
Abstract
In previous work (Betancourt, Genetics 181:1535, 2009), I propagated three large laboratory populations of an RNA phage (MS2) as they adapted to a controlled laboratory environment. These populations were large enough so that evolution might be expected to be mostly repeatable, but they nevertheless fixed different suites of mutations over the course of the experiment. Here, I investigate one possible explanation for these results: epistasis, in which the effect of a mutation depends on its genetic background, may have prevented populations with different initial substitutions from fixing the same set of subsequent mutations. I show that two mutations that previously occurred in different genetic backgrounds are beneficial on either background. This result suggests that sign epistasis—in which a mutation is beneficial on one background, but deleterious on another—is not the cause of different evolutionary trajectories observed in the Betancourt (2009) experiment. However, they can be explained by either magnitude epistasis—in which mutations have stronger or weaker beneficial effects depending on the background—or by the simultaneous fixation of multiple beneficial mutations. Surprisingly, the large populations of the previous experiment showed less parallel evolution than the small populations of this experiment, which lends support to the fixation of multiple beneficial mutations contributing to the patterns seen in both experiments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Epistasis among beneficial mutations has the potential to influence evolutionary trajectories of an adaptively evolving population—i.e., which beneficial alleles are fixed and in what order. Sign epistasis, in which an allele is beneficial on one background but deleterious on another, can make the fate of an allele contingent on which alleles were fixed previously (Welch and Waxman 2005; Weinreich et al. 2005; Weinreich et al. 2006). Even in the less extreme case of magnitude epistasis, where the beneficial effect of an allele is increased or reduced depending on the genetic background, can effectively preclude some evolutionary trajectories. In other words, even if all beneficial alleles are ultimately fixed, magnitude epistasis can determine their order of fixation (Weinreich et al. 2006).
Epistasis between segregating alleles has been the focus of much attention (Coyne et al. 2000; Goodnight and Wade 2000) as it is closely connected to Wright’s (1931) shifting balance theory. However, alleles with epistatic effects do not have to be segregating within a population for epistasis to affect their fates (Welch and Waxman 2005; Weinreich et al. 2005). Consequently, the importance of epistasis to adaptive evolution may depend not on the incidence of epistatic alleles within populations, but on the extent to which fixed beneficial mutants alter the selective context for subsequently appearing mutants. Several lines of evidence suggest that epistatic interactions between mutations may be reasonably common. First, compensatory mutations fixed in genetic backgrounds containing deleterious mutations are probably beneficial only on those backgrounds (Poon and Chao 2005). Second, studies of genetic divergence between species have shown that alleles which are severely deleterious in one species can be fixed on the permissive genetic background of another (Kondrashov et al. 2002; Coyne and Orr 2004; Kern and Kondrashov 2004; Kulathinal et al. 2004; The Chimp Genome Sequencing Consortium 2005). Finally, in experimental tests, pairs of known beneficial mutations frequently show epistasis, either in sign or in magnitude (Mo et al. 1997; Bull et al. 2000; Sanjuan et al. 2004; Weinreich et al. 2006).
Here, I use material generated in a previous study (Betancourt 2009) to explore the role played by sign epistasis in adaptively evolving populations of MS2, an RNA bacteriophage. In that study, three identical phage populations were selected for growth at cold temperature, and each initially fixed a beneficial mutation with a large effect on growth. In this study, I examine the role of those initial substitutions in determining the effects of subsequent mutations. I investigate this by propagating new phage populations, and observing whether mutations previously restricted to one of the backgrounds appear only on that background, or are fixed on the other background as well.
Materials and Methods
Phage Strains
MS2 is a bacteriophage in the genus Levivirus (family Leviviridae). Phage in this genus consists of a single positive strand protein-coding RNA encapsulated by a virion coat. The genome of MS2 is small (~3569 nts) and consists of maturase, coat, lysis, and replicase genes, as well as several regulatory regions, typically consisting of RNA secondary structures of the single stranded RNA chromosome (Klovins et al. 1997).
The MS2 strains used in this experiment were derived from experimental populations, in which large populations (1 × 107 phage each) were adapted to growth at 30°C (under conditions that did not favor host–phage or phage–phage coevolution) and fixed a single mutation with a large beneficial effect (Betancourt 2009). To obtain these strains, I isolated single plaques (clonal derivates of a single phage) and sequenced whole genomes to confirm that their consensus genotypes consisted each of the initial substitutions (G253C and U862C) and no background mutations. Three suitable plaques were identified in this way, two containing G253C [derived from Line 2 of Betancourt (2009)] and one containing U862C [derived from Line 1 of Betancourt (2009)]. For G253C, the three lines were propagated from one of the phage clones (replicates A–C) and two from the other (replicates D and E).
The experiment is essentially similar to that of Betancourt (2009), with a few important differences. First, population sizes were smaller (~1 × 105 phage, instead of 1 × 107), due to the limited starting material (three plaques, see above) available for this experiment. Second, because I was only interested in the first substitution fixed in this experiment, populations were propagated for fewer generations (20 instead of 50 generations). Finally, because the populations in this experiment already contain one strongly beneficial mutation (either G253C or U862C), it seems reasonable to presume that selection in this experiment is weaker than in the Betancourt (2009) experiment.
Serial Passaging
Phage were grown in a 5-ml culture of TOP 10 F’ Escherichia coli cells (Invitrogen) in standard Luria–Bertani medium supplemented with 14 μg/ml tetracycline (Sigma) to maintain the presence of the F-plasmid. Each population was propagated for 20 serial passages [with each passage long enough to encompass attachment, replication and the beginning of lysis for of one infection cycle (data not shown)] at 30°C. For each passage, an initial inoculation of 105 phage was grown in a 1000-fold excess of host cells for 130 min. At the end of this time period, the phage and host cell cultures were centrifuged for 20 min to separate host cells and phage lysate. A portion of the lysate was plated on host cells using the soft agar overlay method to determine phage population size, and another portion (containing ~1 × 105 phage) was used to inoculate the next serial passage. To prevent contamination, three measures were taken: (i) a portion of the E. coli culture used for each passage was set aside as a negative control—i.e., this portion was not infected, but was grown alongside the phage culture and tested to confirm the absence of phage in the initial culture, (ii) phage populations with the same initial substitution were grown at different times and (iii) phage populations grown at the same time (which differed in genotype) were checked for cross-contamination by sequencing.
PCR and Sequencing
Phage RNA was isolated using a standard phenol–chloroform isoamyl alcohol protocol and precipitated in 95% ethanol with 30-mM NaOAc. This RNA was used as a template for a reverse transcription reaction using iScript (BioRad), and the resulting cDNA used as a template for PCR. PCR fragments were directly sequenced using internal primers, and sequences were analyzed using Sequencher (Genecodes). Data are deposited in the Genbank database under accession numbers HQ332484-HQ332512.
For each line, I sequenced phage lysate from the final serial passage to identify substitutions. These “whole genome” sequences, >95% of the genome, include all of the coding regions (approximately from Genbank reference sequence NC 001417 positions of 94–3521) sequenced in both directions. Mutations were considered fixed (or nearly so) if they appeared as single peaks in the chromatograms of these “population” sequences; previous work shows that substitutions detected in this way are in fact at high frequencies when individual clones are resequenced. For two of the lines that appeared to be polymorphic in the phage lysate sequences (i.e., that had double peaks in the chromatograms), I sequenced the portion of genome containing the putative polymorphisms for 8–11 phage clones (each representing an individual phage from the population).
Analysis
To compare the level of parallel evolution in the present experiment and Betancourt (2009), I used a permutation test implemented in R (http://www.r-project.org), permuting mutations between the lines and between the experiments 10,000 times. The degree of parallel evolution was calculated as the fraction of pairs of mutations from different lines that are the same, out of the maximum possible. For this purpose, 1691 and 1692 were counted as the same mutation, as they may have similar functional effects (Betancourt 2009) and as this is conservative.
Results
Review of Previous Results
Of the three lines propagated by Betancourt (2009), two showed a high degree of parallel evolution, sharing two identical (G253C and A1697G) and two very similar mutations [U1691C, C1692U; both affecting a region where the function is mediated by an RNA secondary structure (Betancourt 2009)]. In contrast, there was no overlap with the substitutions found in the third replicate. Since all three lines shared a large population size (107), high mutation rate (1.5 × 10−3–1 × 10−4 per round of infection, Drake 1993; Drake et al. 1998) and similar environments, the degree of difference between them is surprising. Sign epistasis is one potential explanation. That is, it may be that the identity of the initial substitution fixed in each line (G253C or U862C) epistatically determines which new mutations have beneficial effects. In that case, the subsequent mutations that were similar between the two parallel lines (U1691C/U1692C, and A1697G) may not have beneficial on the other background.
Present Experiment
Ten phage populations were propagated, five from phage with the G253C genotype (lines 253A-E) and five with the U862C genotype (lines 862A-E) for 20 phage generations. Sequencing of the whole populations at the end of the experiment showed that nine of the lines had fixed or nearly fixed new mutations (Table 1), and that three of the lines (253E and 862D, E) showed evidence of polymorphism. Resequencing of individual phage in these lines revealed sites containing polymorphisms at intermediate frequencies (Table 2). Overall, the lines showed strong parallel evolution, regardless of genetic background, most of the lines (8/10) contained the U1691C mutation.
Besides the initial substitution, there were two pairs of similar mutations shared between the lines showing a high degree of parallel evolution in the Betancourt (2009) experiment (Line 2: U1691C/Line 3: C1692U, and Line 2: A1697G/Line 3: A1697G). Two of these mutations (U1691C and A1697G) occurred on both genetic backgrounds (summing across both experiments, Table 1). In addition, a substitution similar to the one previously restricted to the U862C background occurs on the G253C background in this experiment (U1741C on the U862C background in Betancourt 2009, U1742C on the G253C background here; both affect the same functional unit and RNA secondary structure).
Other combinations of mutations, however, do show evidence of epistasis. In particular, two pairs show antagonistic epistasis, in which beneficial mutations are either deleterious (i.e., sign epistasis) or less beneficial than expected (i.e., magnitude epistasis) in combination. For example, lines with U862C did not fix G253C, and vice versa, in either experiment—in spite of the fact that each mutation taken separately has large beneficial effects (Betancourt 2009). Mutations at sites 1691 and 1697 show a similar pattern: both occur at high frequency (~50%) in three independent lines [twice in this experiment and once in the Betancourt (2009)], but never appear on the same background. This kind of antagonistic epistasis is similar to that seen in other experiments (e.g., Bull et al. 2000; Sanjuan et al. 2004; Weinreich et al. 2006).
Discussion
To address the question, the current experiment assumes that the evolution observed is adaptive. There are two reasons to think that this assumption is reasonable: First, the fixed mutations were fixed singly, with no evidence of hitchhiking, and in a short time frame in a large census population (phage were propagated for 20 generations at a bottleneck size of 105), they were most likely to be the result of adaptive evolution. Second, all of the mutations in this experiment show some degree of parallel evolution, with phage either in the current experiment or in the Betancourt (2009) experiment, usually considered strong evidence of adaptation (reviewed in Wood et al. 2005).
The present experiment shows that at least three of the mutations that differed between backgrounds in the Betancourt (2.009) experiment are beneficial on either background. Thus, the observation that motivated this experiment—some parallel evolution between populations with the same initial substitution, and divergent evolution between populations with different initial substitutions—does not appear to be due to sign epistasis. The generality of this result is unclear; other experimental systems have shown both similar (Holder and Bull 2001) and contrasting (Weinreich et al. 2006) results. The observation itself may be due to magnitude epistasis between the beneficial mutations or may be a stochastic effect ultimately due to the fixation of genomes containing multiple beneficial mutations (see below).
This study, like other experimental evolution studies on viruses (e.g., Wichman et al. 1999; Crill et al. 2000; Cuevas et al. 2002; Bull et al. 2000; Bollback and Huelsenbeck 2009), shows a high rate of parallel evolution between lines, suggesting virus have a restricted number of ways in which they can adapt to the relevant experimental conditions and a limited number of beneficial mutations. Theoretical work suggests that the restrictions on adaptation in may be especially acute for RNA phage, where the high mutation rate yields strong selection for a compact genome (Holmes 2003; Belshaw et al. 2008; Belshaw et al. 2008). The results of this experiment suggest that nearly all of the mutations that are beneficial under these experimental conditions and that have large enough effects to be seen in a short-term experiment have been observed: Of the 10 possible beneficial mutations seen in this experiment (counting all of the mutations seen in this experiment, whether polymorphic or fixed, Tables 1, 2), nine were observed more than once (Betancourt 2009; this study). Even the single mutation not seen before (U403C) resides in the same codon as two other previously observed mutations [U404C (Betancourt 2009) and C405U, this study)].
Other factors in these experiments—strong selection, large population sizes, high mutation rates, and opportunity for clonal interference—also promote a high level of parallel evolution. That is, under these conditions, one might expect evolution to tend toward being deterministic, with the mutation with the largest beneficial effect being the next fixed. Counterintuitively, however, evolution was more parallel in the current experiment, which had smaller population sizes and weaker selection, than in the Betancourt (2009) experiment (Table 1; proportion parallel mutations averaged over lines, this experiment: 0.66, Betancourt (2009) experiment: 0.25; two-tailed permutation test, P = 0.027). A remaining difference between the experiments (the longer time for which the Betancourt 2009 lines were propagated) might explain this difference, as a longer propagation time allows more time for deleterious mutations to accumulate, reducing the effective size of the population (Peck 1994, Johnson and Barton 2005). However, calculations based on the work of Johnson and Barton (2005) show that most plausible values of the relevant parameters for these experiments do not yield a large difference in effective size.
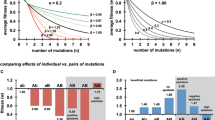
There are two plausible scenarios that might explain a higher degree of parallel evolution in small populations. First, magnitude epistasis between the first substitutions and subsequent mutations might mean that the identity of the best mutation differs depending on the initial substitution (Fig. 1a). This explanation is appealing, as it can explain the observation that motivated this experiment, but not entirely satisfactory, as it suggests that the present study should have shown an effect of genetic background. A strong mutational bias might explain why it did not; the small populations of this study might be restricted to fixing the best frequently occurring mutation (see also Rokyta et al. 2005).

Factors affecting parallel evolution between lines. Shown are contrasts between small and large populations, with hypothetical scenarios that may promote more parallel evolution in the small populations. Beneficial mutations are shown, with the degree of benefit indicated by the number of plus signs. Mutations or combinations of mutations—due to either mutational bias or finite population size—that have not occurred are grayed out
Second, the large populations might have experienced multiple occurrences of beneficial mutations, contributing to their lower degree of parallel evolution (Fig. 1b). That is, if two beneficial mutations occur in the same genome (not necessarily simultaneously), the double mutant might be fitter than the best possible single mutation, causing its loss (Desai et al. 2007). Note that the large populations show clear evidence of simultaneous sweeps of multiple mutations (Betancourt 2009; see also Bollback and Huelsenbeck 2007). But these large populations are likely not so big that they experience every possible combination of multiple beneficial mutations; as a result, they might not always fix the same suites of multiple mutations. Thus, the transition from adaptation due to single mutations to adaptation via the fixation of double mutations may reduce the amount of parallel evolution. This process, unlike epistasis or the accumulation of deleterious mutations, can also explain the different initial substitutions (Desai et al. 2007; see also Betancourt 2009).
It is important to note that the above explanations apply to short time scales such as those seen in this experiment. In the long-term, in the absence of sign epistasis or environmental change, there will be perfect parallel evolution between all lines, with all beneficial mutations fixed (Weinreich et al. 2005). Even in the presence of sign epistasis, populations adapting to the same environment are likely to show strong parallel evolution (Unckless and Orr 2009).
References
Belshaw R, Gardner A, Rambaut A, Pybus OG (2008) Pacing a small cage: mutation and RNA viruses. Trends Ecol Evol 23:188–193
Betancourt AJ (2009) Genomewide patterns of substitution in adaptively evolving populations of the RNA bacteriophage MS2. Genetics 181:1535
Bollback JP, Huelsenbeck JP (2007) Clonal interference is alleviated by high mutation rates in large populations. Mol Biol Evol 24:1367–1406
Bollback JP, Huelsenbeck JP (2009) Parallel genetic evolution within and between bacteriophage species with varying degrees of divergence. Genetics 181:225–234
Bull JJ, Badgett MR, Wichman HA (2000) Big-benefit mutations in a bacteriophage inhibited with heat. Mol Biol Evol 17:942–950
Burch C, Chao L (2000) Evolvability of an RNA virus is determined by its mutational neighbourhood. Nature 406:625–628
Coyne JA, Orr HA (2004) Speciation. Sinauer Associates, Sunderland, MA
Coyne JA, Barton NH, Turelli M (2000) Is Wright’s shifting balance process important in evolution? Evol Int J Org Evol 54:306–317
Crill WD, Wichman HA, Bull JJ (2000) Evolutionary reversals during viral adaptation to alternating hosts. Genetics 154:27–37
Cuevas JM, Elena SF, Moya A (2002) Molecular basis of adaptive convergence in experimental populations of RNA viruses. Genetics 162:533–542
Desai MM, Fisher DS, Murray AW (2007) The speed of evolution and maintenance of variation in asexual populations. Curr Biol 17:385–394
Drake JW (1993) Rates of spontaneous mutation among RNA viruses. Proc Natl Acad Sci USA 90:4171–4175
Drake JW, Charlesworth B, Charlesworth D, Crow JF (1998) Rates of spontaneous mutation. Genetics 148:1667–1686
Gerrish PJ, Lenski RE (1998) The fate of competing beneficial mutations in an asexual population. Genetica 102–103:127–144
Goodnight CJ, Wade MJ (2000) The ongoing synthesis: a reply to Coyne, Barton, and Turelli. Evol Int J Org Evol 54:317–324
Holder KK, Bull JJ (2001) Profiles of adaptation in two similar viruses. Genetics 159:1393–1404
Holmes EC (2003) Error thresholds and the constraints to RNA virus evolution. Trends Microbiol 11:543–546
Johnson T, Barton NH (2005) The effect of deleterious alleles on adaptation in asexual populations. Genetics 162:395–411
Kern AD, Kondrashov FA (2004) Mechanisms and convergence of compensatory evolution in mammalian mitochondrial tRNAs. Nat Genet 36:1207–1212
Kim Y, Orr HA (2005) Adaptation in sexuals versus asexuals: clonal interference and the Fisher-Muller model. Genetics 171:1377–1386
Klovins J, Tsareva NA, de Smit MH, Berzins V, van Duin J (1997) Rapid evolution of translational control mechanisms in RNA genomes. J Mol Biol 265:372–384
Kondrashov AS, Sunyaev S, Kondrashov FA (2002) Dobzhansky-Muller incompatibilities in protein evolution. Proc Natl Acad Sci USA 99:14878–14883
Kulathinal RJ, Bettencourt BR, Hartl DL (2004) Compensated deleterious mutations in insect genomes. Science 306:1553–1554
Mo H, Stamatatos L, Ip JE, Barbas CF, Parren PW, Burton DR, Moore JP, Ho DD (1997) Human immunodeficiency virus type 1 mutants that escape neutralization by human monoclonal antibody IgG1b12. off. J Virol 71:6869–6874
Peck JR (1994) A ruby in the rubbish: beneficial mutations, deleterious mutations and the evolution of sex. Genetics 137:597–606
Poon A, Chao L (2005) The rate of compensatory mutation in the DNA bacteriophage phiX174. Genetics 170:989–999
Poon A, Davis BH, Chao L (2005) The coupon collector and the suppressor mutation: estimating the number of compensatory mutations by maximum likelihood. Genetics 170:1323–1332
Rokyta DR, Joyce P, Caudle SB, Wichman HA (2005) An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus. Nat Genet 37:441–444
Sanjuan R, Moya A, Elena SF (2004) The contribution of epistasis to the architecture of fitness in an RNA virus. Proc Natl Acad Sci USA 101:15376–15379
The Chimp Genome Sequencing Consortium (2005) Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69–87
Unckless RL, Orr HA (2009) Dobzhansky-Muller incompatibilities and adaptation to a shared environment. Heredity 102:214–217
Wahl LM, Krakauer DC (2000) Models of experimental evolution: the role of genetic chance and selective necessity. Genetics 156:1437–1448
Weinreich DM, Watson RA, Chao L (2005) Perspective: sign epistasis and genetic constraint on evolutionary trajectories. Evol Int J Org Evol 59:1165–1174
Weinreich DM, Delaney NF, Depristo MA, Hartl DL (2006) Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 312:111–114
Welch JW, Waxman D (2005) The nk model and population genetics. J Theor Biol 234:329–340
Wichman HA, Badgett MR, Scott LA, Boulianne CM (1999) Different trajectories of parallel evolution during viral adaptation. Science 285:422–424
Wood TE, Burke JM, Rieseberg LH (2005) Parallel genotypic evolution: when evolution repeats itself. Genetica 123:157–170
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Acknowledgments
Thanks to J. Bollback, S. Collins, K. Dyer, H. Kuehne, D. Presgraves, and J. Welch for helpful discussion and to L. Yanoso for technical help. I particularly thank A. Orr and J. Huelsenbeck for helpful discussion and encouragement during the course of this experiment. This work was funded in part by a National Science Foundation (NSF) Doctoral Dissertation Improvement grant to A.J.B. and H. A. Orr.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Betancourt, A.J. Lack of Evidence for Sign Epistasis Between Beneficial Mutations in an RNA Bacteriophage. J Mol Evol 71, 437–443 (2010). https://doi.org/10.1007/s00239-010-9397-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-010-9397-0