
Abstract
The amphipod Crangonyx islandicus is a recently discovered species endemic to Iceland. Populations of C. islandicus are highly structured geographically and genetically. The COI and 16S mitochondrial genes confine six monophyletic groups which have diverged for up to 5 million years within Iceland, and may present two cryptic species. To investigate the potential cryptic species status we analyse here the internal transcribed spacers (ITS1 and ITS2) and compare its variation with the patterns obtained with the mtDNA. The ITS regions present much less divergence among the geographic regions in comparison with the mtDNA, distances based on ITS1 are correlated with the COI distances as well as with geographic distances, but most of the variation is observed within individuals. The variation in the ITS region appears to have been shaped both by homogenization effect of concerted evolution and divergent evolution. A duplication of 269 base pairs is found in the ITS1 of all individuals from the southern populations, its divergence from its paralog appears to predate the split of the different groups within Iceland but some evidence point to rapid diversification after the split. This duplication does not affect the secondary structures found in the 3′ and 5′ ends of the sequence, suggested to have a role in the excision of the ITS1. Compensatory base changes within the ITS2 sequences which have been suggested to be a species indicator were not detected.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Crangonyx islandicus is an endemic amphipod species dwelling in groundwater underneath lava fields in Iceland. Analysis of variation in the mitochondrial COI and 16S genes has revealed cryptic species diversity within C. islandicus (Kornobis et al. 2010). The populations are highly structured geographically and confine at least six monophyletic groups which have diverged within Iceland for up to 5 million years. The geographical patterns of the genetic variation indicate that C. islandicus survived repeated glaciation periods in Iceland in sub-glacial refugia at the tectonic plate boundary, probably facilitated by geothermal activity. As convergent evolution in morphological traits is common among subterranean species, molecular markers have been widely used for detection of cryptic diversity and species delimitation (e.g., Lefébure et al. 2006; Fišer et al. 2008). An application of the species screening thresholds on C. islandicus, developed by Hebert et al. (2004) and Witt et al. (2006), based on the COI variation gave evidence for two provisional species, one with a wide distribution in Iceland, and a second one found in northeastern Iceland (Kornobis et al. 2010).
The mtDNA COI region has been the main marker used in the Barcoding Life initiative (see for review: Savolainen et al. 2005). The use of a sole mtDNA marker may lead to an over- or under-estimation of species diversity (e.g., Roe and Sperling 2007), for example the COI gene displays high levels of variation even among conspecifics in copepod species (Goetze 2003). As single gene trees and species trees have shown incongruencies (see Roe and Sperling 2007), additional DNA barcodes have been proposed such as the ITS2 to identify species and have proved to be highly effective (Yao et al. 2010). The internal transcribed spacer regions (ITS1 and ITS2) of the ribosomal RNA have been commonly used as suitable markers for molecular taxonomy (Tang et al. 2003) including crustacean species (Chu et al. 2001; Ota et al. 2010). Criteria based on the variation within the ITS2 region, such as the occurrence of compensatory base changes (CBC) are highly informative for species identification for plants and fungi (Müller et al. 2007). Variation within the ITS1 has been suggested to be suitable for intraspecies analysis and has been used to assess divergence between populations e.g., in the crustacean Penaeus japonicus (Chu et al. 2001). The ITS2 region has been shown to be less informative for intraspecies variation than ITS1 (Areekit et al. 2009), thus the variation in both regions might be particularly informative for genetic analyses of C. islandicus populations.
Most rRNA genes in prokaryotes and eukaryotes are subject to concerted evolution (Dover et al. 1982; Ganley and Kobayashi 2007, see for review Nei and Rooney 2005), where the member genes are assumed to evolve as an unit in concert within a gene family. Homogenization of the members of the gene family results from unequal cross-overs and gene conversion (see Nei and Rooney 2005) and affects the intraspecific variability (e.g., Bower et al. 2008), opposite to the effect of divergent evolution, following a death–birth process, where duplications or intraspecific paralogs will differ more from each other than interspecific orthologs (Ota and Nei 1994; Ambrose and Crease 2010). The different mechanisms of evolution for the mitochondrial gene COI and the ITS regions are known to have led to different patterns in intraspecific variations for various species (e.g., Hansen et al. 2006; Navajas et al. 1998; Carlini et al. 2009). A difference in variation of nuclear and mitochondrial markers is expected due to the haploidy and uniparental inheritance of the latter, resulting in four times smaller effective population size than for a diploid nuclear marker. In addition, lack of recombination within the mtDNA is expected to reduce the variation due to background selection (Charlesworth et al. 1993) and hitch-hiking (Maynard-Smith and Haig 1974). This difference of mtDNA and nuclear markers may be further augmented when considering a nuclear gene family, with multiple gene copies (Mano and Innan 2008).
The aims of this study are twofold: first, to compare the genetic structures observed in an earlier study for the COI and 16S mitochondrial genes among C. islandicus populations to the patterns of variation for the nuclear ITS1 and ITS2 genes, and second to evaluate the cryptic species diversity within C. islandicus.
Materials and Methods
Molecular Work
The nuclear ITS region was amplified using primers ITS1 F and 28S R (Table A1 in Supplementary Material) from 18 individuals already extracted by Kornobis et al. (2010). Sampling locations of the specimens are presented in Fig. 1. PCR amplifications were performed in 20 μl, containing 0.15 mM dNTPs, 0.1% tween, 0.5 μg/μl of BSA, 1× Taq buffer, 0.35 μM primers, 0.5 unit of Taq (New England Biolabs), and 10–100 ng of template. Amplifications were obtained with the PCR conditions: 94°C 4 min, followed by 39 cycles of 94°C 30 s, annealing temperature ranged from 50 to 60°C depending on the samples, for 45 s, 72°C 1.5 min, with a final elongation step at 72°C for 10 min. PCR products were cut out of the gel and purified using the Nucleospin Extract II Kit (Macherey–Nagel). They were then ligated to TOPO vector (TOPO TA Cloning Kit, Invitrogen) and cloned in chemically competent cells DH5α™-T1R. Plasmids purified using the Nucleospin Plasmid Kit (Macherey–Nagel) were sequenced in both directions and with internal primers (Table A1 in Supplementary Material), one to four clones per individual, using ABI BigDye Terminator v3.1 (Applied Biosystems) and ran on ABI PRISM™ 3100 Genetic Analyser. Raw sequences were checked and edited using BioEdit 7.0.9.0. (Hall 1999). Sequences have been submitted to GenBank under accession number from JN258055 to JN258095.

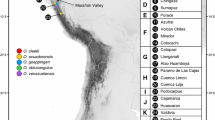
Sampling locations of the groundwater amphipod Crangonyx islandicus in Iceland analysed in this study. Numbers and capital letters refer to sites and different monophyletic groups defined by the COI and 16S variation in Kornobis et al. (2010). The volcanic active zone is displayed in gray. Glaciers are in light gray. Longitudes and latitudes are shown in degrees on the x- and y-axes, respectively
Alignment, Annotation, and Secondary Structure
Complete sequences were trimmed into separate datasets 18S, ITS1, 5.8S and ITS2. 18S, ITS1, and 5.8S were annotated by using annotations available for the amphipod species Diporeia hoyi, as well as by detecting highly conserved regions (18S and 5.8S) in an alignment encompassing other Crangonyctoidean species (in prep.). The ITS2 region was defined using the MCMC analysis available at the ITS2 Database III (Koetschan et al. 2010) choosing the eukaryote model, with maximum E values <0.1 (since the suggested value of E < 0.001 did not lead to successful annotation for all the sequences), and a minimum size of 150 nt (as suggested in the ITS2 annotation software).
The ITS1 and ITS2 sequences were aligned using RNAsalsa 0.8.1 (Stocsits et al. 2009) to produce simultaneously the alignment and secondary structure for each sequence. The consensus structure constrain, necessary as input for a RNAsalsa run, was obtained with RNAalifold (Bernhart et al. 2008). After checking nakedly, no ambiguous parts in the alignments were found. The highly conserved 18S, 5.8S, and 28S regions were aligned by eye in BioEdit (Hall 1999). The program 4SALE (Seibel et al. 2008) was used to plot the consensus secondary structures, and to identify potential cryptic species by searching for CBC in the ITS2 region. Mountain plots (see Hofacker 2003) were drawn to compare the different structures.
Genetic Diversity, Comparison with COI mtDNA Diversity and Population Structure
Variability indices (i.e., segregating sites, phylogenetically informative sites, haplotype, and nucleotide diversity) were summarized using DnaSP v5 (Librado and Rozas 2009), the APE (Paradis et al. 2004), and PEGAS (Paradis 2010) packages implemented in R (R Core Development Team 2005). The relationships between ITS1 haplotypes were characterized by a reduced median network (Bandelt et al. 1995) computed in Network v4.6 (available at http://www.fluxus-engineering.com/sharenet.htm) and redrawn in R.
The optimal model of evolution for each dataset was chosen according to the maximum likelihood tree with respect to substitutions, transition/transversion ratio, proportion of invariant sites, and the gamma distribution parameters selected with PhyML (Guindon and Gascuel 2003) and implemented in the R-package APE. The Akaike Information Criterion (AIC) (Akaike 1974) was used to detect the model which best fitted the data. Two types of genetic distances were calculated, one based on the best fitted model of nucleotide mutations and a second one based on differences in number of tandem repeats and indel events, each considered as a point mutation.
The correlation of the ITS1 and ITS2 pairwise genetic distances, and the genetic distances based on mtDNA COI (Kornobis et al. 2010) were tested with a Mantel test, using the ade4 package (Dray and Dufour 2007) in R. In order to avoid bias in the Mantel test due to different sampling effort between locations, we tested also the correlation on a subset of distances, including solely two clones (when available) per location.
To characterize the structure among populations, a standard AMOVA based on pairwise distances and haplotype frequencies were computed using Arlequin 3.5 (Excoffier and Lischer 2010). The observed variation was partitioned, among mtDNA monophyletic groups, as defined by Kornobis et al. (2010), among individuals within monophyletic group and within individuals. Due to the limited number of samples obtained for ITS1 and ITS2, the only mtDNA monophyletic groups considered were A, A′, D, and E. Results were compared with a corresponding result from an AMOVA based on COI pairwise distances computed in Arlequin. To avoid a bias due to larger samples of the COI than for the ITS1 and ITS2, the AMOVA presents the average of an analysis of ten random samples of the COI sequences where the number of sequences is equal to the number of sequenced clones obtained for the ITS1.
Results
Sequence Variation
In total, 41 complete sequences were obtained for the ITS1 region and 30 for the ITS2 and 5.8S. Forty partial sequences were obtained from the 3′ end of the 18S gene. For the mitochondrial genes, a total of 128 sequences of the COI (658 bp) and 103 of the 16S (414 bp) were previously obtained by Kornobis et al. (2010). Diversity indices are summarized in Table 1 for each DNA region. The ITS1 region is highly variable in length, ranging from 363 to 672 bp. The length variation is mainly due to a 269 bp duplication (called “RII” in this article) solely present in southern populations (Tables 1, 2; Fig. 2), and was identified in a dot matrix view using http://blast.ncbi.nlm.nih.gov. Interestingly, the long version of 672 bp of the ITS1 contains an open reading frame (ORF) of 107 amino acids located in the middle of the sequence (Fig. 2). Minor length variation is also due to variation in number of TGA tandem repeats surrounding RII and CT repeats (Table 2; Fig. 2). The nucleotide diversity of ITS1 and ITS2 nuclear regions were much less variable than observed for the COI and 16S mitochondrial genes but showed more variation than the 18S and 5.8S regions (Table 1). The number of haplotypes for the ITS1 and ITS2 is however large compared to the sample size and the number of haplotypes observed for the COI and 16S sequences. The nucleotide diversity within monophyletic groups, defined by COI and 16S genealogy (Kornobis et al. 2010), is higher for the ITS1 and ITS2 than for the COI (Table 2). Nucleotide diversity among clones within individuals ranges from 0 to 1.7%.

The internal transcribed region of the nuclear rRNA complex in C. islandicus. Location of the ITS1 and ITS2 region is shown with respect to the rRNA genes 18S, 5.8S, and 28S. Regions RI and RII represent a duplicated region. RII is only present in southern populations. The ORF region corresponds to a 107-amino acid open reading frame. Microsatellites are displayed in light gray and are designated as TGA1, TGA2, and CT
Based on the AIC, the best fit model was the HKY (Hasegawa et al. 1985) for the ITS1 and the F84 (Felsenstein and Churchill 1996) for the ITS2. The TN93 model (Tamura and Nei 1993), the most similar substitution model to HKY and implemented in R, was used for pairwise distance calculations for the ITS1.
Secondary Structure of the ITS Regions
Two main secondary structures are observed for the ITS1 within C. islandicus (Fig. 3) due to the occurrence of the 269 bp duplication, otherwise the structures are similar (Fig. 4). The slight differences in length of the helices observed in Fig. 4 can be either explained by single point mutations or variation in the number of repeats in the microsatellites TGA1 and TGA2. The occurrence of the insert in the southern population is not affecting the structure of the common region RI (Figs. 3, 4). The ITS1 secondary structure presents three conserved helices for all individuals studied (Fig. 3). Interestingly, two of these helices are occurring at the 5′ and 3′ ends of the ITS1. One CBC was observed in the ITS1 sequence comparing one clone from one individual from northern Iceland to the other sequences.

The secondary structure of the ITS1 in C. islandicus. The consensus secondary structure obtained with RNAsalsa is displayed for the northern (a) and southern (b) populations. The location of the two microsatellites is indicated by TGA1 and TGA2. The structures framed by broken lines (numbered 1–3) represent the only three helices which are conserved among all individuals studied. The asterisk and the arrow indicate, respectively, the 5′ end of the ITS1 and its orientation (5′–3′). The duplicated region (RII) in b is displayed in light gray

Mountain plot representing the secondary structure of the ITS1 for all Crangonyx islandicus individuals. a Structures of the southern populations. b Structures of the northern populations. The position of the segregating sites is shown by ticks on the x axis in the lower graph. Triangles represent the positions of the phylogenetically informative sites. RI and RII (a) are delimiting the two duplicates. Microsatellites are highlighted by gray regions and annotated TGA1 and TGA2. The dotted line in b corresponds to the duplicated RII
The few variable sites observed in the ITS2 sequences do not lead to noticeable changes in the secondary structures predicted by RNAsalsa. The secondary structure of the ITS2 is characterized by two long helices at the 5′ and 3′ end of the sequence with terminal ramifications. No CBC were observed among the ITS2 sequences.
Geographic Patterns
Most of the variation of the ITS1 and ITS2 is found within individuals (Table 3), or 59 and 75% for the ITS1 and 82 and 93% for the ITS2, depending on how the variation is analyzed. The difference is larger when the variation among individuals within groups is also considered. This is clearly different from the results obtained from the mtDNA COI sequences where only 12.55% of the variation is within sample locations. However, the geographic structure, observed with mtDNA genes, holds to a certain degree when a sample size comparable to the one obtained for the ITS1 and ITS2 was used for the COI (Table 3). When the indel variation (RII and microsatellites) were considered as point mutations, 23% of the ITS1 variation is among groups defined by the mtDNA genealogy (Fig. 5). This value is reduced to 8% when the indels were not considered. Including indels in the partition of the ITS2 variation did not result in larger differentiation among groups (Table 3).

Existence of the large indel (RII) of the ITS1 plotted on the bayesian phylogenetic tree based on mt genes COI and 16S for C. islandicus populations in Iceland (modified from Kornobis et al. 2010). The regions RI and RII of the ITS1 are found in mtDNA lineages A–D (from southern Iceland), whereas mtDNA lineages E and F (from northern Iceland) possess only region RI. The number at each node represents the time to the most recent common ancestor of the monophyletic groups in millions of years. Posterior probability values are displayed in the box. Branches are drawn to scale, with the bar indicating 0.5 nucleotide substitution per hundred sites
The pairwise ITS1 genetic distances increase with geographic distances (Mantel test, P < 0.001, see Fig. 6a) but not for the ITS2 (P = 0.84). This correlation for the ITS1 stands even when the Mantel test was conducted solely on two clones per location (P < 0.001). Although, the ITS1 pairwise genetic distances are much smaller than the COI distances they were correlated (Mantel test, P < 0.05, see Fig. 6b), variation in ITS2 was not related to the COI genetic distances (P = 0.92). Considerable variation is observed in the genetic distances of the ITS1 for a given COI genetic distance (Fig. 6b) which is partly explained by the large variation witnessed within individuals for the ITS1. COI nucleotide variation for the complete dataset is more than three times higher than the ITS2 nucleotide variation and the proportion of informative sites for the COI is also much higher (Table 1). This variation in the COI led to the identification of clear geographic groups, whereas in this study most of the clusters are inferred from the ITS1 variations and are much less geographically structured (Fig. 7). Although, most of the variation in ITS1 is found within individuals, the genetic variation among ITS1 sequences support the pattern of geographic structure observed with the mitochondrial genes (Figs. 5, 7). The E and F mtDNA groups from the North are distinct from the A, A′, B, and C mtDNA groups from the western volcanic zone in central and southwestern Iceland by a single mutation (Figs. 1, 5, and 7). The occurrence of the 269-bp indel (RII in the ITS1) and the length variation of the tandem repeats TGA1 and TGA2 supports further the distinction between the northern and southern regions (Table 2; Fig. 1). Except for one individual, the D group from the eastern volcanic zone in southern Iceland is also different from the central and southwestern populations (A, A′, B, and C) by a single mutation (Fig. 1, 7). Most of the segregating sites in the other regions, 18S, 5.8S, and ITS2, are singletons and their only effect on the network (Fig. 7) are to increase the length of the branches already drawn. Two informative sites were found in the ITS2 region, one mutation which is in all clones of a single individual and a second mutation shared by only two clones from the central and the southwestern regions. A single informative site found in the 5.8S region distinguishes the two different clones from northeastern Iceland from the other sequences.

ITS1 genetic distances versus geographic distances (a) and COI genetic distances (b) within Crangonyx islandicus. Genetic distances are based on number of substitutions per sites, corrected with the TN93 substitution model, and length differences in microsatellites. Variation in RII (duplication) is omitted

Unrooted reduced median network of the ITS1 haplotypes of C. islandicus populations. The size of the pies refers to observed frequencies of haplotypes. Pie shadings refer to groups defined by geographic areas and mtDNA genealogy from Kornobis et al. (2010) and presented in Figs. 1 and 5. The length of the branches are drawn in proportion to the number of mutations between haplotypes
Indel Region in the ITS1
The regions RI and RII of the ITS1 were found in all 26 clones from the southern populations, whereas RII was absent from all 9 clones from the northern populations (Table 2; see Figs. 2, 4). The variation observed in the sequence of the 269-bp indel (RII) is clearly lower than the one observed within RI, but the variation between the two paralogs RI and RII within individuals is considerably higher than within the two regions (Fig. 8).

Pairwise genetic distances within C. islandicus based on ITS1. Boxplot 1 Distances between all individuals for region RI. Boxplot 2–8 present only information obtained from southern populations where RII is found. 2 Distances between individuals for RI. 3 Distances between individuals for RII. 4 Distances between RI and RII within individuals. 5 and 6 Distances between RI and RII within individuals considering the secondary structure of RII, pairwise distances in stem regions (5) and loop regions (6). 7 and 8 Distances between RI and RII within individuals considering the secondary structure of RI, pairwise distances in stem regions (7) and loop regions (8)
Further analyses of the variation within stems and loops and between the RI and RII regions shows distinct patterns. The variation in number of segregating sites in RI is around two times higher in the loops while similar numbers are observed within stem and loop in RII (Table 4, Fisher’s test P = 0.019). Comparison of variation between RI and RII, with respect to stem and loop, showed significantly different values (Table 4, Fisher’s test P = 0.044). The highest ratio of variable sites was found when considering loops in RI and stems in RII. Similarly, genetic distances within clones between RI and RII were higher considering stem regions in RII than loops, whereas the average distances between stem and loop regions in RI did not differ (Fig. 8).
Discussion
The ITS region in C. islandicus defines well-conserved secondary structures which are maintained despite some variation, mainly in length. Three helices are found, two of them occurring at the 5′ and 3′ ends of the ITS1 may have a role in the maturation of the ribosomal RNAs. The secondary structure is known to have an important role in the excision of the ITS regions during the processing of the rRNA genes (van Nues et al. 1995; Côté and Peculis 2001). According to Gottschling and Plötner (2004), a helix structure near the 3′ end of the ITS1 appears to be highly conserved among the eukaryotes secondary structures, though the taxon sampling for these regions is still limited. The duplication RII, occurring in the middle of the ITS1 sequence, is not affecting the secondary structure of the regions near both 5′ and 3′ ends. No variation was observed in the secondary structure of the ITS2 within C. islandicus.
Variation along the ITS regions is determined, as other member of gene families by the relative rates of homogenization due to gene conversion and unequal crossing over, and divergence due to mutation, natural selection, and genetic drift (see Nei and Rooney 2005). The ITS1 is more variable than the ITS2 in this study as observed also in other studies at the intra-specific level (e.g., Areekit et al. 2009). The variable sites are more commonly found in the loop structures, although to a lesser degree in ITS2, possibly indicating different selective pressures along the secondary structures. The ITS1 was found to be highly variable in length, with the occurrence of the two microsatellites at both ends of a large duplicated region. Microsatellites have been suggested to trigger crossing-over (Brandström et al. 2008) and could have played a role in the apparition of the duplicated region RII present in southern population of C. islandicus. Variation in length of the ITS1 has been commonly reported among species (e.g., Ko and Jung 2002; Gamerschlag et al. 2008; Chu et al. 2001) and within species (e.g., Bower et al. 2008; Ko and Jung 2002; van Herwerden et al. 1999). Such length variation has been observed in the amphipod species Gammarus minus (Carlini et al. 2009) and Gammarus tigrinus (Kelly et al. 2006) but to a lesser extent, their ITS1 length ranging, respectively, from 298 to 324 bp and from 550 to 563 bp. It is noticeable that the length of the ITS1 of the southern population of C. islandicus is among the largest found among crustacean species (cf. Chu et al. 2001). Similarly, the ITS2 in C. islandicus is found to be among the largest one by comparison with sequences available in the ITS2 database (Koetschan et al. 2010).
The population genetic structure based on the ITS regions is only partly comparable to the patterns revealed by the mtDNA variation of the barcoding marker COI (see Kornobis et al. 2010). The variation within the ITS regions are only giving a weak support for the main geographic groups obtained from the mtDNA genealogy (Kornobis et al. 2010). The northern populations (mtDNA groups E and F) clustered together, whereas the mtDNA genealogy shows an early divergence of the northeastern group (F) from all the other groups, including E from northern Iceland. Nonetheless, the ITS1 variation is correlated with geographic distances among populations. The small divergence found among the populations of C. islandicus for the ITS1 and ITS2, reflects lower substitution rates than in the mtDNA COI and 16S genes in this species. ITS1 has already been shown to be less informative than the COI for intra- and inter-specific comparisons for amphipods (Kelly et al. 2006; Carlini et al. 2009) and flatworms (Hansen et al. 2003; Hansen et al. 2006). Whether this is due to the homogenization effect of concerted evolution, some other selective constrains, or solely due to increased effective population size of a nuclear marker present in a gene family (Mano and Innan 2008) is not known. The high variation in this study within individuals indicates that the homogenization is not strong, although it may retard the substitutions of the segregating variants within populations. A larger effective population size may result in increased variation within groups and consequently to lower estimates of population subdivision such as the Fst and in longer time to fixation of segregating variants.
In contrast with the study based on the COI and 16S, differentiation among populations observed for the ITS regions does not point to any cryptic species. CBC supporting the occurrence of cryptic speciation (Müller et al. 2007) were not observed in the ITS2 sequences. Two COI species delimitations criteria (Lefébure et al. 2006; Hebert et al. 2004; Witt et al. 2006) applied to the C. islandicus populations led to different conclusions about the new species status of the F group (Kornobis et al. 2010). The fact that no CBC were present among the ITS2 sequences is though not sufficient to reject the hypothesis that C. islandicus is a species complex. In a study by Müller et al. (2007), only 77% of the taxa which did not display any CBC were actually belonging to the same species.
The high divergence between RI and RII in the ITS1, in comparison with the variation within both regions, may indicate that the duplication event might have occurred before the split of the populations in northern and southern Iceland for 1–5 million years ago according to the time estimates based on COI and 16S variation (Kornobis et al. 2010). The duplicated region RII could have been lost in both mtDNA lineages present in northern Iceland. An alternative and more parsimonious explanation is that the duplication event RII occurred 1–1.3 millions years ago, diversified from RI and spread throughout southern Iceland. Accordingly, RII has been shaped by a divergent evolution whereas RI has been conserved by purifying selection. Such a divergent evolution, either due to drift or selection, where the duplication may have acquired a new function and interspecific orthologs are more similar to one another than they are to intra-specific paralogs (Ota and Nei 1994; Nei and Rooney 2005), has an opposite effect than the homogenization caused by concerted evolution (Arnheim 1983). There is some support for the hypothesis of divergent evolution of RII compared to RI. The difference observed between RII and the rest of the ITS1 in the amount of variation relative to the secondary structure supports reduced effect of homogenization. More frequent changes are observed in sites which were in loops of RI but are present in stems of RII (see Fig. 8), such mutations may be favored as they contribute to the structural stability of RII.
Evolution of the ITS regions within C. islandicus appears to be shaped both by concerted and divergent evolution. This makes any interpretations of the geographic patterns and species delimitation within this species more difficult than with the variation obtained by the COI mtDNA sequences. A larger genome wide study allowing to separate the loci specific patterns from the rest of the genome is though needed to understand the evolution of the ITS regions in C. islandicus.
References
Akaike H (1974) A new look at the statistical model identification. IEEE Trans Autom Control AC-19:716–723
Ambrose C, Crease T (2010) Evolution of repeated sequences in the ribosomal DNA intergenic spacer of 32 arthropod species. J Mol Evol 70:247–259
Areekit S, Singhaphan P, Khuchareontaworn S, Kanjanavas P, Sriyaphai T, Pakpitchareon A, Khawsak P, Chansiri K (2009) Intraspecies variation of Brugia spp. in cat reservoirs using complete ITS sequences. Parasitol Res 104:1465–1469
Arnheim N (1983) Concerted evolution of multigene families. In: Nei M, KoehnN RK (eds) Evolution of genes and proteins. Sinauer, Sunderland, pp 38–61
Bandelt HJ, Forster P, Sykes BC, Richards MB (1995) Mitochondrial portraits of human populations using median networks. Genetics 141:743–753
Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF (2008) RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinform 9:474
Bower JE, Dowton M, Cooper RD, Beebe NW (2008) Intraspecific concerted evolution of the rDNA ITS1 in Anopheles farauti sensu stricto (Diptera: Culicidae) reveals recent patterns of population structure. J Mol Evol 67:397–411
Brandström M, Bagshaw AT, Gemmell NJ, Ellegren H (2008) The relationship between microsatellite polymorphism and recombination hot spots in the human genome. Mol Biol Evol 25:2579–2587
Carlini DB, Manning J, Sullivan PG, Fong DW (2009) Molecular genetic variation and population structure in morphologically differentiated cave and surface populations of the freshwater amphipod Gammarus minus. Mol Ecol 18:1932–1945
Charlesworth B, Morgan MT, Charlesworth D (1993) The effect of deleterious mutations on neutral molecular variation. Genetics 134:1289–1303
Chu KH, Li CP, Ho HY (2001) The first internal transcribed spacer (ITS-1) of ribosomal DNA as a molecular marker for phylogenetic and population analyses in crustacea. Mar Biotechnol 3:355–361
Côté C, Peculis B (2001) Role of the ITS2-proximal stem and evidence for indirect recognition of processing sites in pre-rRNA processing in yeast. Nucleic Acids Res 29:2106–2116
Dover G, Coen E, Strachan T (1982) The Dynamics of sequence turnover and concerted evolution of families of rDNA, histone genes and non-coding DNA of Drosophila. Heredity 49:141
Dray S, Dufour AB (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22:1–20
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Felsenstein J, Churchill GA (1996) A hidden Markov model approach to variation among sites in rate of evolution. Mol Biol Evol 13:93–104
Fišer C, Sket B, Trontelj P (2008) A phylogenetic perspective on 160 years of troubled taxonomy of Niphargus (Crustacea: Amphipoda). Zool Scr 37:665–680
Gamerschlag S, Mehlhorn H, Heukelbach J, Feldmeier H, D’Haese J (2008) Repetitive sequences in the ITS1 region of the ribosomal DNA of Tunga penetrans and other flea species (Insecta, Siphonaptera). Parasitol Res 102:193–199
Ganley ARD, Kobayashi T (2007) Highly efficient concerted evolution in the ribosomal DNA repeats: total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res 17:184–191
Goetze E (2003) Cryptic speciation on the high seas; global phylogenetics of the copepod family Eucalanidae. Proc R Soc Lond B 270:2321–2331
Gottschling M, Plötner J (2004) Secondary structure models of the nuclear internal transcribed spacer regions and 5.8S rRNA in Calciodinelloideae (Peridiniaceae) and other dinoflagellates. Nucleic Acids Res 32:307–315
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hall T (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hansen H, Bachmann L, Bakke TA (2003) Mitochondrial DNA variation of Gyrodactylus spp. (Monogenea, Gyrodactylidae) populations infecting Atlantic salmon, grayling and rainbow trout in Norway and Sweden. Int J Parasitol 33:1471–1478
Hansen H, Martinsen L, Bakke TA, Bachmann L (2006) The incongruence of nuclear and mitochondrial DNA variation supports conspecificity of the monogenean parasites Gyrodactylus salaris and G. thymalli. Parasitology 133:639–650
Hasegawa M, Kishino H, Yano T (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Hebert PDN, Stoeckle MY, Zemlak TS, Francis CM (2004) Identification of Birds through DNA Barcodes. PLoS Biol 2:1657–1663
Hofacker IL (2003) Vienna RNA secondary structure server. Nucleic Acids Res 31:3429–3431
Kelly DW, MacIsaac HJ, Heath DD (2006) Vicariance and dispersal effects on phylogeographic structure and speciation in a widespread estuarine invertebrate. Evolution 60:257–267
Ko KS, Jung HS (2002) Three nonorthologous ITS1 types are present in a polypore fungus Trichaptum abietinum. Mol Phylogenet Evol 23:112–122
Koetschan C, Förster F, Keller A, Schleicher T, Ruderisch B, Schwarz R, Müller T, Wolf M, Schultz J (2010) The ITS2 Database III-sequences and structures for phylogeny. Nucleic Acids Res 38:275–279
Kornobis E, Pálsson S, Kristjánsson BK, Svavarsson J (2010) Molecular evidence of the survival of subterranean amphipods (Arthropoda) during Ice Age underneath glaciers in Iceland. Mol Ecol 19:2516–2530
Lefébure T, Douady CJ, Gouy M, Gibert J (2006) Relationship between morphological taxonomy and molecular divergence within Crustacea: proposal of a molecular threshold to help species delimitation. Mol Phylogenet Evol 40:435–447
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Mano S, Innan H (2008) The evolutionary rate of duplicated genes under concerted evolution. Genetics 180:493–505
Maynard-Smith J, Haig J (1974) The hitch-hiking effect of a favourable gene. Genet Res 23:23–35
Müller T, Philippi N, Dandekar T, Schultz J, Wolf M (2007) Distinguishing species. RNA 13:1469–1472
Navajas M, Lagnel J, Gutierrez J, Boursot P (1998) Species-wide homogeneity of nuclear ribosomal ITS2 sequences in the spider mite Tetranychus urticae contrasts with extensive mitochondrial COI polymorphism. Heredity 80:742–752
Nei M, Rooney A (2005) Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39:121–152
Ota T, Nei M (1994) Divergent evolution and evolution by the birth-and-death process in the immunoglobulin VH gene family. Mol Biol Evol 11:469–482
Ota Y, Tanaka K, Hirose M, Hirose E (2010) Description of a new species of Elaphognathia (Isopoda: Gnatheidae) from Japan based on morphological and molecular traits. J Crustac Biol 30:710–720
Paradis E (2010) Pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26:419–420
Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290
R Core Development Team (2005) R: a language and environment for statistical computing. Foundation for Statistical Computing, Vienna
Roe A, Sperling F (2007) Population structure and species boundary delimitation of cryptic Dioryctria moths: an integrative approach. Mol Ecol 16:3617–3633
Savolainen V, Cowan RS, Vogler AP, Roderick GK, Lane R (2005) Towards writing the encyclopedia of life: an introduction to DNA barcoding. Phil Trans R Soc Lond B 1462:1805–1811
Seibel PN, Müller T, Dandekar T, Wolf M (2008) Synchronous visual analysis and editing of RNA sequence and secondary structure alignments using 4SALE. BMC Res Notes 1:91
Stocsits RR, Letsch H, Hertel J, Misof B, Stadler PF (2009) Accurate and efficient reconstruction of deep phylogenies from structured RNAs. Nucleic Acids Res 37:6184–6193
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Tang B, Zhou K, Song D, Yang G, Dai A (2003) Molecular systematics of the Asian mitten crabs, genus Eriocheir (Crustacea: Brachyura). Mol Phylogenet Evol 29:309–316
van Herwerden L, Blair D, Agatsuma T (1999) Intra- and interindividual variation in ITS1 of paragonimus westermani (Trematoda: digenea) and related species: implications for phylogenetic studies. Mol Phylogenet Evol 12:67–73
van Nues RW, Rientjes JMJ, Morre SA, Mollee E, Planta RJ, Venema J, Raue HA (1995) Evolutionarily conserved structural elements are critical for processing of internal transcribed spacer 2 from Saccharomyces cerevisiae precursor ribosomal RNA. J Mol Biol 250:24–36
Witt JDS, Threloff DL, Hebert PDN (2006) DNA barcoding reveals extraordinary cryptic diversity in an amphipod genus: implications for desert spring conservation. Mol Ecol 15:3073–3082
Yao H, Song J, Liu C, Luo K, Han J, Li Y, Pang X, Xu H, Zhu Y, Xiao P, Chen S (2010) Use of ITS2 region as the universal DNA barcode for plants and animals. PLoS One 5:e13102
Acknowledgments
We are grateful to the University of Iceland and the Icelandic Research Council (Rannis) for financial support. We want also to thank the editor and the anonymous reviewer who provided helpful suggestions on the draft manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Kornobis, E., Pálsson, S. Discordance in Variation of the ITS Region and the Mitochondrial COI Gene in the Subterranean Amphipod Crangonyx islandicus . J Mol Evol 73, 34–44 (2011). https://doi.org/10.1007/s00239-011-9455-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-011-9455-2