
Abstract
The diversity of functional genes and the related processes are important issues for conservation biology. This is especially relevant for populations that have suffered from demographic reduction as a consequence of the processes of postglacial colonization. In this perspective, the aims of the present study are (1) to quantify the genetic diversity of functional genes and (2) to disentangle the long- and short-term effects of natural selection that shapes genetic diversity from those of drift, mutation, and allopatric fragmentation. This research was conducted using an extensive genetic polymorphism analysis of populations of longnose dace (Rhinichthys cataractae) living over an area once covered by Pleistocene glaciations. The sequence and diversity of one exon of three genes (MHC IIβ, growth hormone, and trypsin) were jointly analyzed with non-coding nuclear loci from 27 populations; these populations were sampled over four major basins of northeastern North America. The survey revealed a surprisingly low allelic richness, especially for the MHC gene, considering the number of individuals and populations sampled. The results suggest that there is a complex mixture of different evolutionary processes shaping the level of polymorphism among longnose dace. While our study underlines the importance of the short-term effects of neutral processes and the major impact of post-glacial colonization on gene diversity, locally dependent balancing selection was detected on MHC. From this perspective, our results support an understanding of the importance of drift on functional gene diversity but also highlight the transient effects of natural selection on allelic composition, even in populations that show drastic reduction of genetic diversity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Local adaptation is defined as the evolution, through natural selection, of traits that have high fitness in specific habitat conditions. Because functional genes are the foundation of these traits, their genetic diversity is believed to dictate the adaptive capacity of individuals and the persistence of a population through environmental changes (Ford 2002). As such, the identification of the processes that shape the diversity of these genes is a major concern for the prediction of evolutionary trajectories of populations and represents an important issue in conservation biology.
In this regard, genes that are directly associated with individual survival have received significant attention (Suprunova et al. 2004; Ingvarsson et al. 2006). Among gene candidates in this category, those of the major histocompatibility complex (hereafter MHC) have seemingly received the most attention. These genes, which provide the foundation of the immune system of vertebrates, encode several receptors that bind protein fragments of both viral and bacterial antigens and, therefore, reduce the occurrence of infection and disease (Bernatchez and Landry 2003; Fraser and Neff 2009). Despite numerous studies performed in experimental, semi-experimental, or natural environments (reviewed in Bernatchez and Landry 2003), generalizations about the mechanisms driving MHC evolution remains mostly unclear. While many have suggested long-term effects of selection (Sauermann et al. 2001; Miller et al. 2001; Froeschke and Sommer 2005; van Oosterhout et al. 2006; Heath et al. 2006; Wegner 2008; among others), a consensus has not been reached regarding the type of selection involved. The short-term mechanisms driving variation in allele frequencies within and among populations have been as much, if not more, ambiguous. Indeed, a growing number of studies suggest large variations in the relative importance of selection (Slade 1992; Landry and Bernatchez 2001; Miller et al. 2001; Campos et al. 2006; Aguilar and Garza 2007) and stochastic processes (Hayashi et al. 2005; Langefors 2005; Peters and Turner 2008) that shape MHC diversity in natural populations. Although these studies were restricted to MHC genes, the ambiguity among conclusions from different studies (sometimes performed on the same taxa) demonstrates the difficulties of interpreting natural evolutionary mechanisms when using few DNA segments from either a restricted number of populations or a limited spatial scale. The genetic diversity of populations is the result of neutral and/or non-neutral processes acting at different evolutionary time scales. As a result, the effects of natural selection are entangled with both the short-term effects of drift and gene flow and the long-term effects of mutation rate and allopatric fragmentation (Excoffier 2001; Otto 2000), both of which are often locally specific. This is especially relevant for populations living over post-glaciated areas, which are believed to have suffered from drastic reduction of genetic diversity during and since the colonization of these regions (Bernatchez and Wilson 1998; Hewitt 2000).
A joint analysis of sequence, frequency, and genotype diversities from numerous genetic markers observed within several populations located on a wide geographic area would be the most effective way that may help identify the evolutionary processes acting on genes. The objective of this study was to increase understanding of the mechanisms acting on functional genes over a territory once covered by Pleistocene glaciations; to do so, a common Cyprinidae species of North America was adopted to evaluate the differential effects of natural selection and neutral evolutionary processes on targeted coding regions. Level of diversity was then quantified on (1) a single exon from three genes: the MHC class IIβ, the growth hormone (GH), and the trypsin (TRY) digestion enzyme and (2) seven neutral markers (microsatellites) within a set of 27 natural populations sampled in an area over which the postglacial colonization has previously been investigated by Girard and Angers (2006b).
Materials and Methods
Model Species and Sampling
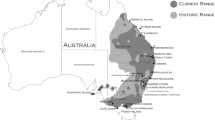
The longnose dace (Rhinichthys cataractae) is a fish species typical of turbulent rivers found in almost all the temperate habitats of North America, from Mexico to the Yukon (Scott and Crossman 1973; Fig. 1a). These fish have little interaction with humans; thus, they are expected to be almost undisturbed by stocking activities or translocations (Scott and Crossman 1973). A total of 542 individuals were sampled by electrofishing (LR-24, Smith-Root) from 27 rivers of the Quebec province (Canada) (Fig. 1b, c, d; Table 1). These rivers are located within four major drainage basins (James Bay, Saint Lawrence River, Outaouais River, and Saguenay River). Sample sizes ranged between 10 and 24 individuals. For each individual, a piece of the caudal fin was removed and stored in 95% ethanol for molecular analyses. In addition, individuals belonging to a closely related species, the blacknose dace (R. atratulus), were analyzed as an outgroup.

Geographic position of populations and their MHC (a), GH (b), and TRY (c) compositions. The current distribution of the longnose dace in North America is presented in gray. In maps, black and gray identifiers represent populations for which Fu or Li test was significant or not, respectively, while white identifiers indicate populations for which the test was not performed (see text). Finally, the minimum spanning network between the longnose dace and blacknose dace (Bd) alleles is shown for each gene. The colors in the trees correspond to those presented in pie charts. The black circles correspond to alleles not found in this study
Molecular Analyses
Functional Genes
We focused our analyses on the second exon of the MHC class IIβ because of its key role in peptide binding (Ono et al. 1993) and its known polymorphism (Garrigan and Edwards 1999; Landry et al. 2001). Primers were designed on the 5′ end of this exon and the 3′ end of the third exon according to conserved sequences observed among several MHC DAB gene sequence data of Danio rerio available on Genbank. By polymerase chain reaction (PCR), the primers 5′-CCAGTGACTACAGTGATATGG-3′ and 5′-TGGAGGTCACATCTGAGG-3′ successfully amplified a segment of approximately 600 bp (which covered the 200 last bp of the second exon, the first 150 bp of the third exon, and the entire intron between them). The following reaction conditions were used: a 12.5-μl reaction aliquot containing 1.5 mmol l−1 of MgCl2, 2.5 nmol l−1 of each dNTP, 0.2 unit of Taq polymerase, 1.25 μl of 10× Taq polymerase buffer (Invitrogen), and approximately 20 ng of DNA. Reaction conditions included an initial denaturation of 10 s at 92°C; followed by 45 cycles combining 15 s at 92°C, 10 s at 54°C, and 30 s at 68°C; and a final extension of 120 s at 68°C. A reverse primer specific to the longnose dace and in closer proximity to the second exon was then designed (5′-AGTGGACACACATGGTGAG-3′), which led to the amplification of a segment of 295 bp (which covered 229 bp on the exon and 66 bp on the intron). The PCR reaction and conditions that optimized this amplification were the same as outlined above, except using an annealing temperature of 50°C. The polymorphism of this locus was then screened using the single strand conformation polymorphism (SSCP) (Orita et al. 1989). The locus was electrophoresed on a 6% nondenaturing gel for 10 h at 20 W in 0.5X TBE. To ensure the reliability of similar migration patterns among populations, the different patterns from each population were sequenced for at least one individual.
No clear information is available on the function and polymorphism of each exon of GH and TRY in fish. Consequently, determining which GH and TRY regions to analyze was not as straightforward as in the case of MHC. For each locus, we used the following procedure to identify an appropriate DNA segment. The longer exons were targeted among the five that constituted each gene. This choice was based on a strict probabilistic assessment that longer exons are more susceptible to accumulating mutations. Primers were thus designed (according to homologous sequences available for other fish species in Genbank) for the fourth (~160 bp) and the fifth (~201 bp) exons of GH, and the second (~160 bp) and third (~260 bp) exons of TRY. GH primers were designed according to conservation sequences found among very divergent species (Cyprinus carpio #X51969 and Hypophthalmichthys molitrix #M94348), and TRY primers were designed according to the genomic sequence #BX539313 of D. rerio. Successful amplifications were performed on longnose dace for all loci except the fourth exon of GH, which was discarded. A SSCP screening was then performed on a total of 12 R. cataractae individuals from four distant populations (populations 1, 8, 19, and 29) to evaluate the polymorphism potential of the three remaining loci. The fifth exon of GH and the third exon of TRY showed polymorphism. No variation was observed for the second exon of TRY and, as such, it was discarded. The PRC conditions of the two loci selected are presented below.
For GH, the primers 5′-CCATTTGTATCTGCACAG-3′ and 5′-TACAGGCATTGACTAAC-3′ successfully amplified a segment of 255 bp that covered the entire fifth exon and 36 bp of the following intron. For TRY, primer pairs 5′-CGTCTGGGTGAGCACAAC-3′ and 5′-TCCCCATCCAGAGATCAGAC-3′ amplified a segment of 222 bp of the third exon. The PCR reactions and conditions that optimized amplification were the same for both sets of primers. The following reaction was used: a 12-5 μl reaction aliquot containing 1.5 mmol l−1 of MgCl2, 2.5 nmol l−1 of each dNTP, 0.2 unit of Taq polymerase, 1.25 μl of 10× Taq polymerase buffer (Invitrogen), and approximately 20 ng of DNA. Reaction conditions included an initial denaturation of 10 s at 92°C: followed by 45 cycles combining 15 s at 92°C, 10 s at 50°C, and 30 s at 68°C; and a final extension of 120 s at 68°C. The polymorphisms of both genes were evaluated on all longnose dace individuals following the same procedure as that used for MHC and as described above. Sequencing was performed for each individual, which showed variation in the migration pattern as explained above in the case of MHC.
The expression of the three coding regions delineated above was confirmed through cDNA amplification on two individuals. For each individual, the total RNA was isolated and purified using the RNeasy Mini Kit (QIAGEN) from 30 mg of muscle tissue previously stabilized in RNA stabilization reagent (RNAlater). Total RNA was then reverse-transcripted into single-strand cDNA using the ProtoScript® First Strand cDNA Synthesis Kit. The resulting cDNA products were used directly in several PCR to confirm the expression of the three genes analyzed. As we were using primers designed in different exons of a given gene, we expected the amplification product that covered an intron in the genomic DNA to be shorter when applied on cDNA. PCRs were performed on cDNA, genomic DNA, and negative templates.
Microsatellite Polymorphism
Populations were screened with seven microsatellite loci (Rhca15b, Rhca16, Rhca20, Rhca23, Rhca31, Rhca34, and Rhca52) according to conditions described by Girard and Angers (2006a, 2008). The polymorphism of the microsatellite markers was evaluated with 6% denaturing urea-polyacrylamide gel electrophoreses.
Statistical Analyses
Sequence Analyses
Long-term effects of natural selection are expected to leave a signal on gene sequences that would be unexpected under neutral evolution. To detect these potential effects, we first performed the McDonald and Kreitman (MK) test (McDonald and Kreitman 1991), available in DNASP 4.10 (Rozas et al. 2003), which evaluates the level of synonymous and non-synonymous substitutions within and between species. Under neutrality, the ratio between among species divergence and within species polymorphism should be equal for synonymous and non-synonymous mutations. Therefore, sequences of each exon were aligned using CLUSTAL W (Thompson et al. 1994). MK test was then performed using three (Genbank #AJ297822, BX539313, and BC092664) and five (Genbank #BC124461.1, AY103492.1, L04805.1, L04808.1, and L04815.1) homologous sequences of D. rerio for TRY and MHC, respectively, and five sequences from C. carpio (Genbank #AJ640136.1, AY553378.1, AY553379.1, AY822624.1, and DQ350436.1) for GH. These outgroup species were chosen because no fixed differences were observed between longnose dace and blacknose dace sequences, which is a condition required to compute the MK test.
Sequences were also analyzed using the HKA tests (Hudson et al. 1987), which explore potential balancing selection effects. The test predicts that, under neutrality, regions of the genome that evolve at high rates will present high levels of polymorphism within species. The test was performed between each pair of genes. For these analyses, the Genbank sequences of D. rerio #NW_001513985.1, NW_001513092.1, and NW_001511399.1 were used for MHC, GH, and TRY homologous sequences, respectively.
Gene Diversity Within Populations
Genetic diversity within populations was calculated using both allele frequencies and sequence information. The Nei’s gene diversity (H E) and the allelic richness (R) according to the rarefaction index of El Mousadik and Petit (1996) were computed using the program FSTAT v.2.9.3 (Goudet 2001). Molecular diversity indices within each sample were evaluated from the observed number of segregating sites (θS) and from the mean number of pairwise differences (θπ) using ARLEQUIN v3.1 (Excoffier 2006).
Short-term effects of natural selection are believed to modify allelic and genotypic diversities in unexpected ways: as such, when natural selection occurs, it is no longer possible to predict the genetic diversity from the demographic history and structure among populations. We evaluated these impacts using several strategies. First, R and H E computed on genes in each populations were correlated to those computed on non-coding microsatellite loci. Under neutrality, a significant positive correlation should be observed. The significance of this correlation was evaluated using a Pearson statistic tested with 999 random permutations of the data. Second, the effects of selection on genes were assessed by (1) examining their deviation from mutation/drift expectations and (2) using the Hardy–Weinberg equilibrium (HWE). These effects were first evaluated on microsatellites in each population. Analyses were performed using the BOTTLENECK program (Cornuet and Luikart 1996): an infinite allele mutation (IAM) model, and a two-phase mutation (TPM) model (the latter used proportions of single-step mutations of 0.25, 0.50, or 0.75 and a multistep change variance of 30 repeats) were employed. The temporal stability of the effective size of each population was further evaluated using the Wilcoxon sign-rank test (Luikart and Cornuet 1998). Deviations from HWE were tested using the exact test of Guo and Thompson (1992) implemented in the software GENEPOP v3.4 (Raymond and Rousset 1995). Populations showing significant departures from mutation/drift and/or HW equilibriums on microsatellites were removed from further analyses because of the impossibility to distinguish natural selection effects from those of local demographic processes in such populations.
In the remaining populations, the accordance with the mutation/drift expectations was evaluated on the three genes with the Ewen–Watterson neutrality test (Watterson 1978), the D of Tajima (1989), and the F of Fu and Li (1993). These analyses were performed overall and within each sample using the program ARLEQUIN v3.1. Subsequently, deviation from HWE was evaluated using the same method that was used for the microsatellites.
Gene Diversity Among Populations
Impacts of natural selection could also be potentially observed on the among populations differentiation, which can be significantly higher (directional selection) or lower (balancing selection) for genes under selection than for neutral markers. Using FSTAT, overall F ST was computed with MHC, GH, TRY, and microsatellites. The neutral expectations of the overall population differentiation for MHC, GH, TRY, and microsatellites were evaluated by the method proposed by Beaumont and Nichols (1996) and implemented in the software FDIST2. The average overall F ST across markers (microsatellites and genes) was used in the simulations. This value was targeted after an exploratory analysis based on the indications of Beaumont that can be found in the FDIST2 package. Accordingly, a robust starting F ST value should yield an output where the observations will be distributed almost equally below and above the average line. Simulations were performed with an infinite allele model. A total of 50,000 simulations were performed to obtain an acceptable compromise between precision and computation time. Each simulation was composed of 100 demes, from which a number of populations were sub-sampled to match the number of populations in the longnose dace dataset. Sample size was defined as the average number of gene copies among the samples. The observed F ST value for each marker was then compared to the simulation intervals according to their H E.
Effects of natural selection among populations were also explored using the Bayesian method implemented in BAYESCAN 2.01 (Foll and Gaggiotti 2008). The approach introduced the effect of selection by decomposing each F ST coefficient into a population component (beta) which is shared by all loci, and a locus-specific component (alpha) which is shared by all populations. Given the data, posterior probabilities were computed over two models for each locus: one that included selection (alpha + beta) and one that exclude it (beta). In this study, posterior probabilities were defined using 50,000 iterations, which were sampled every 10 iterations; each chain was preceded by 50 pilot runs of 5000 iterations and an additional burning period of 10,000 iterations. For each run, the stationarity of the posterior distribution of all parameters through the sampled iterations was assessed using the Geweke statistic (Geweke 1992) available in the boa R package (Smith 2007). Finally, departure from neutrality was assessed using the ratio of the posterior probabilities (posterior odds) of each model. The prior odds of the neutral model were set to 1 (minimum value) and 10 (default value). As explained in the BAYESCAN user manual, prior odds influence both the power of the method and the probability of finding false positives. Increasing the prior odds value will tend to eliminate these errors but does so at the cost of reducing the power of the method.
Results
Sequence Analyses
The analyses performed on total RNA confirmed the transcription of the MHC, GH, and TRY genes. In each case, the PCR products resulting from cDNA amplifications were shorter than those obtained from genomic DNA. As expected, the size of the cDNA amplification products were equal to that of their respective spliced genomic DNA, which strongly suggests that introns were removed and exons joined within the cDNA of all the three genes analyzed.
Only four distinct alleles were observed on the MHC exon among the longnose dace populations. Sequencing replicates (see Method) confirmed that SSCP and sequencing were reliable. These alleles (MHC-I to MHC-IV; Genbank #EU848571–EU848574) were characterized by a total of 11 polymorphic sites. Among them, five were parsimony-informative (125, 126, 135, 139, and 144). Alleles differed by an average of 6.3 substitutions. Ten substitutions resulted in amino acid replacements, and only one substitution was synonymous. The non-synonymous nucleotide diversity (0.032) was twice that of the synonymous nucleotide diversity (0.014). In spite of an over-representation of non-synonymous substitutions in polymorphic versus divergent sites (which provided an NI (neutrality index) of 3.75), the MK test was not significant (G = 1.46; P = 0.23).
Both GH and TRY showed low polymorphisms, which are mostly represented by synonymous substitutions. Again, sequencing replicates confirmed SSCP reliability. The GH exon was close to fixation among the longnose dace populations. Only two alleles (GH-I and GH-II; Genbank #EU836876–EU836877) that differed by a single non-synonymous mutation were observed, which resulted in a nucleotide diversity of 0.004. The MK test was not significant (G = 0.618; P = 0.43). Three alleles were observed on the TRY exon (TRY-I to TRY-III; Genbank #EU836882–EU836884). These alleles diverged by a single synonymous (117) and two non-synonymous (16 and 109) substitutions. The nucleotide diversity of the non-synonymous sites (0.008) was slightly lower than that of synonymous sites (0.013), and the MK test was not significant (G = 0.387, P = 0.533). In addition, none of the HKA tests provided significant results, suggesting that polymorphisms of the MHC, GH, and TRY genes were not under selection.
The minimum spanning network of MHC alleles revealed that MHC-I and MHC-II are closer to blacknose dace alleles (average of 3.5 substitutions) than to MHC-III and MHC-IV (average of 8 substitutions; Fig. 1a). Interestingly, the same pattern was observed for GH and TRY, whereby one allele of each gene showed the identical sequence in both species (Fig. 1b, c).
Microsatellite Diversity Within Populations
Seven populations (9, 12, 13, 18, 26, 27, and 31) were not in accordance with HWE (Table 1). However, only three populations (12, 13, and 18) showed a highly significant deviation (P < 0.001) for more than one locus. Consequently, only these populations were considered as deviating from panmixia and were removed from the further analyses of genetic diversity within populations.
According to the Wilcoxon rank-tests, three populations (3, 6, and 9) were in discordance with mutation–drift equilibrium under three out of four sets of mutation assumptions with P < 0.05. All of the other populations appeared to be in accordance with mutation–drift equilibrium under all mutation assumptions, except populations 16 (IAM) and 26 (TPM with a proportion of single-step mutations of 0.75); for both of these populations, the analysis was significant at the 0.05 threshold. This discordance with demographic equilibrium was, however, not considered as severe as that of the three previous populations. These two populations were thus retained for further analyses.
Gene Diversity Within Populations
Genetic diversity within populations varied significantly among the three functional genes (Table 1). For instance, when R was computed on MHC, GH, and TRY, the resulting average was 2.4, 1.2, and 1.6 alleles by population, respectively. This difference is highly significant (F = 21.19; P < 0.0001). Significant differences were also observed for H E (F = 14.34; P < 0.001), θS (F = 41.27; P < 0.0001), and θπ (F = 30.93; P < 0.0001). GH and TRY were either fixed or in accordance with both HW and mutation–drift equilibriums (Table 1). In contrast, the MHC exon showed an excess of homozygotes (27 and 29), an excess of diversity (8), or both (2 and 14). That most of the populations were at HWE suggests the absence of null alleles for these genes. Results obtained with the D of Tajima were almost never significant, with the exception of two populations for MHC (7 and 24) and one for TRY (26; Table 1). Furthermore, the overall D for MHC (1.34; P = 0.08), GH (−0.40; P = 0.31), and TRY (0.81; P = 0.19) was not significant.
The F of Fu and Li showed high discordances between MHC and the two other genes. Nine populations (hereafter referred to as the “F SIGN subset”) showed a higher molecular diversity between alleles than expected under neutrality. In contrast, 10 populations (the F NS subset) followed the neutral expectation (Table 1). The populations from the F SIGN subset also showed a higher number of non-synonymous substitution differences between each pair of alleles. Indeed, between 1.24 (pop. 15) and 1.75 (pop. 1) non-synonymous substitutions were observed within the F SIGN subset as compared to between 0.27 (pop. 16) and 1.29 (pop. 30) in the F NS one (Fig. 2). The difference between the averages was highly significant (t = 7.591; P < 0.0001). This was not observed within populations (Table 1) or within over all populations for either GH (F = −0.42; P = 0.21) or TRY (F = 2.76; P = 0.14).

Average number of nonsynonymous substitutions between each pair of MHC alleles within populations of F SIGN (gray) and F NS (white) subsets. Average numbers of nonsynonymous substitutions between each pair of MHC alleles within subsets (dashed lines) and overall averages (solid lines) are also presented
The MHC richness was significantly correlated to the richness of microsatellites (r = 0.42; P < 0.05; Fig. 3a). While the correlation remained significant when only the F NS populations were taken into account (r = 0.53; P < 0.05; Fig. 3a), a complete absence of correlation was observed when the F SIGN subset (r = −0.002; P = 0.55) was considered. Similar trends were observed for H E (Fig. 3b). Owing to its low level of diversity, the results of GH were not very informative (Fig. 3c, d); yet it is worth noting that TRY indices of diversity displayed strong correlations with those of microsatellites. In this context, the results of TRY were similar to those observed on MHC on the F NS subset (Fig. 3e, f).

Relationships between genes and microsatellites R and H E within each population. In the MHC figures, white squares and black circles represent populations within F SIGN and F NS subsets, respectively. Correlation values were calculated using all of the populations
Diversity Among Populations
The neutral expectations of the overall population differentiation assessed using the simulation procedures of FDIST2 showed that all microsatellites were within the F ST 0.99 confidence interval, regardless of whether F SIGN and F NS populations were considered together (Fig. 4a) or separately (Fig. 4b, c). When all populations are taken into account, MHC, GH, and TRY all fell within the neutral F ST interval (Fig. 4a). The F ST values computed over the F NS subset for MHC and TRY were also in accordance with neutral expectations (Fig. 4c). Computation could not be performed for GH because it was monomorphic in this population subset. However, while GH and TRY showed an F ST value in the neutral interval in the F SIGN subset, the F ST computed on MHC was clearly below the lower limit of the neutral confidence interval (Fig. 4b).

Average (bold lines), 95% (solid lines), and 99% (dashed lines) confidence intervals for the relationships between F ST and H E according to FDIST2 simulations performed with a sample of 20 (a), 9 (b), and 11 (c) populations. Observed values of the seven microsatellites (empty circles) and the three genes (filled circles) computed overall F SIGN and F NS (a), only F SIGN (b), and only F NS (c) are superimposed on these simulation results. GH result is not presented in c because it was monomorphic within this subsample
The BAYESCAN approach provided the same results when prior odds for the neutral model were set to 1. The approach was not powerful enough to detect selection on any loci when prior odds were set to 10 (results not shown). While F ST of MHC was not considered an outlier when either all populations (Fig. 5a) or only F NS subset (Fig. 5c) were taken into account, its value was above the neutrality maximum threshold when only the F SIGN populations were considered (Fig. 5b). The two other genes remained in accordance with the neutral expectations (Fig. 5a, b, c). The effect of selection on MHC across population subsets is also visible through the posterior probability density of its alpha parameter. Centered at 0 when all populations (Fig. 5d) and F NS subset (Fig. 5f) are considered, the distribution shifts toward an average of −1 when considering only the F SIGN subset (Fig. 5e). According to Foll and Gaggiotti (2008), this negative value is associated with balancing selection. Selection was also diagnosed on two microsatellites (Rhca34 and Rhca52). Interestingly, the F ST values of these two microsatellites were also positioned close to the upper bound of the 0.99 confidence interval defined by FDIST2; this is an additional indication that the two methods converged to the same results.

Posterior odds in favor of the selection model (PO) for each microsatellite (empty circles) and gene (filled circles) according to the BAYESCAN approach (whereby the locus-specific F ST is on the y-axis) computed overall (a), only F SIGN (b), and only F NS (c) subsets. Bold lines correspond to the PO thresholds leading to a false discovery rate of no more than 5%. These thresholds were computed using the R function provided with the BAYESCAN package. GH was removed from the F NS subset analysis because it was monomorphic within this subsample. Panels (d) to (f) correspond to the posterior distributions of the MHC alpha parameter according to BAYESCAN when performed overall (d), only on F SIGN (e), and only F NS (f) subsets
Discussion
In this study, we presented an extensive comparison between neutral markers and coding genes among several populations to determine the effect of the short- and long-term evolutionary processes that shape the diversity of specific functional genes in longnose dace over a territory once covered by Pleistocene glaciations. For all three exons, the allelic richness was extremely low. While these results were not as surprising for GH and TRY, which are probably under the action of directional and/or purifying selection, the few distinct alleles (four alleles) found on the MHC exon were astonishing. Because of the potential importance of co-evolution in host–pathogen systems such as the one involving MHC, allelic diversity is believed to be of prime importance for the viability of populations (Bernatchez and Landry 2003). The MHC allelic richness observed in this study for the longnose dace, while being comparable with a few other studies (see Peters and Turner 2008; and references therein), remains surprisingly low compared with the majority of studies performed on MHC class ΙΙβ (Froeschke and Sommer 2005: 58 individuals, 20 alleles; Aguilar and Garza 2006: 450 individuals, 88 alleles; and Ottovà et al. 2007: 30 individuals, 48 alleles; and others), especially considering the high number of individuals analyzed (542). We acknowledge the fact that some rare MHC alleles may have been missed, as shown by the four populations with slight excesses of homozygotes. Nevertheless, we are confident that the most frequent MHC lineages were detected because (1) amplifications in this study resulted in at least one and no more than two bands in every individual analyzed, and (2) most populations were under HWE. Furthermore, the absence of STOP codons and the exon-coding expression revealed by the total RNA examination confirmed that the segment selected was not part of a pseudogene.
Long-term Processes
The sequence analyses performed in this study revealed few clues about the long-term impacts of natural selection on any of the genes surveyed. While the high ratio of non-synonymous/synonymous substitutions that was observed on MHC could be in accordance with a historical effect of diversifying selection (Hughes and Nei 1988; Froeschke and Sommer 2005; Schad et al. 2005; Aguilar and Garza 2006), the MK test remains non-significant, and the effect of stochastic mutations cannot be rejected. The HKA tests also failed to detect other forms of selection in any of the genes studied. Biological interpretations of these results remain difficult to perform since the restricted number of alleles observed within our population set has likely considerably decreased the power of these analyses.
The incomplete lineage sorting observed between longnose and blacknose daces for all the three genes must also be interpreted with caution. The maintenance of alleles (or allele lineages) through speciation events is often associated with the action of natural selection (Mayer et al. 1992; O’Brien and Yuhki 1999; Bryja et al. 2006). However, conservations of primer sites and ranges of overlap of allelic size between both species were also observed on several microsatellite loci expected to be neutral (Girard and Angers 2006a). Such similarity throughout the genome is not in accordance with selection and thus suggests that other evolutionary processes may have been operating. Potential hybridization between both Rhinichthys species (while never reported to our knowledge), large effective population size, or recent speciation appears more likely to explain incomplete lineage sorting between both the species (Nei 1987; Pamilo and Nei 1988; Takahata 1989).
Short-term Processes
Despite not having detected many alleles, analyses performed to identify short-term selection processes were successful, at least with regard to the MHC exon. One of the most striking results of this study is the separation of populations into two different subsets, which was suggested by the Fu and Li statistic. The genetic and molecular diversities of MHC appeared to be strongly related to those observed on neutral markers in almost half of the sampled populations (F NS populations). We cannot reject the hypothesis that the evolution of the MHC exon in these populations resulted mainly from random effects associated with demographic processes following the postglacial colonization. Campos et al. (2006) found similar results in brown trout (Salmo trutta) populations, where genetic drift had appeared to become the predominant evolutionary force shaping genetic variation and had eroded the effect of selection.
On the other hand, the MHC gene and molecular diversities of nine populations (F SIGN subset) appear to depart from neutral expectations. Significant F tests, lower population differentiation than expected, and the absence of a correlation between the diversity of MHC and microsatellites appear to reflect the effects of a balancing selection process. The fact that these results were not observed on the other genes (especially TRY) and microsatellites increases the plausibility of the hypothesis that specific selective evolutionary processes acted on MHC at least in these populations.
Interestingly, these populations have the highly divergent allele MHC-III in common. The combination of this allele with either MHC-I or MHC-II is clearly the reason why the Fu and Li test was significant in these specific populations. Despite their similar MHC composition, these populations are spread across the entire sampling area (Fig. 1a); this is not expected under neutral evolution considering the strong effects of post-glacial colonization on the geographic organization of the genetic diversity of these populations (Girard and Angers 2006b). The fact that the presence of the MHC-III allele has few relationships with the allopatric fragmentation pattern of the species may indicate the effects of non-neutral mechanisms, which seek to maintain high genetic diversity on this exon by keeping this highly divergent allele.
Nevertheless, the mechanisms behind the maintenance of the MHC-III allele have yet to be identified. In these populations, if selection was apparent on allelic frequencies, then all genotype frequencies were in accordance with neutral expectations. Consequently, if mechanisms such as overdominance, diversifying selection through heterozygote advantage, or disassortative mating did occur, they were not detected. While both small sampling sizes and low number of alleles could have reduced the power to detect these biological mechanisms, such discordance between selectively allelic and neutral–genotypic frequencies has already been observed on MHC (Hayashi et al. 2005). This discordance was interpreted as the result of natural selection on the long-term evolution of MHC, and drift was understood as the main process impacting short-term evolution. However, there is a clear necessity for additional investigations to ensure that current failure to detect either selection or the biological mechanisms guiding selection are not solely due to low gene diversity or sampling sizes.
In conclusion, there is no doubt that the lack of diversity observed considerably reduced our capacity to interpret the shaping mechanisms behind functional genes of longnose dace over the surveyed area. However, the depauperate allelic pool being observed on all genes has likely been influenced by the important diversity reduction during post-glacial colonization, which may have overcome the effects of natural selection. The fact that few alleles were observed (and seemed to evolve neutrally in several populations) may temper the importance of functional gene diversity (especially on MHC) on population viability, as suggested in previous study (Campos et al. 2006; Babik et al. 2009). However, while our study underlines the major impact of post-glacial colonization on the diversity of functional genes, it is noticeable that even with such a low number of alleles, locally dependant balancing selection was detected on MHC; this supports the idea that these specific genes evolve under strong selective evolutionary mechanisms. Nonetheless, the lack of diversity observed so far in this species on both neutral and non-neutral markers over the wide area surveyed is evident and may affect the persistence of these populations in the face of future environmental changes. Considering the significant imprint of the Pleistocene glaciations throughout the northern hemisphere, further studies on functional gene diversity appears appropriate to evaluate and generalize their impacts on the persistence of the numerous species affected by these major disturbances of the past.
References
Aguilar A, Garza JC (2006) A comparison of variability and population structure for major histocompatibility complex and microsatellite loci in California coastal steelhead (Oncorhynchus mykiss Walbaum). Mol Ecol 15:923–937
Aguilar A, Garza JC (2007) Patterns of historical balancing selection on the salmonid major histocompatibility complex class II β gene. J Mol Evol 65:34–43
Babik W, Pabijan M, Arntzen W, Cogalniceanu D, Durka W, Radwan J (2009) Long-term survival of a urodele amphibian despite depleted major histocompatibility complex variation. Mol Ecol 18:769–781
Beaumont MA, Nichols RA (1996) Evaluating loci for use in the genetic analysis of population structure. Proc Roy Soc B 263:1619–1626
Bernatchez L, Landry C (2003) MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol 16:363–377
Bernatchez L, Wilson C (1998) Comparative phylogeography of nearctic and palearctic fishes. Mol Ecol 7:431–452
Bryja J, Galan M, Charbonnel N, Cosson JF (2006) Duplication, balancing selection and trans-species evolution explain the high levels of polymorphism of the DQA MHC class II gene in voles (Arvicolinae). Immunogenetics 58:191–202
Campos JL, Posada D, Moran P (2006) Genetic variation at MHC, mitochondrial and microsatellite loci in isolated populations of Brown trout (Salmo trutta). Conserv Genet 7:515–530
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
Excoffier L (2001) Analysis of population subdivision. In: Balding DJ, Bishop MJ, Cannings C (eds) Handbook of statistical genetics. Wiley, London, pp 271–308
Excoffier L (2006) Arlequin: an integrated software for population genetics data analysis Version 3.1. Distributed by the author, Computational and Molecular Population Genetics Lab, University of Berne, Bern. http://cmpg.unibe.ch/software/arlequin3/
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180(2):977–993
Ford MJ (2002) Applications of selective neutrality tests to molecular ecology. Mol Ecol 11:1245–1262
Fraser BA, Neff BD (2009) MHC class IIB additive and non-additive effects on fitness measures in the guppy. J Fish Biol 75:2299–2312
Froeschke G, Sommer S (2005) MHC Class II DRB Variability and parasite load in the Stripped mouse (Rhabdomys pumilio) in the Southern Kalahari. Mol Biol Evol 22:1254–1259
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Garrigan D, Edwards SV (1999) Polymorphism across an exon-intron boundary in an avian MHC class II B gene. Mol Biol Evol 16:1599–1606
Geweke J (1992) Evaluating the accuracy of sampling-based approaches to the calculation of posterior moments. In: Berger JO, Bernardo JM, Dawid AP, Smith AFM (eds) Bayesian statistics. Oxford University Press, New York, pp 169–194
Girard P, Angers B (2006a) Characterization of microsatellite loci in longnose dace (Rhinichthys cataractae) and interspecific amplification in five other Leuciscinae species. Mol Ecol Notes 6:69–71
Girard P, Angers B (2006b) The impact of post-glacial marine invasions on the genetic diversity of an obligate freshwater fish, the longnose dace (Rhinichthys cataractae), on the Quebec peninsula. Can J Fish Aquat Sci 63:1429–1438
Girard P, Angers B (2008) Assessment of power and accuracy of methods for detection and frequency-estimation of null alleles. Genetica 134:187–197
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices Version 2.9.3. Distributed by the author, Population genetics Lab, University of Lausanne, Lausanne. http://www2.unil.ch/popgen/softwares/fstat.htm
Guo SW, Thompson EA (1992) Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics 48:361–372
Hayashi K, Yoshida H, Nishida S, Goto M, Pastene LA, Kanda N, Baba Y, Koike H (2005) Genetic variation of the MHC DQB locus in the finless porpoise (Neophocaena phocaenoides). Zool Sci 23:147–153
Heath DD, Shrimpton JM, Hepburn RI, Jamieson SK, Brode S, Docker MF (2006) Population structure and divergence using microsatellite and gene locus markers in Chinook salmon (Oncorhynchus tshawytscha) populations. Can J Fish Aquat Sci 63:1370–1383
Hewitt G (2000) The genetic legacy of the quaternary ice ages. Nature 405:907–913
Hudson RR, Kreitman M, Aguadé M (1987) A test of neutral molecular evolution based on nucleotide data. Genetics 116:153–159
Hughes AL, Nei M (1988) Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335:167–170
Ingvarsson PK, Garcia MV, Hall D, Luquez V, Jansson S (2006) Clinal variation in phyB2, a candidate gene for day-length-induced growth cessation and bud set, across a latitudinal gradient in European aspen (Populus tremula). Genetics 172:1845–1853
Landry C, Bernatchez L (2001) Comparative analysis of population structure across environments and geographical scales at major histocompatibility complex and microsatellite loci in Atlantic salmon (Salmo salar). Mol Ecol 10:2525–2539
Landry C, Garant D, Duchesne P, Bernatchez L (2001) ‘Good genes as heterozygosity’: the major histocompatibility complex and mate choice in Atlantic salmon (Salmo salar). Proc R Soc B 268:1279–1285
Langefors AH (2005) Adaptive and neutral genetic variation and colonization history of Atlantic salmon. Environ Biol Fishes 74:297–308
Luikart G, Cornuet JM (1998) Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol 12:228–237
Mayer WE, O’hUigin C, Zaleska-Rutcynska Z, Klein J (1992) Trans-species origin of Mhc-DRB polymorphism in the chimpanzee. Immunogenetics 37:12–23
McDonald JH, Kreitman M (1991) Adaptive protein evolution at the Adh locus in Drosophila. Nature 351:652–654
Miller KM, Kaukinen KH, Beacham TD, Withler RE (2001) Geographic heterogeneity in natural selection on an MHC locus in sockeye salmon. Genetica 111:237–257
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
O’Brien SJ, Yuhki N (1999) Comparative genome organization of the major histocompatibility complex: lessons from the Felidae. Immunol Rev 167:133–144
Ono H, O’Huigin C, Vincek V, Klein J (1993) Exon-intron organization of fish major histocompatibility complex class II beta genes. Immunogenetics 38:223–234
Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T (1989) Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphism. Proc Natl Acad Sci USA 86:2766–2770
Otto SP (2000) Detecting the form of selection from DNA sequence data. Trends Genet 16:526–529
Ottovà E, Imkovà AT, Morand S (2007) The role of major histocompatibility complex diversity in vigour of fish males (Abramis brama L.) and parasite selection. Biol J Linnean Soc 90:525–538
Pamilo P, Nei M (1988) Relationships between gene trees and species trees. Mol Biol Evol 5:568–583
Peters MB, Turner TF (2008) Genetic variation of the major histocompatibility complex (MHC class II β gene) in the threatened Gila trout, Oncorhynchus gilae gilae. Conserv Genet 9:257–270
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Sauermann U, Nurnberg P, Bercovitch FB, Berard JD, Trefilov A, Widdig A, Kessler M, Schmidtke J, Krawczak M (2001) Increased reproductive success of MHC class II heterozygous males among free-ranging rhesus macaques. Hum Genet 108:249–254
Schad J, Ganzhorn JU, Sommer S (2005) MHC constitution and parasite burden in the Malagasy mouse lemur, Microcebus murinus. Conserv Genet 5:299–309
Scott WB, Crossman EJ (1973) Freshwater fishes of Canada. Bulletin/Fisheries Research Board of Canada 184, Ottawa
Slade RW (1992) Limited MHC polymorphism in the southern elephant seal: implications for MHC evolution and marine mammal biology. Proc Roy Soc B 249:163–171
Smith BJ (2007) Boa: an R package for MCMC output convergence assessment and posterior inference. J Stat Softw 21(11):1–37
Suprunova T, Krugman T, Fahima T, Chen G, Shams I, Karol A, Nevo E (2004) Differential expression of dehydrin genes in wild barley, Hordeum spontaneum, associated with resistance to water deficit. Plant Cell Environ 27:1297–1308
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Takahata N (1989) Gene genealogy in three related populations: consistency probability between gene and population trees. Genetics 122:957–966
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
van Oosterhout C, Joyce DA, Cummings SM (2006) Evolution of MHC class IIB in the genome of wild and ornamental guppies, Poecilia reticulata. Heredity 97:111–118
Watterson GA (1978) The homozygosity test of neutrality. Genetics 88:405–417
Wegner KM (2008) Historical and contemporary selection of teleost MHC genes: did we leave the past behind? J Fish Biol 73:2110–2132
Acknowledgments
The authors wish to thank Kim-Pascale St-Pierre, Pascale Gibeau, Roland Vergilino, Isabelle Bouthillier, Isabelle Gaudet, Geneviève Roy, Simon Vervaet, and Bénédicte Poncet for the field and lab works. The authors are also grateful to Louis Bernatchez and two anonymous reviewers for their very helpful comments, and to Karyn Gray and Anna Lee-Popham for proofreading. This study was supported by an NSERC research grant to BA. PG was financially supported by the FQRNT, the Fonds Étienne-Magnin, the Faculté des Études Supérieure of Université de Montréal, and the Département des Sciences Biologiques.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Girard, P., Angers, B. The Functional Gene Diversity in Natural Populations over Postglacial Areas: The Shaping Mechanisms Behind Genetic Composition of Longnose Dace (Rhinichthys cataractae) in Northeastern North America. J Mol Evol 73, 45–57 (2011). https://doi.org/10.1007/s00239-011-9456-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-011-9456-1