al. 2002). An increased inflation parameter has the effect of making the inflation operator stronger and in turn increases the granularity of clusters (Enright et al. 2002). To determine if varying inflation parameters would have an effect on our fungal phylogeny, six different inflation values were chosen (I = 1.2, 1.5, 1.8, 2.0, 4, and 6) and in turn yielded six individual phylogenomic datasets. For comparative purposes, a seventh phylogenomic dataset was built by locating homologous families using a previously described randomized BlastP approach (Creevey et al. 2004; Fitzpatrick et al. 2006; Pisani et al. 2007).
Due to computational constraints, only gene families with less than 200 members were analyzed (Table 1). Gene families were aligned using the multiple sequence alignment software Muscle v3.7 (Edgar 2004) with the default settings. Using the default settings, misaligned or fast evolving regions of alignments were removed with Gblocks (Castresana 2000). Permutation tail probability (PTP) tests (Archie 1989; Faith and Cranston 1991) were performed on each alignment to ensure that the presence of evolutionary signal was better than random (P < 0.05). Optimum models of protein evolution were selected using Modelgenerator (Keane et al. 2004) and these were used to reconstruct maximum likelihood phylogenies in Phyml v3.0 (Guindon and Gascuel 2003). Bootstrap (BP) resampling was performed 100 times on each alignment, and majority rule consensus (threshold of 70%) trees were reconstructed.
Reconstruction of Single and Multi-gene Supertrees
Gene families were partitioned based on the criteria whether they were SG families or multi-gene families (have more than one representative from any one species).
SG families were the underlying data in our matrix representation with parsimony (Baum 1992; Ragan 1992) (MRP) supertree. After removing gene families that failed the PTP test, we were left with 4,753, 6,678, 7,757, 8,341, 11,641, 13,347, and 9,336 trees as source data for our seven different phylogenomic datasets (Table 1). MRP trees were reconstructed for each phylogenomic dataset using the supertree software CLANN version 3.1.4b (Creevey and McInerney 2005). BP resampling (100 replicates) was performed on each dataset. Supertree nodes with less than 50% BP support were collapsed.
Both singlegene and multi-gene families were used to reconstruct supertrees using gene tree parsimony (Page 1998; Slowinski and Page 1999) implemented in the software DupTree version 1.48 (Wehe et al. 2008). After removal of gene families that failed the PTP test, we were left with 13,759, 19,789, 21,876, 22,788, 27,735, 30,012, and 23,026 trees as souce data for our seven different phylogenomic datasets (Table 1). For each phylogenomic dataset, BP resampling (100 replicates) was performed and nodes with less that 50% BP support were collapsed.
Heads or Tails (HorT) test
To assess the possible effects, multiple sequence alignment quality may have on our phylogenomic supertrees, and alignments were performed in reverse residue order and scored using the HorT test (Landan and Graur 2007). Alignments with a sum-of-pairs score >90% were retained for supertree analysis. Due to computational constraints, this analysis was only performed on SG families.
Supermatrix Analysis
Examining our phylogenomic datasets derived using different clustering cutoffs (I = 1.2, 1.5, 1.8, 2, 4, 6, and randomized criteria), we could not locate a SG family that was universally distributed in all genomes used in this study (Supplementary file 1). Instead of using universally distributed genes, we located gene families with a wide phyletic range, we define these as a single-copy gene family found in more than half of the genomes analyzed. We chose the families (87 in total) from the phylogenomic dataset derived with an inflation value of 1.2 (I1.2). These 87 gene families were individually aligned, misaligned, or fast evolving regions of alignments were removed with Gblocks (default settings) and concatenated together to yield an alignment exactly 12,267 amino acids in length. A Bayesian phylogeny was reconstructed using PhyloBayes implementing the CAT+Γ models (Lartillot and Philippe 2008). A posterior consensus tree was obtained by pooling trees of two independent runs; the analysis was stopped when the observed discrepancy across bipartitions (maxdiff) was less than 0.15.
In Silico Prion Analysis
A recent bioinformatic/proteome analysis of S. cerevisiae found more than 200 proteins contain putative prions domains, of these 29 passed rigorous biochemical and genetic assays and were classified as potential prions (Alberti et al. 2009). Using the HMMER ver 3.0 package (http://hmmer.org/), we scored the presence or absence of these 29 proteins in each fungal genome used in this analysis. A bidirectional database search with a cutoff E-value = 10−5 was performed. We consider proteins located by this bidirectional strategy as orthologs. Orthology assignments were manually checked for species represented in the yeast genome order browser (YGOB) (Byrne and Wolfe 2005) and the Candida genome order browser (CGOB) (Fitzpatrick et al. 2010). Manually curated orthology databases are not currently available for the remaining fungal species used in this analysis. If an ortholog could not be located in a genome, a tblastn search was performed to insure mis-annotation was not responsible. All putative orthologs were screened by a previously described hidden Markov model (HMM) (Alberti et al. 2009) to determine if the ortholog contained a candidate prion domain or not.
Proteins located in a one-way phmmer search are considered homologs. For completeness, all homologs were also screened for prion domains by the HMM.
Results and Discussion
The choice of Markovian Clustering (MCL) Inflation Value Does Not Have a Significant Impact on Phylogenetic Supertree Reconstruction
SG families were located using a BlastP database search followed by a MCL technique, a random BlastP-based search only strategy to locate SG families was also employed (see methods). To determine the possible effects, the MCL inflation (I) value may have on our phylogenomic analysis, a selection of I values were chosen (ranging from 1.2 to 6) generating seven individual SG phylogenomic datasets (Table 1). An I value of 1.2 yielded the smallest dataset with 5,489 gene families accounting for 63,727 individual protein coding genes while the largest dataset was obtained with an I value set to 6 (15,555 families and 150,406 protein coding genes) (Table 1). Maximum likelihood phylogenies were reconstructed for each single-copy family in each phylogenomic dataset. Branches with less than 70% BP support were collapsed. These 70% majority rule consensus trees were the input data for our single-copy supertree analyses. Branches on the resultant supertrees with less than 50% support are not considered to be significant and were also collapsed. For brevity, we refer to supertrees derived from the dataset with an inflation value of 1.2 as the I1.2 supertree, we use a similar nomenclature for all other datasets (I1.5, I1.8, I2, I4, and I6), the supertree derived from the random BlastP strategy is referred to as the Random supertree.
Overall the resultant SG-derived supertrees are relatively congruent with one another (Fig. 1 and Supplementary Fig. 1). The branching order of some clades do differ slightly however. For example, the phylogenetic order of some of Aspergilli clades differ depending on which supertree is considered (Fig. 1 and Supplementary Fig. 1). Five of the seven supertrees (I1.2, I1.5, I1.8, I2, and I6) infer a sister group relationship between (A. flavus, A. oryzae, and A. terreus) and (A. carbonarius, A. niger) (63, 74, 74, 80, and 92% BP, respectively, Supplementary Fig. 1a–d, f). I4 and the random supertrees differ slightly as they do not infer this sister group relationship and instead infer a sister group relationship between A. nidulans and (A. carbonarius, A. niger) (Supplementary Fig. 1e, g).

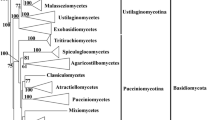
Majority rule (50%) consensus phylogeny of seven phylogenetic supertrees derived from single-gene (SG) families. Each phylogenetic supertree was derived from a different underlying set of gene families. The composition of the genes in each dataset is dependent on the inflation value (I) used by the MCL software while clustering genes into families. Branches that received less than 50% BP support in the underlying supertrees were collapsed. Phyla, subphyla, and class clades are labelled. The Chytridiomycota and Mucoromycotina phyla have been selected as the outgroup. The Basidiomycota and Ascomycota form monophyletic clades and together form the Dikarya subkingdom. V. polyspora has undergone a whole genome duplication (WGD) but does not form a monophyletic clade with the other species that have also undergone a WGD
Another minor topological difference occurs at the base of the clade for the genomes that have undergone a whole genome duplication (WGD) (Fig. 1). Six of the supertrees infer that within the WGD clade C. glabrata lies closer to the base of the WGD clade than S. castelli does, while I1.2 infers the reverse (Supplementary Fig. 1a). Just outside the WGD clade, four of the supertrees (I1.2, I1.5, I2, and I4) infer a sister group relationship between Zygosaccharomyces rouxii and Vanderwaltozyma polyspora (Supplementary Fig. 1a–e), the remaining three supertrees infer an unresolved clade containing these two species and the WGD clade (Supplementary Fig. 1c, f, g). This inference is surprising as V. polyspora has undergone a WGD (Scannell et al. 2007) and we expected it to form a monophyletic clade with the other WGD species. Six of the SG supertrees infer a sister group relationship between the Saccharomycotina and Pezizomycotina to the exclusion of the Taphrinomycotina (Fig. 1). The one exception is the I2 supertree which infers a sister group relationship between the Taphrinomycotina and Saccharomycotina (70% BP support, Supplementary Fig. 1d).
There are a number of minor incongruences among the phylogenetic relationships of the Basidiomycete species (Fig. 1). All the supertrees successfully reconstruct the main Basidiomycete subphyla (Fig. 1 and Supplementary Fig. 1). There are some topological differences pertaining to the sister group relationships among these however. Four supertrees infer a sister group relationship among the Pucciniomycotina and Agaricomycotina (Fig. 1 and Supplementary Fig. 1a–c and g), the remaining three infer a trichotomy between the Pucciniomycotina, Agaricomycotina, and Ustilagomycotina (Supplementary Fig. 1d–f). There is also minor conflicts relating to the branching orders within the subclass Agaricomycetidae (Fig. 1 and Supplementary Fig. 1).
From the results presented here it is evident that while the number and composition of SG families vary with increasing inflation values (Table 1), the resultant phylogenetic supertrees are relatively congruent (Fig. 1). Our results show that supertrees derived from 60,372 protein coding genes are comparable to those derived from 140,745 protein coding genes. Strongly supported clades are constant in all supertrees. Incongruences do occur, but generally these clades are weakly supported. Denser sampling of some species particularly among the Basidiomycetes should help improve consistency across all supertrees presented here. Therefore, for the fungal dataset utilized here, the MCL inflation value does not strongly influence our reconstruction of the FTOL. However, we do feel it is worthwhile deriving multiple supertrees from different underlying gene family data especially when a controversial inference is made. Interestingly, the random BlastP strategy employed in previous phylogenomic analyses (Fitzpatrick et al. 2006; Holton and Pisani 2010; Pisani et al. 2007) lacks the MCL clustering step, however, for the fungal dataset analyzed here this approach produces genome phylogenies that are comparable to those that have undergone a MCL clustering step (Fig. 1 and Supplementary Fig. 1).
Effect of Alignment Orientation on Phylogenetic Supertree Reconstruction
Accurate multiple sequence alignment is a fundamental step in recovering a reliable phylogeny (Mullan 2002; Wong et al. 2008). In theory, the order in which residues are aligned (i.e., amino-to-carboxy or carboxy-to-amino direction) should yield identical sequence alignments. However, a recent study has shown that this is seldom true (Landan and Graur 2007). A method termed “heads or tails” (HorT) has been developed to score the level of agreement/disagreement between gene families that have been aligned either from the amino-to-carboxy or carboxy-to-amino direction (Landan and Graur 2007). Gene families that display large discrepancies between their heads and tails alignments may yield incongruent phylogenies.
To examine the possible effect alignment orientation may have on our fungal supertrees, we reconstructed supertrees where the underlying sequences have been aligned in the carboxy-to-amino direction (“tails”) (Supplementary Fig. 2) and compared them to our original supertrees (Supplementary Fig. 1) which are derived from alignments aligned in the amino-to-carboxy direction (“heads”).
Overall we found that the resultant supertrees are congruent with one another regardless of alignment orientation (Supplementary Fig. 3). For brevity, we refer to supertrees derived from the dataset with an inflation value of 1.2 with underlying gene families aligned from N to C termius as the H1.2 supertree, and from C to N terminus as T1.2. We use a similar nomenclature for all other datasets (H1.5, T1.5, etc.).
Looking at individual supertrees with the same underlying datasets, we do see a number of small incongruences (which will not be listed in detail). For example, H1.2 (Supplementary Fig. 1a) and T1.2 (Supplementary Fig. 2a) disagree regarding the placement of the Taphrinomycotina clade (Supplementary Fig. 3a). H1.2 places this clade at the base of the Ascomycota (Supplementary Fig. 1a, 75% BP) and a sister group relationship between the Saccharomycotina and Pezizomycotina (81% BP). T1.2 fails to confidently infer the basal Ascomycota relationship of the Taphrinomycotina clade but does support it weakly (Supplementary Fig. 2a, 49% BP). Similarly, the placement of Allomyces macrogynus also conflicts between both supertrees (Supplementary Fig. 3a). H1.2 places it at the base of the Chytridiomycota/Mucoromycotina clade (55% BP, Supplementary Fig. 1a) wheras T1.2 infers it is more closely related to the Ascomycetes and Basidiomycetes (96% BP Supplementary Fig. 2a). Another incongruence relates to the base of the WGD species clade (Supplementary Fig. 3a). H1.2 places S. castelli closer to the base of the WGD clade relative to C. glabrata (64% BP, Supplementary Fig. 1a) conversely T1.2 places C. glabrata closer to the base (61% BP, Supplementary Fig. 2a). Overall we observe incongruences between H and T supertrees when clades are weakly supported. Based on our observations, strongly supported clades in one supertree are normally strongly supported in the other regardless of the orientation in which the underlying gene families have been aligned. This may be due to the fact that we only use conserved blocks for phylogenetic analyses (see methods) therefore avoiding some of the pitfalls associated with poorly aligned regions. It would be interesting to see if 100% congruence could be achieved between supertrees by utilizing different alignment software/methods.
Using the HorT method, we also excluded pairs of alignments that did not share 90% column similarity between one another. This step resulted in up to 37.2% of multiple sequence alignments being removed from individual datasets (Supplementary file 3). Examining each dataset (I1.2, I1.5, I2, I4, I6, and Random), we see that the resultant supertrees generated from the alignments that pass the HorT test are 100% congruent with one another regardless if they are aligned from the amino-to-carboxy or carboxy-to-amino direction (not shown).
However, the utilization of gene families that pass the HorT test does not lead to a consensus regarding the branching pattern of major clades when individual datasets are compared to one another (Supplementary Fig. 4). For example, only 3 of HorT supertrees reconstruct a monophyletic Ascomycota clade (Supplementary Fig. 4). Similarly only 3 of the HorT supertrees reconstruct the Saccharomycotina lineage (Supplementary Fig. 4). Therefore, based on our analysis, the removal of alignments that fail to pass the HorT criteria (>90% column similarity) reduces our ability to infer the evolutionary history of the fungal species considered here. The use of reliable alignments in a phylogenomic analysis should be encouraged, however, and alignments that passed a lower column similarity cutoff (>80% for example) may have improved the ability of our supertrees to infer robust fungal evolutionary relationships and warrants further investigation.
Reconstructing the Fungal Genome Phylogeny Using Both Single- and Multi-gene Families
Rigorous phylogenomic analyses attempt to use all relevant phylogenetic data. The MRP supertrees presented here (Fig. 1 and Supplementary Fig. 1) are derived from SG families. This approach minimizes the analysis of gene families that contain paralogs. The removal of paralogous families is a conservative approach but results in only a fraction of the fungal gene set being represented in our genome phylogeny, ranging from a low of ~6% for the I1.2 dataset (~60,000 genes) to a high of ~14% for the I6 dataset (~140,000 genes) (Table 1).
In an attempt to use all available data, we also reconstructed genome phylogenies using both single- and multi-gene families with the gene tree parsimony method (Page 1998; Slowinski and Page 1999). This approach significantly increased the number of underlying genes analyzed (e.g., 430,945 genes, ~43% of dataset) in the I1.2 datasets and 664,849 (~66% of the dataset) in the I6 dataset, Table 1. Genes that were not included in our analysis either belonged to a gene family that lacked phylogenetic signal (failed PTP test, Table 1) or were members of a gene family with less than 4 taxa.
Overall the resultant single/multigene genome (SMG) phylogenies are highly congruent with one another (Fig. 2 and Supplementary Fig. 5). Major phyla, subphyla, and classes are consistently recovered regardless of the underlying gene families (Table 1). As with the SG genome phylogenies, there are minor topological differences between individual trees. For example, two SMG phylogenies (I14 and I6) fail to place Ashbya gossypii beside the (Lachancea thermotolerans, Kluyveromyces waltii, and S. kluyveri) clade and instead infer a sister group relationship between A. gossypii and S. kluyveri (Fig. 2 and Supplementary Fig. 5e, f). Similarly, all but one SMG phylogeny (Random) places C. guilliermondii next to C. lusitaniae at the base of the CTG clade and instead infers a sister group relationship with the (Pichia stipitis, Debaromyces hansenii) clade; however, this inference is poorly supported (52% BP, Supplementary Fig. 5g). Five of the SMG infers a sister group relationship between the Basidiomycete subphyla Pucciniomycotina and Agaricomycotina while the remaining two (I4 and I6) conflicts with this topology and infers a sister group relationship between the Ustilagomycotina and Pucciniomycotina (Supplemental Fig. 5e, f).

Majority rule (50%) consensus phylogeny of seven phylogenetic supertrees derived from single- and multi-gene (SMG) families. Each phylogenetic supertree was derived from a different underlying set of gene families. The composition of the genes in each dataset is dependent on the inflation value (I) used by the MCL software while clustering genes into families. Branches that received less than 50% BP support in the underlying supertrees were collapsed. Phyla, subphyla, and class clades are labelled. The Chytridiomycota and Mucoromycotina phyla have been selected as the outgroup. The Basidiomycota and Ascomycota form monophyletic clades and together form the Dikarya subkingdom
There is universal agreement regarding the sister group relationships within the Pezizomycotina subphylum. All of the SMG phylogenies infer a strongly supported sister group relationship between the Sordariomycetes/Leotiomycetes and Dothideomycetes classes to the exclusion of the Eurotiomycetes (Fig. 2 and Supplementary Fig. 5). This is interesting as phylogenies derived from multi-gene families alone (excluding SG families) fail to confidently reconstruct this relationship (not shown). There is also universal agreement regarding the placement of V. polyspora within a monophyletic WGD clade (Fig. 2).
Different Phylogenomic Approaches Reconstruct the FTOL
As well as using all available single and multi-gene families to reconstruct supertrees, we also reconstructed a fungal phylogeny using a supermatrix approach. Initially, we had intended to use genes that were single-copy and universally distributed in all fungal species. Surprisingly, we failed to locate a SG family that met these criteria. This highlights some of the difficulties associated with locating robust phylogenetic markers; however, we feel that a database search strategy followed by careful human annotation steps would uncover universally distributed single-copy genes. As a compromise to manually curating our gene sets, we selected 87 gene families that were found to be present in at least half of the fungal species used in this analysis. The average number of genes per family was ~73. Conserved blocks from these genes families were concatenated together to give an alignment containing 12,267 aligned amino acid positions. Concatenation without alignment trimming would have yielded an alignment with 77,348 amino acids, meaning we have removed ~84% of amino acid positions. Interestingly, alignment trimming with a more liberal method [trimAl (Capella-Gutierrez et al. 2009)] yielded an alignment with 17,973 sites. Further analysis is required to determine if significant differences would occur in the resultant phylogenies. However, due to computational constraints, we reconstructed a Bayesian supermatrix phylogeny (BSP) based on the alignment that had been stripped using Gblocks (Fig. 3).

Supermatrix Bayesian phylogeny (BSP) derived from 87 genes distributed across the fungal kingdom. The Chytridiomycota and Mucoromycotina phyla have been selected as the outgroup. The Basidiomycota and Ascomycota form monophyletic clades and together form the Dikarya subkingdom. Phyla, subphyla, and class clades are labelled. Numbers on individual nodes represent Bayesian posterior probabilities (BPP). Nodes without numbers received a BPP of 1. V. polyspora has undergone a whole genome duplication (WGD) but does not form a monophyletic clade with the other species that have also undergone a WGD
Overall the topologies of SG, SMG and BSP genome phylogenies are in good agreement with one another (Fig. 4). We have already discussed some of the discrepancies that occur between SG and SMG phylogenies depending on the MCL clustering value used to derive gene families. When comparing SG and SMG genome phylogenies, we will consider the consensus trees (i.e., Figs. 1 and 2) to be representative. All three genome phylogenies correctly recover the Ascomycota, Basidiomycota, and Chytridiomycota phyla and infer the Dikarya (Ascomycota and Basidiomycota) subkingdom (Blackwell et al. 2006; Galagan et al. 2005b; Guarro et al. 1999; James et al. 2006a; Liu et al. 2006; Marcet-Houben and Gabaldon 2009) (Figs. 1, 2, 3, and 4).

Majority rule consensus phylogeny of the single-, multi-gene, and supermatrix phylogenies illustrating the degree of congruence. The original phylogenies are displayed in Figs 1, 2, and 3. The Chytridiomycota and Mucoromycotina phyla have been selected as the outgroup. Branches that are not supported by at least 2 of the species phylogenies are collapsed. Phyla, subphyla, and class clades are labelled
Phylogenetic Relationships Among the Chytridiomycota and Mucoromycotina
The Chytridiomycota is generally considered the most basal fungal phylum (Guarro et al. 1999; James et al. 2006a; Liu et al. 2006; Steenkamp et al. 2006) although some studies have shown the base of the fungal tree to be paraphyletic (Blackwell et al. 2006). Our phylogenies strongly support a sister group relationship between the Chytridiomycota and Mucoromycotina however (Figs. 1, 2, 3, and 4). This inference agrees with another whole genome-based study (Marcet-Houben and Gabaldon 2009). We cannot rule out the possibility that this sister group relationship is an artifact of long-branch attraction, however, as both these phyla are poorly sampled at the genome level (Supplementary file 1). Previous analysis have shown the Chytridiomycota to be paraphyletic (James et al. 2006b; Lutzoni et al. 2004; Steenkamp et al. 2006), our genome phylogenies actually infer a monophyletic Chytridiomycota clade (Figs. 1, 2, and 3). Closer inspection shows this inference does not have strong support, however, only 4 of our SG supertrees places A. macrogynus beside the (Spizellomyces punctatus, Batrachochytrium dendrobatidis) clade. Recent phylogenetic analysis has proposed that A. macrogynus belongs to a new phylum separate from the Chytridiomycota termed the Blastocladiomycota (James et al. 2006b). The addition of extra Blastocladiomycota species to our dataset may confer with this inference as the monophyly of the Chytridiomycota is poorly supported.
Phylogenetic Relationships Among the Ascomycota
All three genome phylogenies recover the three Ascomycota subphyla (Pezizomycotina, Saccharomycotina, and Taphrinomycotina, Figs. 1, 2, 3, and 4). Until recently the phylogenetic relationships between these three subphyla were uncertain with some analyses placing Saccharomycotina and Taphrinomycotina as sister clades (Baldauf et al. 2000; Diezmann et al. 2004) while others inferred a sister group relationship between Pezizomycotina and Saccharomycotina (Fitzpatrick et al. 2006; Kuramae et al. 2006; Philippe et al. 2004; Robbertse et al. 2006). Recently a comprehensive phylogenomic analysis of 113 nuclear genes by Liu et al. (2009) has shown that the Taphrinomycotina are a monophyletic clade and branch as a sister group to a (Pezizomycotina, Saccharomycotina) clade. All our genome phylogenies agree with this topological arrangement (Figs. 1, 2, 3, and 4).
Phylogenetic Relationships Among the Saccharomycotina
Within the clade that contains C. albicans and close relatives (CTG clade) there is some incongruence regarding the relationships among D. hansenii, P. stipitis, and C. guilliermondii (Fig. 4). The SG supertree and BSP infer a sister group relationship between D. hansenii and C. guilliermondii (Figs. 1 and 3) in agreement with previous phylogenetic analysis derived from concatenated mitochondrial proteins (Jung et al. 2010). Conversely, the SMG phylogeny infers a sister group relationship between D. hansenii and P. stipitis in agreement with previous phylogenomic (Fitzpatrick et al. 2010; Jeffries et al. 2007) and phylogenetic studies (Suh et al. 2006).
Both SMG and SGD phylogenies infer a sister group relationship between P. pastoris and the CTG clade (Figs. 1 and 2), this agrees with previous supermatrix-derived phylogenies (De Schutter et al. 2009). Our BSP phylogeny places P. pastoris near the base of Saccharomycotina clade (1.0 Bayesian posterior probability (BPP), Fig. 3), however, based on our literature searches we were could not find any published support for this inference.
Regarding the Saccharomycetaceae clade SG and SMG, phylogenies recover a monophyletic Lachancea genus clade (S. kluyveri, L. thermotolerans, and K. waltii) (Figs. 1 and 2). A. gossypii and K. lactis are from different genera (Eremothecium and Kluyveromyces, respectively) but are inferred as sister taxa to one another and in turn to the Lachancea clade (Figs. 1 and 2). This topology is supported by other phylogenetic studies (Diezmann et al. 2004; Kuramae et al. 2006; Marcet-Houben and Gabaldon 2009). The BSP phylogeny does not infer this close relationship and instead places A. gossypii and K. lactis at the base of a polyphyletic Saccharomycetaceae clade (Fig. 3). Our SG phylogeny places C. glabrata closer to the base of the WGD clade relative to S. castellii (Fig. 1). Previous syntenic analysis (Scannell et al. 2006) and phylogenomic analysis (Fitzpatrick et al. 2006) have shown that this inference is unreliable and may be the result of compositional biases (Fitzpatrick et al. 2006). Our BSP phylogeny infers a sister group relationship between C. glabrata and S. castelli (Fig. 3). Both SG and BSP phylogenies also infer a sister group relationship between Z. rouxii and V. polyspora. This inference is surprising as V. polyspora has undergone a WGD (Scannell et al. 2007) and we expected it to form a monophyletic clade with the other WGD species. The failure to accurately reconstruct this inference may be due to hidden paralogy in our SG phylogenomic datasets. Conversely, the SMG phylogeny places V. polyspora at the base of a monophyletic WGD clade (Fig. 2). Therefore, the use of multi-gene families in a supertree context may help overcome the problems of hidden paralogy associated with supertrees derived from SG families.
Phylogenetic Relationships Among the Pezizomycotina
Within the Pezizomycotina, well-defined class clades are evident (Sordariomycetes, Dothideomycetes, Eurotiomycetes, and Leotiomycetes) (Figs. 1, 2, 3, and 4). Presently, the relationships among these classes are unclear as different phylogenetic analyses have proposed conflicting evolutionary scenarios (Fitzpatrick et al. 2006; Lutzoni et al. 2004; Robbertse et al. 2006; Schoch et al. 2009). All our phylogenies infer a sister group relationship between the Sordariomycetes and Leotiomycetes species (Figs. 1, 2, 3, and 4), this sister group relationship is supported by previous analyses (Fitzpatrick et al. 2006; James et al. 2006a; Kuramae et al. 2006; Lumbsch et al. 2005; Schoch et al. 2009). Our SG supertree fails to infer sister group relationships between the Sordariomycetes/Leotiomycetes, Dothideomycetes and Eurotiomycetes clades and instead infers a trichotomy at the base of the Pezizomycotina clade (Fig. 1). However, the SMG and BSP phylogenies place the Dothideomycetes and Eurotiomycetes as sister clades (Figs. 2 and 3). This relationship is supported by previous phylogenomic (Fitzpatrick et al. 2006; Robbertse et al. 2006) and phylogenetic analyses (Schoch et al. 2009) but alternative topologies have also been suggested (James et al. 2006a; Lutzoni et al. 2004). However, based on the wealth of data utilized in out SMG supertree analysis, we are confident the inference of Dothideomycetes and Eurotiomycetes as sister clades to be correct.
Phylogenetic Relationships Among the Aspergilli
Previous phylogenetic analysis has shown that A. nidulans belongs to the subgenus Nidulantes and is divergent from the other Aspergilli species used in this analysis (Peterson 2008). Our SMG phylogeny is congruent with this view as it places A. nidulans at the base of the Aspergillus clade (Fig. 2). However, the SG phylogeny places it within the Aspergillus clade (Fig. 1). The BSP phylogeny places it as the sister group of A. niger, A. carbonarius (1.00 BPP, Fig. 3). The addition of genome sequences from species closely related Nidulantes would help resolve these topological incongruences. However, based on previous phylogenomic analyses (Peterson 2008) and the high level of congruence observed across our SMG phylogenies we are confident that A. nidulans is divergent from the remaining Aspergilli used in this analysis (Fig. 2).
Phylogenetic Relationships Among the Basidiomycota
Our genome phylogenies successfully recover monophyletic clades for the three Basidiomycota subphyla (Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina). The phylogenetic relationships among these three subphyla are uncertain, although cytological (Lutzoni et al. 2004) and concatenated phylogenies (Hibbett 2006; James et al. 2006a) suggest a sister group relationship between the subphyla Ustilaginomycotina and Agaricomycotina. Our SG and SMG phylogenies both suggest that Agaricomycotina is more closely related to the Pucciniomycotina clade than to the Ustilaginomycotina clade (Figs. 1 and 2). This inference is not universal in the SG supertrees, however, as only four of the seven datasets (I1.2, I1.5, I1.8, and Random) recover this relationship (Fig. 1); furthermore, BP support for three of these inferences is quite low (I1.2 = 51%, I1.5 = 58%, and I1.8 = 57%, Supplementary Fig. 1a–c). The BSP phylogeny actually infers a sister group relationship between the Agaricomycotina and Ustilaginomycotina clades although this topology is not strongly supported (0.88 BPP, Fig. 3). Based on our data, we cannot confidently resolve the relationships among the three Basidiomycete subphyla but expect additional taxon sampling in the future would increase our ability to resolve these relationships. This data should be soon available as the Joint Genome Initiative is currently sequencing 30 Basidiomycete genomes for the SAP Community proposal that aims to sequence a diverse assemblage of saprotrophic Basidiomycota (http://gp-edge2.jgi-psf.org:1080/programs/fungi/fungal-projects.jsf).
Phylogenomic Distribution of Yeast Prion-Like Proteins in the Fungal Kingdom
Applying bioinformatics, genetics, biochemical, and cell biology techniques, Alberti et al. (2009) recently identified an array of new potential prion proteins in S. cerevisiae. When combined with the list of already confirmed yeast prions, this brings the total number of proteins with potential prion-forming ability in this organism to approximately 30. We scored the presence/absence of putative orthologs/homologs of these prion candidates throughout the fungal kingdom (methods) and mapped them onto our FTOL (Fig. 5). Accession numbers for all putative orthologs/homologs are provided in Supplementary material (Supplementary file 4). Where possible we manually checked orthology assignments using genome order browsers (GOBs). Currently, manually curated GOBs are only available for species closely related to C. albicans and S. cerevisiae, respectively (Byrne and Wolfe 2005; Fitzpatrick et al. 2010). The use of these GOBs allowed us to identify 29 additional orthologs that were not detected by our bidirectional database search strategy (Supplemental file 4). We also located 15 additional orthologs using a tblastn strategy (Supplemental file 4). A previous analysis investigated the evolution of four yeast prions [PSI +], [URE3], [RNQ+], and [NU+] in 21 fungal species (19 Ascomycetes and 2 Basidiomycetes) (Harrison et al. 2007). Our analysis builds on this previous work in terms of the number of genomes and putative prions analyzed. It should be noted that we have searched for yeast prion-like proteins in this study; therefore, we are underestimating the number of potential prions in the fungal kingdom, as prions from evolutionary distant species may have unique prion domain characteristics.

Phylogenomic distribution of yeast prion-like proteins in the Fungal kingdom. Phylogenetic tree is a modified version of Fig. 4. 29 putative yeast prions are represented in a heat map. Presence and absence of prions have been determined
Figure 5 demonstrates that there is a wide-ranging distribution of potential yeast prion orthologs across the FTOL. Sup35, Ure2, Rnq1, New1, Swi1 Cyc8, and Mot3 constitute the group of yeast prions that have accumulated most experimental evidence to suggest that they can form and propagate as prions. Indeed, unequivocal prion proof in the form of in vitro formation of infectious protein particles has been obtained for Sup35, Ure2, and Rnq1 (Brachmann et al. 2005; King and Diaz-Avalos 2004; Patel and Liebman 2007; Tanaka et al. 2004). The conservation of these 7 well-characterized prions varies dramatically across the FTOL and subsets of the remaining 22 can be classified as exhibiting distribution patterns akin to one or other of these confirmed prions. The most dramatic and restricted ortholog distribution is for Rnq1, where orthologs are only found in 13 species and restricted to a monophyletic clade that contains close relative of S. cerevisiae (Fig. 5). Currently, the only confirmed in vivo role for Rnq1 appears to be in aiding the appearance of other prions, if so then it appears that only a small number of fungal species have retained this capacity. A similar prion domain containing ortholog distribution to Rnq1 is observed for Pgd1 and Ybl081w that could be suggestive of a similar prion-templating only function for these putative prion proteins. The putative prion protein Sap30 has an even narrower conserved prion domain range that indicates that this protein is worthy of assessment for heterologous prion-templating ability in S. cerevisiae.
The two most extensively studied yeast prion-forming proteins, Sup35 and Ure2, show a very different distribution in conservation of their prion-forming domains. The prion domain in Sup35 is much more widely conserved throughout the FTOL compared to the Ure2 prion domain. This difference presumably reflects the importance of the Q/N-rich domain in enhancement of protein function and/or prion-forming ability for each protein, respectively. Currently, there is a lively debate in the yeast prion field as to whether the [PSI +] prion is a “disease” of yeast or provides a potential benefit to yeast cells in times of stress (for recent reviews, see Lindquist 2009; Wickner et al. 2010). The conservation pattern of the Sup35 prion domain depicted in Fig. 5 does suggest that there is a significant selection pressure for the maintenance of this Q/N-rich region. While the data does not suggest an obvious mechanism for this selection pressure it could be used to inform and identify specific members of the FTOL for further analysis regarding the ability of the respective Sup35 proteins to form prions or to assess enhancement of protein function by the presence of the Q/N-rich domain.
What is the selection pressure for maintenance of potential prion-forming domains through evolution? This remains an open question that needs to be addressed on a case-by-case basis to any protein with orthologs harboring a conserved (Q/N-rich) prion domain. Given the observed conservation pattern of prion domains across the FTOL it is highly likely that some Q/N-rich domains have been retained due to enhancement of protein function, while others due to the retention for prion-forming ability. The extent of which is more prevalent remains to be determined.
Conclusion
We have reconstructed the FTOL using three independent approaches. Overall the resultant phylogenies are congruent with one another and successfully recover the major fungal phyla, subphyla, and classes. We have shown that the underlying gene families used to reconstruct the FTOL do not have a major effect on phylogenomic inferences, nor does the direction that that these families are aligned. Topological differences do occur, but these are mainly in poorly sampled or supported clades. For the first time in fungal phylogenomics, we have utilized multi-gene families to reconstruct the FTOL. The use of multi-gene families allows us to use all relevant phylogenetic data. With the advent of next generation sequencing, the taxonomical diversity and number of fungal genomes are expected to increase rapidly over the coming years. This oncoming deluge of genome data should help further resolve the FTOL. The phylogenomic FTOL presented here should provide a basis for future comparative fungal genomic analyses.
We have also mapped the presence and absence of yeast prion-like proteins onto the FTOL. The distribution of orthologs with conserved putative prion domains varies greatly depending on the protein in question. Some yeast prion protein orthologs are present in the majority of species in the FTOL while others are restricted to only a few species within a particular grouping. The difference in distribution is reflective of the in vivo role of the particular putative prion protein as well as the importance of the Q/N-rich domain to protein function or prion-forming ability.
References
Alberti S, Halfmann R, King O, Kapila A, Lindquist S (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137:146–158
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res (Online) 25:3389–3402
Archie JW (1989) A randomization test for phylogenetic information in systematic data. Syst Zool 38:251–278
Baldauf SL, Roger AJ, Wenk-Siefert I, Doolittle WF (2000) A kingdom-level phylogeny of eukaryotes based on combined protein data. Science 290:972–977
Barrett M, Donoghue MJ, Sober E (1991) Against consensus. Syst Zool 40:486–493
Baum BR (1992) Combining trees as a way of combining data sets for phylogenetic inference, and the desirability of combining gene trees. Taxon 41:3–10
Begerow D, John B, Oberwinkler F (2004) Evolutionary relationships among beta-tubulin gene sequences of basidiomycetous fungi. Mycol Res 108:1257–1263
Blackwell M, Hibbett DS, Taylor JW, Spatafora JW (2006) Research Coordination Networks: a phylogeny for kingdom Fungi (Deep Hypha). Mycologia 98:829–837
Brachmann A, Baxa U, Wickner RB (2005) Prion generation in vitro: amyloid of Ure2p is infectious. EMBO J 24:3082–3092
Bullerwell CE, Forget L, Lang BF (2003) Evolution of monoblepharidalean fungi based on complete mitochondrial genome sequences. Nucleic Acids Res 31:1614–1623
Butler G, Rasmussen MD, Lin MF, Santos MA, Sakthikumar S, Munro CA, Rheinbay E, Grabherr M, Forche A, Reedy JL, Agrafioti I, Arnaud MB, Bates S, Brown AJ, Brunke S, Costanzo MC, Fitzpatrick DA, de Groot PW, Harris D, Hoyer LL, Hube B, Klis FM, Kodira C, Lennard N, Logue ME, Martin R, Neiman AM, Nikolaou E, Quail MA, Quinn J, Santos MC, Schmitzberger FF, Sherlock G, Shah P, Silverstein KA, Skrzypek MS, Soll D, Staggs R, Stansfield I, Stumpf MP, Sudbery PE, Srikantha T, Zeng Q, Berman J, Berriman M, Heitman J, Gow NA, Lorenz MC, Birren BW, Kellis M, Cuomo CA (2009) Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459(7247):657–662
Byrne KP, Wolfe KH (2005) The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res 15:1456–1461
Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Creevey CJ, McInerney JO (2005) Clann: investigating phylogenetic information through supertree analyses. Bioinformatics 21:390
Creevey CJ, Fitzpatrick DA, Philip GK, Kinsella RJ, O’Connell MJ, Pentony MM, Travers SA, Wilkinson M, McInerney JO (2004) Does a tree-like phylogeny only exist at the tips in the prokaryotes? Proc Biol Sci 271:2551–2558
Dagan T, Martin W (2006) The tree of one percent. Genome Biol 7:118
De Schutter K, Lin YC, Tiels P, Van Hecke A, Glinka S, Weber-Lehmann J, RouzÈ P, Van de Peer Y, Callewaert N (2009) Genome sequence of the recombinant protein production host Pichia pastoris. Nat Biotechnol 27:561–566
Delsuc F, Brinkmann H, Philippe H (2005) Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet 6:361–375
Diezmann S, Cox CJ, Schonian G, Vilgalys RJ, Mitchell TG (2004) Phylogeny and evolution of medical species of Candida and related taxa: a multigenic analysis. J Clin Microbiol 42:5624–5635
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792
Eernisse D, Kluge A (1993) Taxonomic congruence versus total evidence, and amniote phylogeny inferred from fossils, molecules, and morphology. Mol Biol Evol 10:1170–1195
Enright AJ, Van Dongen S, Ouzounis CA (2002) An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30:1575
Faith DP, Cranston PS (1991) Could a cladogram this short have arisen by chance alone? On permutation tests for cladistic structure. Cladistics 7:1–28
Faris JD, Zhang Z, Lu H, Lu S, Reddy L, Cloutier S, Fellers JP, Meinhardt SW, Rasmussen JB, Xu SS, Oliver RP, Simons KJ, Friesen TL (2010) A unique wheat disease resistance-like gene governs effector-triggered susceptibility to necrotrophic pathogens. Proc Natl Acad Sci USA 107:13544–13549
Fitzpatrick DA, Logue ME, Stajich JE, Butler G (2006) A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol Biol 6:99
Fitzpatrick DA, Logue ME, Butler G (2008) Evidence of recent interkingdom horizontal gene transfer between bacteria and Candida parapsilosis. BMC Evol Biol 8:181
Fitzpatrick DA, O’Gaora P, Byrne KP, Butler G (2010) Analysis of gene evolution and metabolic pathways using the Candida Gene Order Browser. BMC Genomics 11:290
Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, Lee SI, Basturkmen M, Spevak CC, Clutterbuck J, Kapitonov V, Jurka J, Scazzocchio C, Farman M, Butler J, Purcell S, Harris S, Braus GH, Draht O, Busch S, D’Enfert C, Bouchier C, Goldman GH, Bell-Pedersen D, Griffiths-Jones S, Doonan JH, Yu J, Vienken K, Pain A, Freitag M, Selker EU, Archer DB, Penalva MA, Oakley BR, Momany M, Tanaka T, Kumagai T, Asai K, Machida M, Nierman WC, Denning DW, Caddick M, Hynes M, Paoletti M, Fischer R, Miller B, Dyer P, Sachs MS, Osmani SA, Birren BW (2005a) Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438:1105–1115
Galagan JE, Henn MR, Ma LJ, Cuomo CA, Birren B (2005b) Genomics of the fungal kingdom: insights into eukaryotic biology. Genome Res 15:1620–1631
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG (1996) Life with 6000 genes. Science 274(546):563–567
Guarro J, Gene J, Stchigel AM (1999) Developments in fungal taxonomy. Clin Microbiol Rev 12:454–500
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Harrison LB, Yu Z, Stajich JE, Dietrich FS, Harrison PM (2007) Evolution of budding yeast prion-determinant sequences across diverse fungi. J Mol Biol 368:273–282
Hawksworth DL (1991) The fungal dimension of biodiversity: magnitude, significance, and conservation. Mycol Res 95:641–655
Hawksworth DL (2001) The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res 109:1422–1432
Heckman DS, Geiser DM, Eidell BR, Stauffer RL, Kardos NL, Hedges SB (2001) Molecular evidence for the early colonization of land by fungi and plants. Science 293:1129–1133
Hibbett DS (2006) A phylogenetic overview of the Agaricomycotina. Mycologia 98:917–925
Hibbett DS, Binder M, Bischoff JF, Blackwell M, Cannon PF, Eriksson OE, Huhndorf S, James T, Kirk PM, Lücking R (2007) A higher-level phylogenetic classification of the Fungi. Mycol Res 111:509–547
Holton TA, Pisani D (2010) Deep genomic-scale analyses of the metazoa reject Coelomata: evidence from single- and multigene families analyzed under a supertree and supermatrix paradigm. Genome Biol Evol 2:310–324
James TY, Kauff F, Schoch CL, Matheny PB, Hofstetter V, Cox CJ, Celio G, Gueidan C, Fraker E, Miadlikowska J, Lumbsch HT, Rauhut A, Reeb V, Arnold AE, Amtoft A, Stajich JE, Hosaka K, Sung GH, Johnson D, O’Rourke B, Crockett M, Binder M, Curtis JM, Slot JC, Wang Z, Wilson AW, Schussler A, Longcore JE, O’Donnell K, Mozley-Standridge S, Porter D, Letcher PM, Powell MJ, Taylor JW, White MM, Griffith GW, Davies DR, Humber RA, Morton JB, Sugiyama J, Rossman AY, Rogers JD, Pfister DH, Hewitt D, Hansen K, Hambleton S, Shoemaker RA, Kohlmeyer J, Volkmann-Kohlmeyer B, Spotts RA, Serdani M, Crous PW, Hughes KW, Matsuura K, Langer E, Langer G, Untereiner WA, Lucking R, Budel B, Geiser DM, Aptroot A, Diederich P, Schmitt I, Schultz M, Yahr R, Hibbett DS, Lutzoni F, McLaughlin DJ, Spatafora JW, Vilgalys R (2006a) Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 443:818–822
James TY, Letcher PM, Longcore JE, Mozley-Standridge SE, Porter D, Powell MJ, Griffith GW, Vilgalys R (2006b) A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 98:860–871
Jeffries TW, Grigoriev IV, Grimwood J, Laplaza JM, Aerts A, Salamov A, Schmutz J, Lindquist E, Dehal P, Shapiro H (2007) Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat Biotechnol 25:319–326
Jung PP, Friedrich A, Souciet JL, Louis V, Potier S, de Montigny J, Schacherer J (2010) Complete mitochondrial genome sequence of the yeast Pichia farinosa and comparative analysis of closely related species. Curr Genet 56:507–515
Keane TM, Naughton TJ, McInerney JO (2004) ModelGenerator: amino acid and nucleotide substitution model selection. National University of Ireland. http://bioinf.nuim.i.e/software/modelgenerator
King CY, Diaz-Avalos R (2004) Protein-only transmission of three yeast prion strains. Nature 428:319–323
Kluge AG (1989) A concern for evidence and a phylogenetic hypothesis of relationships among Epicrates (Boidae, Serpentes). Syst Biol 38:7–25
Kuramae EE, Robert V, Snel B, Weifl M, Boekhout T (2006) Phylogenomics reveal a robust fungal tree of life. FEMS Yeast Res 6:1213–1220
Landan G, Graur D (2007) Heads or tails: a simple reliability check for multiple sequence alignments. Mol Biol Evol 24:1380–1383
Lartillot N, Philippe H (2004) A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol 21:1095–1109
Lartillot N, Philippe H (2008) Improvement of molecular phylogenetic inference and the phylogeny of Bilateria. Philos Trans R Soc Lond B Biol Sci 363:1463–1472
Lartillot N, Brinkmann H, Philippe H (2007) Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol Biol 7(Suppl 1):S4
Lindquist S (2009) Protein folding sculpting evolutionary change. Cold Spring Harb Symp Quant Biol 74:103–108
Liu YJ, Hodson MC, Hall BD (2006) Loss of the flagellum happened only once in the fungal lineage: phylogenetic structure of kingdom Fungi inferred from RNA polymerase II subunit genes. BMC Evol Biol 6:74
Liu Y, Leigh JW, Brinkmann H, Cushion MT, Rodriguez-Ezpeleta N, Philippe H, Lang BF (2009) Phylogenomic analyses support the monophyly of Taphrinomycotina, including Schizosaccharomyces fission yeasts. Mol Biol Evol 26:27–34
Lumbsch HT, Schmitt I, Lindemuth R, Miller A, Mangold A, Fernandez F, Huhndorf S (2005) Performance of four ribosomal DNA regions to infer higher-level phylogenetic relationships of inoperculate euascomycetes (Leotiomyceta). Mol Phylogenet Evol 34:512–524
Lutzoni F, Kauff F, Cox CJ, McLaughlin D, Celio G, Dentinger B, Padamsee M, Hibbett D, James TY, Baloch E, Grube M, Reeb V, Hofstetter V, Schoch C, Arnold AE, Miadlikowska J, Spatafora J, Johnson D, Hambleton S, Crockett M, Shoemaker R, Sung G, Lucking R, Lumbsch T, O’Donnell K, Binder M, Diederich P, Ertz D, Gueidan C, Hansen K, Harris R, Hosaka K, Lim Y, Matheny B, Nishida H, Pfister D, Rogers J, Rossman A, Schmitt I, Sipman H, Stone J, Sugiyama J, Yahr R, Vilgalys R (2004) Assembling the fungal tree of life: progress, classification, and evolution of subcellular traits. Am J Bot 91:1446–1480
Marcet-Houben M, Gabaldon T (2009) The tree versus the forest: the fungal tree of life and the topological diversity within the yeast phylome. PLoS One 4:e4357
McKintosh E, Tabrizi SJ, Collinge J (2003) Prion diseases. J Neurovirol 9:183–193
Mullan LJ (2002) Multiple sequence alignment—the gateway to further analysis. Brief Bioinform 3:303–305
Page RD (1998) GeneTree: comparing gene and species phylogenies using reconciled trees. Bioinformatics 14:819–820
Patel BK, Liebman SW (2007) “Prion-proof” for [PIN+]: infection with in vitro-made amyloid aggregates of Rnq1p-(132–405) induces [PIN+]. J Mol Biol 365:773–782
Peterson SW (2008) Phylogenetic analysis of Aspergillus species using DNA sequences from four loci. Mycologia 100:205–226
Philippe H, Snell EA, Bapteste E, Lopez P, Holland PW, Casane D (2004) Phylogenomics of eukaryotes: impact of missing data on large alignments. Mol Biol Evol 21:1740–1752
Pisani D, Cotton JA, McInerney JO (2007) Supertrees disentangle the chimerical origin of eukaryotic genomes. Mol Biol Evol 24:1752–1760
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144
Ragan MA (1992) Matrix representation in reconstructing phylogenetic relationships among the eukaryotes. Biosystems 28:47–55
Robbertse B, Reeves JB, Schoch CL, Spatafora JW (2006) A phylogenomic analysis of the Ascomycota. Fungal Genet Biol 43(10):715–725
Saupe SJ (2007) A short history of small s: a prion of the fungus Podospora anserina. Prion 1:110–115
Scannell DR, Byrne KP, Gordon JL, Wong S, Wolfe KH (2006) Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts. Nature 440:341–345
Scannell DR, Frank AC, Conant GC, Byrne KP, Woolfit M, Wolfe KH (2007) Independent sorting-out of thousands of duplicated gene pairs in two yeast species descended from a whole-genome duplication. Proc Natl Acad Sci USA 104:8397–8402
Schoch CL, Sung GH, Lopez-Giraldez F, Townsend JP, Miadlikowska J, Hofstetter V, Robbertse B, Matheny PB, Kauff F, Wang Z (2009) The Ascomycota tree of life: a phylum-wide phylogeny clarifies the origin and evolution of fundamental reproductive and ecological traits. Syst Biol 58:224
Si K, Choi YB, White-Grindley E, Majumdar A, Kandel ER (2010) Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 140:421–435
Slowinski JB, Page RD (1999) How should species phylogenies be inferred from sequence data? Syst Biol 48:814–825
Steenkamp ET, Wright J, Baldauf SL (2006) The protistan origins of animals and fungi. Mol Biol Evol 23:93–106
Suh SO, Blackwell M, Kurtzman CP, Lachance MA (2006) Phylogenetics of Saccharomycetales, the ascomycete yeasts. Mycologia 98:1006–1017
Tanaka M, Chien P, Naber N, Cooke R, Weissman JS (2004) Conformational variations in an infectious protein determine prion strain differences. Nature 428:323–328
Wehe A, Bansal MS, Burleigh JG, Eulenstein O (2008) DupTree: a program for large-scale phylogenetic analyses using gene tree parsimony. Bioinformatics 24:1540–1541
Wickner RB (1994) [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264:566–569
Wickner RB, Shewmaker F, Edskes H, Kryndushkin D, Nemecek J, McGlinchey R, Bateman D, Winchester CL (2010) Prion amyloid structure explains templating: how proteins can be genes. FEMS Yeast Res 10:980–991
Wong KM, Suchard MA, Huelsenbeck JP (2008) Alignment uncertainty and genomic analysis. Science 319:473–476
Yuan X, Xiao S, Taylor TN (2005) Lichen-like symbiosis 600 million years ago. Science 308:1017–1020
Acknowledgments
We wish to acknowledge the financial support of the Irish Health Research Board (HRB). We also wish to acknowledge the SFI/HEA Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support. EMMT was funded through a SFI-UREKA summer school and would like to thank Dr James McInerney and Dr Davide Pisani. He also wishes to thank his academic advisor Prof. Silvia Restrepo.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic Supplementary material.
Rights and permissions
About this article
Cite this article
Medina, E.M., Jones, G.W. & Fitzpatrick, D.A. Reconstructing the Fungal Tree of Life Using Phylogenomics and a Preliminary Investigation of the Distribution of Yeast Prion-Like Proteins in the Fungal Kingdom. J Mol Evol 73, 116–133 (2011). https://doi.org/10.1007/s00239-011-9461-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-011-9461-4