
Abstract
The full-length element of the non-LTR retrotransposon R2 is here characterized in three European isopteran species: the more primitive Kalotermes flavicollis (Kalotermitidae), including two highly divergent mitochondrial lineages, and the more derived Reticulitermes lucifugus and R. urbis (Rhinotermitidae). Partial 3′ sequences for R. grassei and R. balkanensis were also analyzed. The essential structural features of R2 elements are conserved in termites. Phylogenetic analysis revealed that termite elements belong to the same clade and that their phylogeny is fully compatible with the phylogeny of their host species. The study of the number and the frequency of R2 insertion variants in four R. urbis colonies suggests a greatly reduced, or completely absent, recent element activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transposable elements (TEs) form the major fraction of many eukaryote genomes (Biémont and Vieira 2006; Cordaux and Batzer 2009) and their interactions with the host genome range from parasitism to domestication with complex time-dependent co-evolutionary patterns (Kidwell and Lisch 2001; Bowen and Jordan 2002; Feschotte and Pritham 2007; Venner et al. 2009).
Among retrotransposable elements (Class I; Wicker et al. 2007), the non-LTR retrotransposons of the R superclade specifically insert into the 28S (R1, R2, R4, R5, R6, R9, and RT families) or the 18S (R7 and R8 families) ribosomal genes (Kojima et al. 2006; Gladyshev and Arkhipova 2009 and references therein). R2 elements, in particular, have been found in many lineages of arthropods (Jakubczak et al. 1991; Burke et al. 1998), but also in other animal phyla, such as Cnidaria, Plathyhelmintes, Echinodermata, and Chordata (Kojima and Fujiwara 2004, 2005; Kojima et al. 2006). All R2 elements recognize the same highly conserved target sequence (5′-AAGG↓TAGC-3′) with only few exceptions (Kojima et al. 2006; Bunikis and Barbour 2005).
The 28S rDNA copies inserted with R2 are obviously not functional; however, since eukaryotic genomes typically harbor hundreds of rDNA units, the inactivation of some repeats may not cause relevant damage to the host. The proportion of R2 inserted units, yet, can vary greatly between and within species, as well as among generations (Bunikis and Barbour 2005; Zhang and Eickbush 2005; Madalena et al. 2008).
R2 elements have a single open reading frame (ORF) flanked by two untranslated sequences of variable length. The protein product shows a central reverse transcriptase (RT) domain. Other highly conserved regions are DNA-binding motifs (Burke et al. 1999) at the N-terminus and an endonuclease domain at the C-terminus. With these exceptions, very limited sequence conservation is observed among R2 elements. The protein’s N-terminal domain can contain one, two, or three cysteine-histidine (zinc-finger) motifs (Kojima and Fujiwara 2005). When three zinc fingers are present, the motifs are arranged as: CCHH; CCHC; and CCHH. The C-terminal end of the R2 protein includes also a CCHC zinc-finger motif (Yang et al. 1999). Some R2 elements end with simple sequence repeats, due to RT addition of non-templated nucleotides (Luan and Eickbush 1995).
R2 inserts through a target primed reverse transcription mechanism (Christensen et al. 2006) and if the synthesis of R2 first strand is incomplete, a 5′ truncated copy—still able to insert—is produced. The truncation variants can be used to monitor R2 activity and its role in the rDNA dynamics (Pérez-González and Eickbush 2001). In general terms, the appearance of a new variant shows that a transposition event has occurred, while its disappearance indicates that rDNA turnover mechanisms have eliminated the inserted unit. Recently, it was also shown that high rates of R2 retrotransposition lead to the rapid loss of pre-existing R2 elements through the deletion of loci upstream the new insertion (Zhang et al. 2008; Zhou and Eickbush 2009). The length of the rDNA array can be restored through inter-chromosomal or sister-chromatid unequal crossing-overs followed by the positive selection of longer variants.
The study of transposition in Drosophila simulans has shown that the level of R2 activity is variable between different isofemale lines (Zhang and Eickbush 2005). Moreover, retrotransposition activity is unrelated with either the rDNA locus size or the number of uninserted units; instead, it depends on the distribution of R2 inserted units within the rDNA locus (Eickbush et al. 2008; Zhou and Eickbush 2009).
R2 phylogeny built on the C-terminal half of reverse transcriptase shows the presence of 11 sub-clades grouped in four main clades, consistent with the number of zinc-finger motifs at the N-terminal end (Burke et al. 1998; Kojima and Fujiwara 2004, 2005). On the other hand, R2 phylogeny shows striking discrepancies with host phylogeny. Elements from distant species are often found in the same sub-clade, while elements from related species lie in different (sub-)clades. Some species also host multiple lineages of R2 (Burke et al. 1993; Kojima and Fujiwara 2004). Inconsistencies between R2 and hosts phylogenies could be explained either by horizontal transfer or by the coexistence of paralogous R2 elements, followed by one or more lineage extinctions. Yet, no evidence for horizontal transfer has been obtained, therefore supporting R2 vertical transmission and an origin as ancient as the divergence of diblastic–triblastic protostomes (Kojima and Fujiwara 2005; Kojima et al. 2006).
Termites are of interest from both a practical (pest control) and a theoretical point of view. The latter aspect involves their population social structure (eusociality). In termite colonies, only some individuals are capable of reproduction, while the majority of the members specialize in non-reproductive tasks. Termites therefore parallel some groups of hymenopterans, but many features differentiate the two cases: termites are in fact hemimetabolous, diplo-diploid insects, and parthenogenesis, if present, is accessory to amphigonic reproduction (Matsuura and Nishida 2001).
Termite reproduction may affect genome dynamics and evolution. The presence of few reproducers in hundreds or thousands of sterile individuals makes the effective population size of a colony significantly smaller than its absolute size. The ensuing bottlenecks and high level of within-colony inbreeding, also due to neotenic reproductives, may favor genetic drift, and consequently genetic diversification among colonies of the same species (Vargo and Husseneder 2009).
R2 elements are here characterized from species of the European genera Reticulitermes (Holmgren) (Rhinotermitidae; subterranean termites) and Kalotermes Hagen (Kalotermitidae; drywood termites). The former represents a derived isopteran clade while the latter belongs to the so-called “lower termites”. Complete R2 sequences were obtained for R. urbis Bagnères, Uva and Clément, R. lucifugus (Rossi), and two lineages of K. flavicollis (Fabricius). For R. grassei (Clément) and R. balkanensis (Plateaux and Clément), the sequencing was limited to the 3′ portion of the element. Furthermore, isopteran R2 phylogeny and the element activity in R. urbis colonies were investigated.
Materials and Methods
DNA Isolation and R2 Amplification/Sequencing
R2 elements were isolated and characterized from five species of European termites: Reticulitermes urbis (Bagnacavallo, Italy), R. lucifugus (Castel Porziano, Italy), R. grassei (Ychoux, France), R. balkanensis (Marathon, Greece), and Kalotermes flavicollis. The latter species includes two highly divergent mitochondrial lineages (Velonà et al. 2010a) here identified as K. flavicollis 1 (Crete, Greece) and K. flavicollis 2 (Feniglia—Italy), respectively.
Total DNA was extracted from single termite heads with a CTAB protocol (Doyle and Doyle 1987). The presence of R2 elements was tested using one of the forward degenerate primers described in Kojima and Fujiwara (2005), coupled with a 28SB-R reverse primer (Supplementary Table S1), located 178 bp downstream of the element insertion site.
PCR was performed in a 50 μl mixture using TaKaRa LA Taq™ with GC Buffer kit (TaKaRa Bio Inc., Shiga, Japan), with reaction conditions set as follows: initial denaturation at 94°C for 2 min; 35 cycles composed by denaturation at 94°C for 30 s, annealing at 50°C for 30 s, extension at 70°C for 3–10 min (approximately 2 min per kbp of the required product); final extension at 70°C for 12 min. Amplicons were purified either directly or following agarose gel electrophoresis, with Wizard® SV Gel and PCR Clean-up System (Promega, Madison, WY, USA), following the manufacturer’s protocol. Purified products were ligated into a pGEM-T Easy Vector (Promega, Madison, WY, USA). The DNA sequence was determined (Macrogen, Inc., Seoul, South Korea) using the same primers that were utilized to amplify the elements. The sequence of R2 5′ end was obtained by primer walking (see Supplementary Table S1 for primer list); primers were designed with the Primer3 software (Rozen and Skaletsky 2000). For each species, the sequence of a complete R2 element was obtained from partial sequences of an average of four different clones combined in a consensus sequence.
Alignments and consensus sequences were obtained with the software CLC Sequence Viewer 6 (CLC bio A/S, http://www.clcbio.com). Open reading frames were identified through the ORF Finder tool at NCBI (http://www.ncbi.nlm.nih.gov/gorf/gorf.html), using the standard genetic code.
Nucleotide and amino acid p-distances (i.e., the number of substitutions over the length of the aligned sequences) were computed with MEGA version 4 (Tamura et al. 2007).
R2 Phylogeny
For R2 phylogeny, the 3′ end of the amino acid sequences (440–530 aa, depending on the species) were added to the alignment of Kojima and Fujiwara (2005), to which all most recent Genbank available R2-encoded proteins were added: Blattella germanica (EF014490; Kagramanova et al. 2007), Amblyomma americanum, Boophilus microplus, Ixodes scapularis, Argas monolakensis (AY682792-AY682794, and AY682796; Bunikis and Barbour 2005); Nematostella vectensis (available from Kojima et al. 2006); Rhynchosciara americana (FJ461304; Madalena et al. 2008); Triops cancriformis (EU854578; Mingazzini et al. 2011). The SLACS retrotransposon of Trypanosoma brucei (CAA34931; Aksoy et al. 1990) was used as an outgroup.
The minimum evolution tree was obtained with MEGA version 4 (Tamura et al. 2007), with bootstrap values determined after 5,000 replicates. The Bayesian tree was calculated with MrBayes 3.1.2 (Ronquist and Huelsenbeck 2003); convergence was reached after 2 million generations. Trees were sampled every 1,000 generations and the first 200 trees were discarded as burn-in after graphic visualization.
R2 Activity Analysis
R2 activity was evaluated on individuals of different castes derived from four R. urbis colonies (Table 1). Fourteen specimens were sampled from a mature colony of about 1,000 individuals, collected in Bagnacavallo (Italy). The other three colonies were selected from a group of 20 colonies, derived from different Bagnacavallo mature colonies, bred in the laboratory to obtain the offspring of known parents. Following Ghesini and Marini (2009), a female and male nymph (their sex was assessed by checking the width of the seventh sternite) and 20 workers were placed in a Petri dish (Ø 9 cm) containing a substrate of moistened sand and a piece of fir (Picea abies) wood. Two years after their settlement, the 11 surviving colonies were screened to exclude those where no juveniles were born, those where one or both reproductive deriving from nymphs had died, and those where reproductives deriving from workers had developed. In the three colonies where the two parents and their offspring could be found, they were separated from workers and preserved in 100% ethanol for further analysis.
Therefore, a total of 51 individuals, belonging to one mature colony (named M) and three incipient colonies (hence called: 1, 2, and 3), were analyzed (Table 1). R2 activity was examined by scoring the number and the frequency of 5′-truncated variants, as described in Pérez-González and Eickbush (2001): R2 5′ ends were PCR amplified coupling a 28S-anchored forward primer and reverse primers annealing within the R2 sequence (RIN1–5, Supplementary Table S1). PCR products were separated by gel electrophoresis (1.5% agarose) and Southern blotted onto a positively charged nylon membrane. Then, filters were hybridized with R2 probes obtained by PCR amplification (see Supplementary Table S2 for the primer list). Probe labeling and signal detection were performed using the DIG High Prime DNA Labeling and Detection Starter Kit I and CDP-Star system (Roche Diagnostics GmbH, Mannheim, Germany). Stringency washes were done at 65°C, in a 0.1× SSC, 0.1% SDS solution. Truncation variants were considered the same when scored positive bands differed by ±10 bp.
Results
The complete R2 element sequence was obtained for Reticulitermes urbis (R2Ru), R. lucifugus (R2Rl), and for the two lineages of Kalotermes flavicollis (R2Kf1, R2Kf2). The 3′ portion of the element, extending over the last 2291 bp and containing the complete RT domain, was determined for both R. grassei (R2Rg) and R. balkanensis (R2Rb). Sequences were deposited in Genbank under accession numbers GU949554–GU949559.
R2 Structure
The structure of the R2Ru, R2Rl, R2Kf1, and R2Kf2 elements is consistent with the structure of known R2 elements (Fig. 1).

Schematic drawings of sequenced R2 elements in Reticulitermes spp. and Kalotermes flavicollis (UTR = untranslated region; ORF = open reading frame; CCHH, CCHC = zinc-fingers; RT, C-MYB, and EN = retrotranscriptase, c-myb, and endonuclease domains)
A 1-bp deletion of the 28S gene occurs at the R2 5′ insertion of R. urbis and R. lucifugus analyzed clones, while in K. flavicollis the corresponding clones do not show any deletion. R2 ends with a very short poly-(A) tail and the 28S gene shows a 2-bp deletion in all species (Fig. 2).

28S gene and R2 nucleotide sequences at the 5′ junction (a) and at the 3′ junction (b) of R2 elements. The number of clones identical to each sequence is shown in the last column. A sequence of R. lucifugus uninserted 28S gene (Genbank Accession no. GU826352) is shown for comparison
The length and structure of the complete element are very similar among the different species of the genus Reticulitermes as well as between the two K. flavicollis lineages (Fig. 1; Table 2). In the K. flavicollis sequences, the beginning of the ORF is unclear because two methionine initiation codons, separated by 123 bp, can be found at 5′ end of the region. Henceforth, it will be assumed that the ORF begins with the first methionine residue.
Some clear-cut differences exist between the elements of Reticulitermes spp. and those of K. flavicollis. While the K. flavicollis elements are shorter than those of Reticulitermes spp., mainly due to their UTRs, the ORFs are longer (Fig. 1; Table 2). The RT coding domain of Reticulitermes spp. elements, when compared with K. flavicollis one, shows a 6-bp deletion (between RT nucleotides 390–391) and a 6-bp insertion (between RT nucleotides 495–496) so that the total RT length turns out to be the same in the two genera. Three well-conserved zinc-finger motifs of the CCHH–, CCHC–, and CCHH–type are present in the N-terminal region of the ORF in all four full-length elements (Table 3).
Nucleotide p-distance between R. urbis and R. lucifugus complete elements is 0.027 ± 0.002, while nucleotide p-distance between the complete elements of the two K. flavicollis lineages is 0.045 ± 0.003.
Limited to the ORF region, the p-distance between the two Reticulitermes spp. elements is 0.028 ± 0.003; on the whole, there are 102 substitutions: 22 (21.6%) at the first codon position, 19 (18.6%) at the second, and 61 (59.8%) at the third codon position. Amino acid sequence comparison reveals 44 substitutions, resulting in a p-distance of 0.037 ± 0.005. The p-distance between the ORFs of the two Kalotermes flavicollis lineages is 0.044 ± 0.003, corresponding to 173 substitutions: 54 (31.2%) at the first codon position, 44 (25.4%) at the second, and 75 (43.3%) at the third codon position. In K. flavicollis, R2Kf1 and R2Kf2 protein products differ for 103 amino acid substitutions, corresponding to a p-distance of 0.079 ± 0.007.
Nucleotide and amino acid p-distances between the RT coding domains of Reticulitermes species indicate R2Rg as the most divergent element; distance values between the two genera are even ten-fold higher (Table 4). Moreover, within the genus Kalotermes and between the two genera, amino acid distances are slightly higher than nucleotide distances, while within the genus Reticulitermes amino acid distances are generally slightly lower than nucleotide distances (Table 4).
R2 Phylogeny
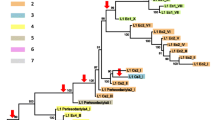
Bayesian and minimum evolution trees (Fig. 3) show the same terminal branching pattern. The four main R2 clades identified by Kojima and Fujiwara (2005) are well recognizable, as well as their sub-clades. The addition of tick sequences produces a new sub-clade in clade D, as already shown by Mingazzini et al. (2011). Minor differences between the two dendrograms occur in the internal branching pattern of a few sub-clades (A2, B1, D2, and D4), corresponding to weakly supported nodes.

Bayesian dendrogram (left, -lnL = 47107.91) and minimum evolution dendrogram (right, SBL = 23.65). Posterior probability or bootstrap values >50 are shown near the corresponding nodes. The scale bar indicates genetic distance. A-D refer to clades/subclades following Kojima and Fujiwara (2005)
As expected on the basis of zinc-fingers number, the sequences of termite elements are included in the clade A and they build up the sub-clade A2, together with the elements from the cockroach Blattella germanica, the hymenopterans Apis mellifera and Nasonia vitripennis (element B), and the spider Hasarius adansoni. Interestingly, isopteran elements are closely related, their relationship being fully compatible with the phylogeny of the examined species. Sequences from Reticulitermes spp. and from Kalotermes flavicollis form two distinct clusters; moreover, R2 from R. urbis and R. balkanensis are closely related and well distinguished from those of R. lucifugus and R. grassei (Fig. 3).
R2 Truncated Variants
On the whole, 14 R2 insertions were identified in R. urbis (Fig. 4): the full-length element insertion and 13 truncated element variants. The complete element and nine of the truncated variants are present in all individuals; the number of insertions per specimen ranges from 10 to 14.

(a) Schematic drawing of the complete element of R. urbis and positions of the Southern blots probes. (b) Insertion profiles of mature colony (M) and incipient colonies (1–3); each vertical bar indicates the position of a truncation; numbers flanking each line represent the number of individuals sharing the corresponding profile (W workers, S soldiers, P male and female parents). Lengths in bp of each truncation variant are shown below
All the individuals from colony M (the mature colony) exhibit the same insertion profile, with 11 truncated elements and the full-length R2. No differences between castes can be observed. Within each of the three incipient colonies, formed by a parental couple and their offspring, the queen and the king share the same insertion profile. The progeny shows the same pattern found in the parents, except in colony 2, where two juveniles lack an insertion that is present in their parents.
The insertion profile exhibits some degree of differentiation among the colonies: while 10 truncated elements are common to all colonies, there is a R2 variant that is shared by three colonies (M, 2, and 3), one that is shared by two colonies (2 and 3), and another one that is present only in colony 3.
Discussion
R2 Structure
Non-LTR R2 elements have been isolated in two isopteran lineages: the drywood termite K. flavicollis (Kalotermitidae) and the subterranean termite Reticulitermes spp. (Rhinotermitidae). Complete R2 element length ranges from 4474 to 4482 bp in K. flavicollis to 5020–5021 bp in R. urbis and R. lucifugus, respectively. The latter are among the longest R2 elements sequenced so far, their length being exceeded only by the elements of Danio rerio (5183 bp) and N. vitripennis B (5028 bp) (Burke et al. 1999; Kojima and Fujiwara 2004).
The basic structural features are the same in all the termite elements and correspond to those found in all known R2 elements: there is a single ORF, flanked by untranslated regions, that contains a reverse transcriptase domain, an endonuclease domain, and DNA-binding motifs (Burke et al. 1999; Yang et al. 1999; Eickbush and Jamburuthugoda 2008). In particular, termite elements share the presence of three zinc-finger motifs in the N-terminal portion of the ORF. This feature is evidence of their belonging to the R2 A clade (Kojima and Fujiwara 2005), a hypothesis confirmed by phylogenetic analysis.
The ORFs of R2 elements from termites are longer than most known ORFs; in particular, the two K. flavicollis elements appear to include the longest ORFs found so far (1307–1309 aa). It is not always straightforward, however, to identify the exact beginning of the ORF in R2 elements and in K. flavicollis elements, protein might actually be 42 aa shorter: there are two methionine initiation codons, separated by 123 bp, so that the ORF might begin with either. The presence of two or more methionine residues upstream of the zinc-fingers is quite common: for example, in the elements of Drosophila mercatorum, Limulus polyphemus, and Danio rerio (Burke et al. 1998; Malik and Eickbush 1999; Kojima and Fujiwara 2004). In other instances, such as in the elements of some Drosophila species, no methionine can be found upstream of the zinc finger motifs. This suggests that the translation initiates upstream of the stop codon preceding the ORF, and that such stop codon is bypassed (George and Eickbush 1999).
Termite elements have a 258 aa long RT. In the other complete R2 elements sequenced so far, the RT ranges in length between 258 (Porcellio scaber) and 268 aa (Ciona intestinalis B).
Termite R2 elements end with a poly-(A) tail, formed by three As in Reticulitermes spp. and two in K. flavicollis. Poly-(A) tails are present in many R2 elements, owing to the capability of the RT of adding non-templated nucleotides before the reverse transcription actually begins (Luan and Eickbush 1995).
The insertion site of termite elements is the same as in the majority of the other organisms studied so far: 5′-AAGG↓TAGC-3′. At the R2 5′ insertion, a 1-bp deletion of the 28S gene is observed in Reticulitermes spp., while in K. flavicollis no deletions are detected. In correspondence of R2 3′ insertion, there is a 2-bp deletion of the 28S gene: such deletions are caused by R2 insertion when the cleavage of the top strand is shifted in relation to the cleavage of the bottom strand (Luan et al. 1993).
The nucleotide p-distance between R. urbis and R. lucifugus elements is similar in the whole element (0.027 ± 0.002), in the ORF (0.028 ± 0.003), and in the RT (0.030 ± 0.006), whereas the distance between the two Kalotermes elements is lower in the RT (0.023 ± 0.005) than in the whole element (0.045 ± 0.003) or in the ORF (0.044 ± 0.003). As a comparison, the sequence variability among D. melanogaster, D. simulans, and D. yakuba (melanogaster subgroup; A.N. X51967; U64957; U64961) RT domains ranges from 0.042 ± 0.007 to 0.106 ± 0.010, fairly above the values observed within termite genera. Both in Reticulitermes spp. (except for the RT coding domain) and in K. flavicollis, amino acid p-distances are higher than the corresponding nucleotide p-distances, owing to a higher proportion of substitutions at the first and second codon positions. This holds, actually, even for the above considered Drosophila RT domains.
R2 Phylogeny
Minimum evolution and Bayesian trees here obtained identify the same R2 clades and sub-clades found by Kojima and Fujiwara (2005); differences in nodal support values are probably due to the addition of new sequences to the alignment. Dendrograms agree in placing termite elements in clade A, sub-clade A2, together with the element from another heterometabolous insect, Blattella germanica. The element from Forficula auricularia, the other heterometabolous insect in which R2 has been isolated, belongs to the D clade, so that in heterometabolous insects at least two R2 clades are present.
R2 phylogeny has been shown to be inconsistent with the host phylogeny (Kojima and Fujiwara 2004), even though some congruencies have been observed at the “local” scale: within the observed sub-clades, R2/host phylogenies are compatible (Kojima and Fujiwara 2005). A remarkable exception is the R2 lineage in the Drosophila genus, whose cladogenesis has been shown to strictly follow that of the host species since 60 Myr ago (Lathe and Eickbush 1997). Isopteran R2 elements are grouped in the same cluster and are also in close relationship with the element found in B. germanica. This agrees with the observation of a “local” congruence with the host phylogeny, considering that the cockroaches are the sister clade of termites (Davis et al. 2009). Although based on a relatively limited taxon sampling (four species versus at least ten European lineages), Reticulitermes R2 phylogeny fully overlaps that of host species. Therefore, the sample reported here suggests that R2 is strictly vertically transmitted in the Reticulitermes genus from about 24 Myr (Velonà et al. 2010b).
Discrepancies between R2 and hosts phylogenies are a common occurrence. In transposable elements, similar situations can be explained either by horizontal transfer or by the existence of paralogous lineages (Cummings 1994; Silva et al. 2004). In the case of R2, as the “horizontal transfer” model has been ruled out (Kojima and Fujiwara 2005; this paper), the second explanation seems to be more adequate. Interestingly, the “paralogous lineages” model somehow parallels the “library hypothesis”, used to explain the evolutionary history of tandem repeat families in related genomes (reviewed in Plohl et al. 2008). In brief, if a collection of different repeats was present in an ancestor’s genome, it is possible that in each derived taxon only one of these repeats has become predominant, the others being reduced to few copies or gone extinct. This dynamics explains the existence of repetitive sequence taxon-specific profiles, not necessarily correlated with taxon phylogeny. Applied to R2 evolutionary history, a pattern like the one expected under the library model would explain why local congruence between R2 and host phylogeny is not always observed.
R2 Activity
The truncated variants generated during transcription can be used to monitor R2 activity (Pérez-González and Eickbush 2001), albeit two potential limitations should be taken into account: (i) this approach does not score new insertions if they are of the same length as a preexisting variant, (ii) if a length variant is present in multiple copies, the elimination of a copy is not detected (Pérez-González and Eickbush 2003). Notwithstanding, this method has proved to give reliable estimates of R2 activity (see for example Zhou and Eickbush 2009 and reference therein; Mingazzini et al. 2011).
The analysis of truncated variants of R. urbis element was carried out on 14 individuals taken from a mature colony, involving workers and soldiers, and on three incipient colonies, each formed by a reproductive couple and its offspring.
In the mature colony, all individuals shared the same truncation profile; thus, no difference between workers and soldiers was found. Within each of the three incipient colonies no new insertions were detected with respect to the parental insertion profile, so that there was no evidence of R2 activity in that generation. On the other hand, an exception to the apparently frozen insertion pattern occurs: two juveniles of colony 1 lacked a truncated variant that was scored in both parents and in the siblings. The loss of the same variant in more than one individual of the filial generation requires further investigations: indeed, the same pattern could be either due to a heterozygous parental condition or to an effective insertion loss, occurred at an early stage of gametogenesis, due to rDNA tandem repeats turnover mechanisms (Nei and Rooney 2005; Zhang et al. 2008).
On the whole, comparisons of R2 activity in R. urbis, both within and between colonies, indicate very low or no transposition capacity at all; this parallels data on some D. simulans lines, explained by a putative cellular mechanism inducing R2 dormancy (Zhang and Eickbush 2005). In this view, also considering the potentially inbred condition of their genome, termites may represent an interesting system to analyze mechanisms influencing R2 dynamics.
References
Aksoy S, Williams S, Chang S, Richards FF (1990) SLACS retrotransposon from Trypanosoma brucei gambiense is similar to mammalian LINEs. Nucleic Acids Res 18:785–792
Biémont C, Vieira C (2006) Junk DNA as an evolutionary force. Nature 443:521–524
Bowen NJ, Jordan IK (2002) Transposable elements and the evolution of eukaryotic complexity. Curr Issues Mol Biol 4:65–76
Bunikis J, Barbour AG (2005) Ticks have R2 retrotransposons but not the consensus transposon target site of other arthropods. Insect Mol Biol 14:465–474
Burke WD, Eickbush DG, Xiong Y, Jakubczak J, Eickbush TH (1993) Sequence relationship of retrotransposable elements R1 an R2 within and between divergent insect species. Mol Biol Evol 10:163–185
Burke WD, Malik HS, Lathe WCIII, Eickbush TH (1998) Are retrotransposons long-term hitchhikers? Nature 392:141–142
Burke WD, Malik HS, Jones JP, Eickbush TH (1999) The domain structure and retrotransposition mechanism of R2 elements are conserved throughout arthropods. Mol Biol Evol 16:502–511
Christensen SM, Ye J, Eickbush TH (2006) RNA from the 5′ end of the R2 retrotransposon controls R2 protein binding to and cleavage of its DNA target site. Proc Natl Acad Sci USA 103:17602–17607
Cordaux R, Batzer MA (2009) The impact of retrotransposons on human genome evolution. Nat Rev Genet 10:691–703
Cummings MP (1994) Transmission patterns of eukaryotic transposable elements: arguments for and against horizontal transfer. Trends Ecol Evol 9:141–145
Davis RB, Baldauf SL, Mayhew PJ (2009) Eusociality and the success of termites: insights from a supertree of dictyopteran families. J Evol Biol 22:1750–1761
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem Bull 19:11–15
Eickbush TH, Jamburuthugoda VK (2008) The diversity of retrotransposons and the proprieties of their reverse transcriptases. Virus Res 134:221–234
Eickbush DG, Ye J, Zhang X, Burke WD, Eickbush TH (2008) Epigenetic regulation of retrotransposons within the nucleolus of Drosophila. Mol Cell Biol 28:6452–6461
Feschotte C, Pritham E (2007) DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet 41:331–368
George JA, Eickbush TH (1999) Conserved features at the 5′ end of Drosophila R2 retrotransposable elements: implications for transcription and translation. Insect Mol Biol 8:3–10
Ghesini S, Marini M (2009) Caste differentiation and growth of laboratory colonies of Reticulitermes urbis (Isoptera, Rhinotermitidae). Insect Soc 56:309–318
Gladyshev EA, Arkhipova IR (2009) Rotifer-specific R9 retrotransposable elements generate an exceptionally long target site duplication upon insertion. Gene 448:145–150
Jakubczak JL, Burke WD, Eickbush TH (1991) Retrotransposable elements R1 and R2 interrupt the rDNA genes of most insects. Proc Natl Acad Sci USA 88:3295–3299
Kagramanova AS, Kapelinskaya TV, Korolev AL, Mukha DV (2007) R1 and R2 retrotransposons of German cockroach Blattella germanica: a comparative study of 5′- truncated copies integrated into the genome. Mol Biol 41:546–553
Kidwell MG, Lisch DR (2001) Perspective: transposable elements, parasitic DNA, and genome evolution. Evolution 55:1–24
Kojima KK, Fujiwara H (2004) Cross-Genome screening of novel sequence-specific Non-LTR retrotransposons: various multicopy RNA genes and microsatellites are selected as targets. Mol Biol Evol 21:207–217
Kojima KK, Fujiwara H (2005) Long-term inheritance of the 28S rDNA-specific retrotransposon R2. Mol Biol Evol 22:2157–2165
Kojima KK, Kuma K, Toh H, Fujiwara H (2006) Identification of rDNA-specific non-LTR retrotransposons in Cnidaria. Mol Biol Evol 23:1984–1993
Lathe WC III, Eickbush TH (1997) A single lineage of R2 retrotransposable elements is an active, evolutionarily stable component of the Drosophila rDNA locus. Mol Biol Evol 14:1232–1241
Luan DD, Eickbush TH (1995) RNA template requirements for target DNA-primed reverse transcription by the R2 retrotransposable element. Mol Cell Biol 15:3882–3891
Luan DD, Korman MH, Jakubczak JL, Eickush TH (1993) Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell 72:595–605
Madalena CRG, Andrioli LPM, Gorab E (2008) Ribosomal RNA gene insertions in the R2 site of Rhynchosciara (Diptera: Sciaridae). Chromosome Res 16:1233–1241
Malik HS, Eickbush TH (1999) Retrotransposable elements R1 and R2 in the rDNA units of Drosophila mercatorum: abnormal abdomen revisited. Genetics 151:653–665
Matsuura K, Nishida T (2001) Comparison of colony foundation success between sexual pairs and female asexual units in the termite Reticulitermes speratus (Isoptera: Rhinotermitidae). Popul Ecol 43:119–124
Mingazzini V, Luchetti A, Mantovani B (2011) R2 dynamics in Triops cancriformis (Bosc, 1801) (Crustacea, Branchiopoda, Notostraca): turnover rate and 28S concerted evolution. Heredity (in press) (doi:10.1038/hdy.2010.86)
Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39:121–152
Pérez-González CE, Eickbush TH (2001) Dynamics of R1 and R2 elements in the rDNA locus of Drosophila simulans. Genetics 158:1557–1567
Pérez-González CE, Burke WD, Eickbush TH (2003) R1 and R1 retrotransposition and deletion in the rDNA loci on the X and Y chromosomes of Drosophila melanogaster. Genetics 165:675–685
Plohl M, Luchetti A, Meštrović N, Mantovani B (2008) Satellite DNAs between selfishness and functionality: structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene 409:72–82
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana, Totowa, pp 365–386
Silva JC, Loreto EL, Clark JB (2004) Factors that affect the horizontal transfer of transposable elements. Curr Issues Mol Biol 6:57–72
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA 4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Vargo EL, Husseneder C (2009) Biology of subterranean termites: insights from molecular studies of Reticulitermes and Coptotermes. Annu Rev Entomol 54:379–403
Velonà A, Ghesini S, Luchetti A, Marini M, Mantovani B (2010a) Starting from Crete, a phylogenetic re-analysis of the genus Reticulitermes in the Mediterranean area. Mol Phylogenet Evol 56:1051–1058
Velonà A, Luchetti A, Mantovani B (2010a). Colony structure of Kalotermes flavicollis (Isoptera, Kalotermitidae): Preliminary results from two molecular markers. XVI° World congress of the international union for the study of social insects (IUSSI), Copenhagen, Denmark, 8–14th August, p 143
Venner S, Feschotte C, Biémont C (2009) Dynamics of transposable elements: towards a community ecology of the genome. Trends Genet 25:317–323
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Gen 8:973–982
Yang J, Malik HS, Eickbush TH (1999) Identification of the endonuclease domain encoded by R2 and other site-specific, non-long terminal repeat retrotransposable elements. Proc Natl Acad Sci USA 96:7847–7852
Zhang X, Eickbush TH (2005) Characterization of active R2 retrotransposition in the rDNA locus of Drosophila simulans. Genetics 170:195–205
Zhang X, Zhou J, Eickbush TH (2008) Rapid R2 retrotransposition leads to the loss of previously inserted copies via large deletions of the rDNA locus. Mol Biol Evol 25:229–237
Zhou J, Eickbush TH (2009) The pattern of R2 retrotransposition activity in natural populations of Drosophila simulans reflects the dynamic nature of the rDNA locus. PLoS Genetics 5:1–11
Acknowledgments
This study was supported by Canziani fund, Università di Bologna, and Fondazione del Monte, Bologna, to BM.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ghesini, S., Luchetti, A., Marini, M. et al. The Non-LTR Retrotransposon R2 in Termites (Insecta, Isoptera): Characterization and Dynamics. J Mol Evol 72, 296–305 (2011). https://doi.org/10.1007/s00239-011-9430-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-011-9430-y