
Abstract
Bluetongue virus (BTV) is a midge-borne member of the genus Orbivirus that causes an eponymous debilitating livestock disease of great agricultural impact and which has expanded into Europe in recent decades. Reassortment among the ten segments comprising the double-stranded (ds) RNA genome of BTV has played an important role in generating the epidemic strains of this virus in Europe. In this study, we investigated the dynamics of BTV genome segment evolution utilizing time-structured data sets of complete sequences from four segments, totalling 290 sequences largely sampled from ruminant hosts. Our analysis revealed that BTV genome segments generally evolve under strong purifying selection and at substitution rates that are generally lower (mean rates of ~0.5–7 × 10−4 nucleotide substitutions per site, per year) than vector-borne positive-sense viruses with single-strand (ss) RNA genomes. These also represent the most robust estimates of the nucleotide substitution rate in a dsRNA virus generated to date. Additionally, we determined that patterns of geographic structure and times to most recent common ancestor differ substantially between each segment, including a relatively recent origin for the diversity of segment 10 within the past millennium. Together, these findings demonstrate the effect of reassortment to decouple the evolutionary dynamics of BTV genome segments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bluetongue virus (BTV) is a midge-borne double-strand (ds) RNA virus of the family Reoviridae (genus Orbivirus) that is the pathogenic agent of Bluetongue disease in ruminants. Bluetongue disease is responsible for considerable morbidity and mortality in livestock, especially sheep (MacLachlan 1994), although cattle may serve as the main reservoir (Barrat-Boyes and MacLachlan 1994; Schwartz-Cornil et al. 2008). Importantly, BTV is vectored by several species of Culicoides midges, and although domestic livestock are known hosts, wild ruminants may also serve as natural reservoir hosts of BTV (Wilson and Mellor 2008).
Bluetongue disease was first observed during the importation of Merino sheep into South Africa (Spreull 1905). BTV subsequently spread throughout Africa and into other continents via the movement of infected animals in the livestock trade, and through the expansion of the insect vector range (Wilson et al. 2008), which currently limits BTV to tropical and temperate regions (i.e., between latitudes 35°S and 40°N) (Mellor et al. 2008). Interestingly, significant episodic changes in the geographic distribution of BTV have been reported in Europe since 1998 (Mellor and Wittmann 2002; Purse et al. 2005; Maan et al. 2008; Saegerman et al. 2008), often involving the introduction of new BTV strains from different regions (e.g., north Africa; Maan et al. 2008). Intriguingly, it is believed that the recent emergence of Bluetongue in northern Europe may have been driven by climatic changes that increased the overwinter persistence of BTV in the vector populations and/or the northward expansion of a BTV-competent vector (Culicoides imicola; Purse et al. 2008; Wilson et al. 2008).
The BTV genome consists of 10 linear segments of dsRNA that encode seven structural proteins (VP1–VP7) and three nonstructural proteins (NS1–NS3). Segments range in size from 822 to 3954 nt, constituting a genome approximately 19 kb in size (Mertens et al. 2005). Previous experimental and computational studies have found that the BTV genome evolves rapidly via the reassortment of genome segments, producing antigenic shifts (Oberst et al. 1987; Ramig et al. 1989; de Mattos et al. 1996; Pierce et al. 1998; Bonneau et al. 2001), and through the accumulation of nucleotide substitutions generated during virus replication (Kowalik and Li 1991; Jenkins et al. 2002; Hanada et al. 2004). Reassortment clearly plays a major role in the generation of genomic diversity. Specifically, while segments (Seg)-2 and 6 accurately predict overall BTV serotype (Mertens et al. 1989), phylogenetic analysis reveals that Seg-3 and -10 display varying levels of reassortment (de Mattos et al. 1996; Pierce et al. 1998; Maan et al. 2007). Interestingly, Seg-3, which encodes the inner capsid protein VP3, is the most conserved segment in the BTV genome and hence may accurately reflect the geographic origin of BTV strains by dividing them into the eastern and western “topotypes” (Pritchard et al. 1995; Maan et al. 2008; Nomikou et al. 2009).
In contrast, less is known about the evolutionary processes that shape patterns of nucleotide substitution across the BTV genome. Among RNA viruses as a whole, vector-borne viruses are thought to evolve under relatively strong selective constraints, imposed by the necessity of replication in two distantly related host species; this, in turn, reduces the rate of nucleotide substitution, particularly at nonsynonymous sites (Chare and Holmes 2004; de Mattos et al. 1996; Holmes 2009; Jenkins et al. 2002; Woelk and Holmes 2002). As a case in point, a recent study of the evolution of BTV Seg-3 suggested that very strong purifying selection reduces the rate of substitution at first and second codon positions (Nomikou et al. 2009). Previous estimates of the nucleotide substitution rate in BTV have ranged from 2.2 × 10−3 to 4.2 × 10−4 nucleotide substitutions per site, per year (subs/site/year), which are similar to those observed in other RNA viruses (Kowalik and Li 1991; Jenkins et al. 2002; Hanada et al. 2004). However, these comparative studies were strongly biased toward single-stranded (ss) RNA viruses such that the evolutionary dynamics of viruses with dsRNA genomes, such as BTV, have received far less attention.
Given the increasing impact of BTV on European livestock populations and the emergence of new BTV strains in Europe (Batten et al. 2008; Maan et al. 2008), revealing the evolutionary and epidemiological dynamics of epidemic BTV is of great importance. These analyses are particularly relevant in the context of vaccination against BTV, which has already been shown to affect the diversity of BTV in unexpected ways, including the generation of epidemic strains via reassortment among vaccine and naturally circulating BTV variants (Batten et al. 2008). Additionally, understanding the evolutionary dynamics of BTV will provide more general information on the factors that shape the genetic diversity of dsRNA viruses. To address these issues, we applied Bayesian Markov chain Monte Carlo (MCMC) coalescent analyses to the largest data set of complete BTV segment sequences assembled to date. From this, we estimated both rates of nucleotide substitution and Time to the Most Recent Common Ancestor (TMRCA) for each genomic segment of BTV. Importantly, these findings provide valuable information on the evolutionary processes shaping BTV genetic diversity, including the extent and impact of reassortment in the evolutionary history of this virus.
Materials and Methods
Data Preparation
Data sets of complete sequences for 8 of the 10 segments of the BTV genome (Seg-2, -3, -5, -6, -7, -8, -9, and -10) were compiled from GenBank (Table 1). Importantly, all sequences gathered were required to have known geographic origins and isolation dates, as geographic and temporal data are of central importance when investigating the evolutionary dynamics of rapidly evolving viruses in space and time. The isolation dates for all sequences used span the interval 1959 to 2007. To reduce the effect of small sample sizes, only segments with more than 20 sequences were considered. Furthermore, all sequences from BTV strains known to have been highly cultured were excluded, as were sequences that have been artificially manipulated (e.g., vaccine strains). Importantly, although the majority of BTV sequences analyzed here are from ruminant hosts and not from midge vectors, this will not affect our estimates of evolutionary dynamics due to the obligate nature of the vector–host transmission cycle in BTV, in which such processes as transmission bottlenecks and differential host selective pressures will affect all viral lineages alike. For each segment data set, sequence alignments were created using the MUSCLE program (Edgar 2004) and then adjusted manually using the Se–Al alignment editing software (Rambaut 1996) to maintain reading frame integrity. In total, 412 BTV sequences isolated over a 40-year sampling period were collected and examined (the exact time span of the sequences for each segment are given in the Table 1). In the Seg-2 alignment, a number of gaps due to insertions and deletions among the different serotypes were observed and included. As previous studies found evidence for intra-segment recombination within members of the Reoviridae (Parra et al. 2004; Anthony et al. 2009), a bootscan/rescan recombination test (Martin et al. 2005) implemented in the RDP3 beta 34 package (Heath et al. 2006) was performed on the Seg-2, -3, -6, and -10 alignments; no significant evidence for intra-segment recombination between BTV serotypes was found. All sequence alignments are available from the authors upon request.
Estimating Evolutionary Dynamics
Rates of molecular evolution (i.e., nucleotide substitutions per site, per year) and the TMRCA in years were estimated for each segment using the Bayesian MCMC approach implemented in the BEAST package (Drummond and Rambaut 2007). In each case, the model of nucleotide substitution that best fit the data set was determined using ModelTest v3.7 (Posada and Crandall 1998; Posada and Crandall 2001). Consequently, the data were analyzed under a General Time Reversible (GTR) model of nucleotide substitution with a gamma distribution (Γ 4) model of site variation (i.e., GTR + Γ 4), as well as a model that assigned a different substitution rate to each codon position (i.e., GTR + CP) and which seems a particularly good description of RNA virus evolution (Shapiro et al. 2006). To be as comprehensive as possible, we also estimated substitution rates using both strict and relaxed (i.e., uncorrelated lognormal; Drummond et al. 2006) molecular clocks. Since inferring demographic history (i.e., rates of population growth and decline) was not the aim of this study, we utilized the most general Bayesian skyline coalescent prior (Drummond et al. 2005), which allows for both constant and complex changes in population size through time. For the analysis under each model, the MCMC was run for a sufficient number of generations to ensure convergence of all parameters. The program Tracer v1.4 (available at http://tree.bio.ed.ac.uk/software/tracer/) was used to inspect posterior distributions and estimate evolutionary parameters of interest. Critically, we limited further analyses to four segments (Seg-2, -3, -6, and -10), as the MCMC did not converge for the remaining segments (Seg-5, -7, -8 and -9 data not shown). The most likely explanation for the nonconvergence of the MCMC was insufficient temporal information in the segment alignments, as the nonconvergent data sets generally comprised fewer sequences sampled during a shorter interval of time when compared to those segments for which the MCMC did converge (Table 1).
As a conservative test of the extent of temporal structure in the four segment data sets for which substitution rates were estimated, we performed a regression analysis of root-to-tip distances against sampling times. This required that we first estimated maximum likelihood phylogenies for each segment using the GTR + CP substitution model and tree-bisection-reconnection (TBR) branch-swapping in PAUP v4.10beta (Swofford 2001). Phylogenies were also estimated from sub-sets of sequences for each segment to identify possible temporal structure in local sub-trees within complete segment phylogenies. Regressions were performed with Path-O-Gen v1.1 (Drummond et al. 2003) and optimal roots that maximize the correlation between root-to-tip distances and sampling times were used in all cases.
Phylogenetic Analysis of Geographic Structure
The posterior set of trees produced from the BEAST analysis was also used to estimate the Maximum Clade Credibility (MCC) phylogeny for each of the BTV segments, including the posterior probabilities for the inferred evolutionary relationships. To determine the extent of geographic structure in BTV populations, we estimated the strength of association between phylogenetic relationships and sampling locations for the four segments that converged in the BEAST analysis: Seg-2, -3, -6, and -10. We then used the parsimony score (PS; Slatkin and Maddison 1989) and association index (AI; Wang et al. 2001) statistics to determine the extent of geographical association with sampling locations across the entire tree, as well as the maximum monophyletic clade size (MC; Parker et al. 2008) statistic to assess the association with particular sampling locations. Every sequence for each BTV segment was coded as coming from one of up to 23 countries (Seg-2: 15 countries; Seg-3: 19; Seg-6: 23; and Seg-10: 21). Using the program BaTS v0.90 beta (Parker et al. 2008), distributions of the three statistics (PS, AI, and MC) were calculated from the posterior samples of BEAST trees and compared to null distributions generated by randomizing the geographic origins of BTV sequences and recalculating across the tree sample to determine the significance of the empirical distributions for each segment (Parker et al. 2008). The first 10% of sampled trees were discarded as burn-in and 1,000 randomizations were performed to estimate null distributions for the statistics of interest for each segment individually.
Estimation of Selection Pressure
To investigate the selection pressures acting on the different segments of the BTV genome (i.e. Seg-2, -3, -6, and -10), we estimated the mean number of nonsynonymous substitutions (d N) and synonymous substitutions (d S) per site (ratio d N/d S using the Single Likelihood Ancestor Counting (SLAC; Pond and Frost 2005c) method implemented in the HYPHY platform (Pond and Frost 2005a) accessed through the Datamonkey web-server (http://www.datamonkey.org; Pond and Frost 2005b). Estimates for d N/d S were made from phylogenies inferred using the Neighbor-Joining algorithm with distances corrected under the GTR substitution model. The entire open reading frame of the NS3 protein in Seg-10 (positions 20–709) was used as the other protein encoded on Seg-10, NS3a, is merely a truncated version of NS3 that shares the same reading frame. The remaining segments (Seg-5, -7, -8, and -9) were not analyzed because their sample sizes were too small to provide accurate estimates of selection pressure or the MCMC did not converge when analyzed in BEAST.
Results and Discussion
Evolutionary Rate of BTV Genomic Segments
Our Bayesian coalescent estimates of evolutionary dynamics in BTV indicate that Seg-2, -3, -6, and -10 evolve at mean rates of between 0.52 and 6.9 × 10−4 substitutions per site, per year (95% HPD = 0.23–10.7 × 10−4; Table 2). Importantly, these rate estimates for each segment were robust and similar regardless of the molecular clock model employed (Table 2). Interestingly, these substitution rates are, on average, lower than those previously estimated for vector-borne, positive-sense ssRNA viruses (e.g., 24.2–3.3 × 10−4 subs/site/year for the Flavivirus and Alphavirus genera; Hanada et al. 2004), though they are comparable to rates for vector-borne, negative-sense ssRNA viruses (e.g., 12.3–0.7 × 10−4 subs/site/year for the Nairovirus, Orthobunyavirus, Phlebovirus, and Vesiculovirus genera; Hanada et al. 2004). Notably, when compared to other dsRNA viruses within the family Reoviridae, BTV appears to evolve less rapidly than the nonvector-borne members of this family (e.g., 19.3–8.4 × 10−4 subs/site/year for the Rotavirus and Orthoreovirus genera, formerly named Reovirus and Orthoreo, respectively; Hanada et al. 2004). This is consistent with the hypothesis that strong purifying selection, imposed by obligate cycling through hosts, reduces the effective substitution rate of vector-borne viruses (Coffey et al. 2008). Furthermore, it is possible that the intracellular encapsulation of the dsRNA genome within a transcriptional core that protects against host defences acting to eliminate dsRNA might also protect against cell factors that mutate ssRNA and so reduce the observed mutation rates, and thus substitution rates, of dsRNA virus genomes (Duffy et al. 2008). Finally, it is also possible that the relatively low overall substitution rates observed for BTV reflect a reduced viral replication associated with persistent infection in the ruminant reservoir hosts during its enzootic cycle (Stott et al. 1992; Takamatsu et al. 2003); although, the exact mechanism of over-wintering in ruminants has not been determined either experimentally or in the field (Takamatsu et al., 2003; Saegerman et al., in press).
Importantly, our rate estimates for individual BTV genome segments allow us to better understand the influence of biological function on viral evolutionary dynamics. To determine how differential selection pressures may have affected these substitution rates, we also estimated the rate of nonsynonymous (d N) to synonymous (d S) substitutions per site, which can be used to identify signatures of purifying (d N/d S < 1) or positive (d N/d S > 1) selection. Surprisingly, we found that Seg-6, which encodes for the VP5 protein that contributes to the outer capsid protein, evolves at a significantly lower rate (5.23 × 10−5 subs/site/year; Table 2) than the other segments, and slower than any rate estimated for a dsRNA virus to date. The d N/d S analyses indicate that strong purifying selection may be reducing the overall substitution rate of Seg-6 (d N/d S = 0.032; Table 2) compared to other segments. This contrasts with the far higher substitution rate (4.37 × 10−4 subs/site/year) and higher d N/d S ratio (0.122) estimated for Seg-2, which also encodes for a capsid protein (VP2). Importantly, VP2 determines BTV serotype and is the most variable segment in the viral genome, whereas VP5 plays a smaller role in capsid construction. Interestingly, Seg-10, which encodes the NS3/NS3a proteins, has a low d N/d S ratio (0.060) similar to Seg-6, but a much higher substitution rate (6.94 × 10−4 subs/site/year) similar to that of Seg-2. This might be explained by functional constraints acting on NS3/NS3a, as these proteins are required for virion assembly and the release of virus particles from infected cells. Finally, Seg-3, which encodes an inner capsid protein (VP3), evolves under very strong purifying selection (d N/d S = 0.006) and less rapidly (1.90 × 10−4 subs/site/year) than Seg-10, the other segment not involved with determining serotype analyzed here. Overall, these results suggest that there may be no general difference in the evolutionary rates of internal versus surface (i.e., serotype determining) genomic segments of BTV. Hence, while the serotype-determining protein (i.e., VP2 encoded by Seg-2) might be the target of periodic positive selection (relatively high substitution rate and d N/d S value), the remainder of the BTV segments seemingly evolve under purifying selection (sometimes strongly so), and the relationship between selection pressure and substitution rate is not consistent across BTV genome segments.
To determine whether our estimates of substitution rate were robust, we repeated the BEAST analysis with the same parameters as described above but removing those sequences isolated prior to 1980 (maximum time span 1980–2007). This serves as a very conservative control for the accidental inclusion of highly cultured BTV isolates in our data sets. The HPD intervals of the substitution rate estimates in this case overlapped with those of the complete data sets, indicating that very old BTV sequences did not introduce biases in our substitution rate estimates (Table S1). Notably, the mean substitution rates estimated from the full and reduced data sets were very similar, with the sole exception of Seg-3, which had higher mean estimates from the reduced data set (4.79 × 10−4 subs/site/year).
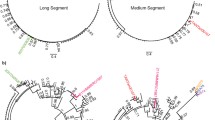
The variation in substitution rates among the BTV genomic segments is puzzling, particularly the extremely low rate of Seg-6, which is approximately 1/10th the rate of Seg-2, -3, and -10. To assess the reliability of our rate estimates, we plotted regressions of root-to-tip distances and sampling times for each segment, which serve as conservative measures of temporal structure in genetic diversity. Importantly, the low correlation (r 2 < 0.05) in the regression for Seg-6 (Fig. 1) is indicative of little temporal structure over the sampling interval that is consistent with low substitution rates (this is also true when the Seg-6 phylogeny is divided into two sub-trees; r 2 = 0.023 and 0.051, respectively). This is visually apparent, for example, by the lack of divergence between Seg-6 sequences sampled in the United States in 1982 and India in 2003 (Fig. 1, accession numbers AY855277 and AJ783904-6). In contrast, regressions performed on the two well-supported topotype clades of Seg-3 reveal a much stronger correlation between root-to-tip distance and sampling time (i.e., r 2 = 0.888 and 0.570 for eastern and western topotypes, respectively), indicative of stronger temporal structure in the Seg-3 data. Thus, barring possible laboratory errors, our tests suggest that the substitution rate of Seg-6 is indeed lower than the other segments and that the variation in substitution rates between segments is not artifactual.

Regression of root-to-tip distance against sampling time for sequences of BTV Seg-6. Distances taken from maximum likelihood phylogenies were regressed and plotted using Path-O-Gen v1.1 (Drummond et al. 2003). The full data set Seg-6 phylogeny was divided into two sub-trees, comprising 36 and 27 sequences, respectively, that were analyzed individually (indicated by dashed boxes on the phylogeny). All phylogenies were preferentially rooted to provide maximum correlation between sampling time and root-to-tip distance; although, the phylogeny displayed is midpoint rooted for clarity
Rate variation of this magnitude may be the product of three general evolutionary scenarios. First, mutation rates may vary between segments, although this is highly unlikely as all segments replicate using the same RNA polymerase. Second, extremely strong purifying selection may reduce the substitution rate of Seg-6 at both synonymous and nonsynonymous sites, suggesting that the rates estimated for the other segments are the result of a more rapid baseline substitution rate. However, the nature of any purifying selection on synonymous sites remains unclear. Third, positive selection, coupled with hitch-hiking at linked neutral sites, may elevate the observed substitution rates for Seg-2, -3, and -10 above the baseline substitution rate estimated for Seg-6, although this is not obviously reflected in mean d N/d S ratios. Importantly, additional sampling and genome sequencing for all segments of circulating BTV strains will be needed to both validate and explain these greatly contrasting evolutionary dynamics.
Age of Genetic Diversity of BTV Segments
Our Bayesian MCMC analysis also allowed us to estimate the TMRCA of the sequences gathered for each BTV genome segment (see Table 2). These TMRCA estimates reveal dramatic differences in the history of individual BTV segments, in turn suggesting that frequent reassortment has effectively de-coupled the demographic histories of the segments. Specifically, Seg-10 has a mean TMRCA of 517 years (95% HPD = 163–970), whereas Seg-6 has a much older TMRCA (mean = 28310, HPD = 12910–49170), although this is largely a function of very low substitution rate in this segment, and Seg-2 and -3 have intermediate TMRCA estimates (Seg-2: mean = 4295, HPD = 1637–7624; Seg-3: mean = 2192, HPD = 843–3836). Interestingly, though the very recent TMRCA of Seg-10 (mean of 517 years) is an order of magnitude more recent than the TMRCA for Seg-2 (4295 years), the two segments have similarly rapid substitution rates (4.37 × 10−4 and 6.94 × 10−4 subs/site/year for Seg-2 and -10, respectively). Overall, the much older TMRCA estimates for Seg-2, -3, and -6 suggest that the reduced genetic diversity of Seg-10 is not likely to have been caused by a bottleneck or other demographic event, since such events affect the diversity of the entire genome, but more likely a localized selective sweep. Furthermore, the nearly two orders of magnitude difference between the TMRCAs of Seg-10 and -6 provide clear evidence that reassortment has shuffled segments with very different evolutionary histories.
Phylogeography and Reassortment of BTV
Previous studies have found considerable geographic structure in BTV populations, with an especially strong east versus west division apparent in the genetic diversity of Seg-3 (Gould and Pritchard 1990; Pritchard et al. 1995; Maan et al. 2008; Nomikou et al. 2009). Our analysis also identified apparent geographic structure in the genetic diversity of BTV, as demonstrated by the MCC phylogenies for each segment (e.g., Seg-3 and Seg-10 in Figs. 2 and 3, respectively). To assess the strength of this structure in a more quantitatively rigorous fashion, we calculated summary statistics describing the correlation between the geographic and phylogenetic relationships of the sequences for each segment from the posterior distribution of genealogies generated in our coalescent analysis of BTV evolutionary dynamics (Table 3). This analysis revealed the presence of significant geographic structure in the diversity of all BTV segments studied (i.e., Seg-2, -3, -6, and -10) when sequences are considered by country (AI: P < 0.001; PS: P < 0.001; Table 3). Significant structure was also found for all segments when the sequences were grouped by continental region (i.e. Africa, the Americas, Asia, Australia, and Europe; AI: P < 0.001; PS: P < 0.001; Table 3). Interestingly, sequences from two continents showed consistent signatures of population structure in all four segments, the Americas (MC: P < 0.020 for all segments) and Europe (MC: P < 0.003). In contrast, sequences from Asia and Africa had significant signatures of population structure for only two segments, Seg-10 (MC: P < 0.001 for sequences from both continents) and Seg-6 (MC: P < 0.001 for Asia and P > 0.050 for Africa). Combined, these results suggest that the BTV populations in Europe and the Americas may serve as sink populations while BTV in Asia and Africa may more often act as source populations. This hypothesis is supported by previous reports of African BTV strains expanding into Europe (e.g., Maan et al. 2008).

MCC phylogeny of 39 BTV Seg-3 sequences calculated from the posterior distribution of trees generated by Bayesian MCMC coalescent analysis in BEAST (Drummond and Rambaut 2007). Posterior probabilities ≥0.70 are indicated in italics above branches of interest, while those branches defining the “East” and “West” topotypes (Pritchard et al. 1995) are identified. Branches are colored to indicate continent of origin, with countries identified on the right-hand side of the phylogeny. The timescale, indicated below the phylogeny, is calibrated to the Common Era (C.E.)

MCC phylogeny of 132 BTV Seg-10 sequences calculated from the posterior distribution of trees generated by Bayesian MCMC coalescent analysis in BEAST (Drummond and Rambaut 2007). Posterior probabilities ≥0.70 are indicated in italics above branches of interest, while the continental origins of sequences are indicated by color, with countries identified on the right-hand side of the phylogeny. Importantly, the timescale, indicated below the phylogeny, is calibrated to the Common Era (C.E.) and the mean Time to the Most Recent Common Ancestor (TMRCA) of Seg-10 is 517 years (95% HPD 163–970), which is substantially more recent than the TMRCA estimates for Seg-2 (95% HPD 1637–7624), Seg-3 (95% HPD 843–3836), and Seg-6 (12910–49170)
Although few BTV viral isolates were represented in the sequence data sets for all segments, which precludes rigorous statistical tests of topological congruence between segment phylogenies, we observed marked topological inconsistencies in the MCC phylogenies of Seg-2, -3, -6, and -10. This is supported by the different patterns of geographic structure and TMRCA estimates between these segments, and which together confirm the presence of recurrent reassortment events in the history of BTV. For example, the phylogeny of Seg-3 (Fig. 2) supports a previously described division of BTV isolates into “east” and “west” topotypes (e.g., Maan et al. 2008; Nomikou et al. 2009), whereas the phylogeny of Seg-10 (Fig. 3) displayed no such division. Additionally, the phylogenies of Seg-2 and -6 (Figures S1 and S2, respectively) do not display the “east” and “west” divisions seen in Seg-3, nor do they have the strong geographic structure of Seg-10. This might be a result of different selection pressures affecting the evolution of each segment (e.g., possible host immune selection acting on the diversity of Seg-2, which largely determines serotype). Interestingly, most of the geographic structure in all segments dates to within the most recent millennia, including the possible selective sweep in Seg-10, which suggests that global movements of livestock associated with European colonization during the last 500 years may have played an important role in the current distribution of BTV genetic diversity.
In sum, we show that the evolutionary process in BTV is shaped by a combination of relatively low rates of nucleotide substitution, itself a function of strong purifying selection, and frequent inter-segment reassortment that effectively decouples their evolutionary dynamics. Notably, our estimates of substitution rate reveal substantial variation in evolutionary dynamics between BTV segments, and that Seg-6 may have the lowest substitution rate of any dsRNA virus estimated to date, although this will need to be confirmed by analysis of a larger sample of sequences. Finally, our results suggest that Seg-10 may have undergone a selective sweep within the past millennium, and that the diversity of the three other segments is significantly older. Determining the cause of this putative sweep, as well as the variation in substitution rate among segments, should clearly be foci of future research. Future investigations would also benefit from the genome-scale analysis of additional BTV isolates rigorously sampled in both time and space.
References
Anthony SJ, Maan N, Maan S, Sutton G, Attoui H, Mertens PPC (2009) Genetic and phylogenetic analysis of the non-structural proteins NS1, NS2 and NS3 of epizootic haemorrhagic disease virus (EHDV). Virus Res 145:211–219
Barratt-Boyes SM, MacLachlan NJ (1994) Dynamics of viral spread in bluetongue virus infectedcalves. Vet Microbiol 40:361–371
Batten CA, Maan S, Shaw AE, Maan NS, Mertens PP (2008) A European field strain of bluetongue virus derived from two parental vaccine strains by genome segment reassortment. Virus Res 137:56–63
Bonneau KR, Mullens BA, MacLachlan NJ (2001) Occurrence of genetic drift and founder effect during quasispecies evolution of the VP2 and NS3/NS3A genes of bluetongue virus upon passage between sheep, cattle, and Culicoides sonorensis. J Virol 75:8298–8305
Chare ER, Holmes EC (2004) Selection pressures in the capsid genes of plant RNA viruses reflect mode of transmission. J Gen Virol 85:3149–3157
Coffey LL, Vasilakis N, Brault AC, Powers AM, Tripet F, Weaver SC (2008) Arbovirus evolution in vivo is constrained by host alternation. Proc Natl Acad Sci USA 105:6970–6975
de Mattos CC, de Mattos CA, MacLachlan NJ, Giavedoni LD, Yilma T, Osburn BI (1996) Phylogenetic comparison of the S3 gene of United States prototype strains of bluetongue virus with that of field isolates from California. J Virol 70:5735–5739
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:e214
Drummond AJ, Pybus OG, Rambaut A (2003) Inference of viral evolutionary rates from molecular sequences. Adv Parasitol 54:331–358
Drummond AJ, Rambaut A, Shapiro B, Pybus OG (2005) Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 22:1185–1192
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PloS Biol 4:699–710
Duffy S, Shackelton LA, Holmes EC (2008) Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet 9:267–276
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Gould AR, Pritchard LI (1990) Relationships amongst bluetongue viruses revealed by comparisons of capsid and outer coat protein nucleotide sequences. Virus Res 17:31–52
Hanada K, Suzuki Y, Gojobori T (2004) A large variation in the rates of synonymous substitution for RNA viruses and its relationship to a diversity of viral infection and transmission modes. Mol Biol Evol 21:1074–1080
Heath L, van der Walt E, Varsani A, Martin DP (2006) Recombination patterns in aphthoviruses mirror those found in other picornaviruses. J Virol 80:11827–11832
Holmes EC (2009) The evolution and emergence of RNA viruses. Oxford University Press, Oxford
Jenkins GM, Rambaut A, Pybus OG, Holmes EC (2002) Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J Mol Evol 2:156–165
Kowalik TF, Li JKK (1991) Bluetongue virus evolution: sequence analysis of the genomic S1 segments and major core protein VP7. Virol 181:749–755
Maan S, Maan NS, Samuel AR, Rao S, Attoui H, Mertens PP (2007) Analysis and phylogenetic comparisons of full-length VP2 genes of the 24 bluetongue virus serotypes. J Gen Virol 88:621–630
Maan S, Maan NS, Ross-Smith N, Batten CA, Shaw AE, Anthony SJ, Samuel AR, Darpel KE, Veronesi E, Oura CA, Singh KP, Nomikou K, Potgieter AC, Attoui H, van Rooij E, van Rijn P, De Clercq K, Vandenbussche F, Zientara S, Breard E, Sailleau C, Beer M, Hoffman B, Mellor PS, Mertens PPC (2008) Sequence analysis of bluetongue virus serotype 8 from the Netherlands 2006 and comparison to other European strains. Virol 377:308–318
MacLachlan NJ (1994) The pathogenesis and immunology of bluetongue virus infection of ruminants. Comp Immunol Microbiol Infect Dis 17:197–206
Martin DP, Posada D, Crandall KA, Williamson C (2005) A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res Hum Retroviruses 21:98–102
Mellor PS, Wittmann EJ (2002) Bluetongue virus in the mediterranean basin 1998–2001. Vet J 164:20–37
Mellor PS, Carpenter S, Harrup L, Baylis M, Mertens PP (2008) Bluetongue in Europe and the Mediterranean Basin: history of occurrence prior to 2006. Prev Vet Med 87:4–20
Mertens PP, Pedley S, Cowley J, Burroughs JN, Corteyn AH, Jeggo MH, Jennings DM, Gorman BM (1989) Analysis of the roles of bluetongue virus outer capsid proteins VP2 and VP5 in determination of virus serotype. Virol 170:561–565
Mertens PP, Maan S, Samuel A, Attoui H (2005) Family Reoviridae. In: Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (eds) Virus taxonomy: Classification and nomenclature. Eighth report of the international committee on the taxonomy of viruses. Elsevier Academic Press, Oxford, pp 466–483
Nomikou K, Dovas CI, Maan S, Anthony SJ, Samuel AR, Papanastassopoulou M, Maan NS, Mangana O, Mertens PP (2009) Evolution and phylogenetic analysis of full-length VP3 genes of Eastern Mediterranean bluetongue virus isolates. PloS ONE 4:e6437
Oberst RD, Stott JL, Blanchard-Channell M, Osburn BI (1987) Genetic reassortment of bluetongue virus serotype 11 strains in the bovine. Vet Microbiol 15:11–18
Parker J, Rambaut A, Pybus OG (2008) Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect Genet Evol 8:239–246
Parra GI, Bok K, Martinez M, Gomez JA (2004) Evidence of rotavirus intragenic recombination between two sublineages of the same genotype. J Gen Virol 85:1713–1716
Pierce CM, Balasuriya UB, MacLachlan NJ (1998) Phylogenetic analysis of the S10 gene of field and laboratory strains of bluetongue virus from the United States. Virus Res 55:15–27
Pond SLK, Frost SDW (2005a) HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679
Pond SLK, Frost SDW (2005b) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533
Pond SLK, Frost SDW (2005c) Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Posada D, Crandall KA (2001) Selecting the best-fit model of nucleotide substitution. Syst Biol 50:580–601
Pritchard LI, Gould AR, Wilson WC, Thompson L, Mertens PP, Wade-Evans AM (1995) Complete nucleotide sequence of RNA segment 3 of bluetongue virus serotype 2 (Ona-A). Phylogenetic analyses reveal the probable origin and relationship with other orbiviruses. Virus Res 35:247–261
Purse BV, Mellor PS, Rogers DJ, Samuel AR, Mertens PP, Baylis M (2005) Climate change and the recent emergence of bluetongue in Europe. Nat Rev Microbiol 3:171–181
Purse BV, Brown HE, Harrup L, Mertens PP, Rogers DJ (2008) Invasion of bluetongue and other orbivirus infections into Europe: the role of biological and climatic processes. Rev Sci Tech 27:427–442
Rambaut A (1996) Se-Al: sequence alignment editor. http://evolve.zoo.ox.ac.uk/
Ramig RF, Garrison C, Chen D, Bell-Robinson D (1989) Analysis of reassortment and superinfection during mixed infection of vero cells with bluetongue virus serotypes 10 and 17. J Gen Virol 70:2595–2603
Saegerman C, Bolkaerts B, Baricalla C, Raes M, Wiggers L, de Leeuw I, Vandenbuscche F, Zimmer J-Y, Haubruge E, Cassart D, Clercq KD, Kirschvink N The impact of naturally-occurring, trans-placental bluetongue virus serotype-8 infection on reproductive performance in sheep. Veterin J. doi:10.1016/j.tvjl.2009.11.012 (in press)
Saegerman C, Berkvens D, Mellor PS (2008) Bluetongue epidemiology in the European Union. Emerg Infect Dis 14:539–544
Schwartz-Cornil I, Mertens PP, Contreras V, Hemati B, Pascale F, Bréard E, Mellor PS, MacLachlan NJ, Zientara S (2008) Bluetongue virus: virology, pathogenesis and immunity. Vet Res 39:e46
Shapiro B, Rambaut A, Drummond AJ (2006) Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol 23:7–9
Slatkin M, Maddison WP (1989) A cladistic measure of gene flow measured from phylogenies of alleles. Genet 123:603–613
Spreull J (1905) Malarial catarrhal fever (bluetongue) of sheep in South Africa. J Comp Pathol Ther 18:321–337
Stott JL, Blanchard-Channell M, Stott ML, Barratt-Boyes SM, Maclachlan NJ (1992) Bluetongue virus tropism for bovine lymphocyte sub-populations. In: Walton TE, Osburn BI (eds) Bluetongue African Horse Sickness and related orbiviruses. CRC Press, Boca Raton, pp 781–787
Swofford DL (2001) PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates, Sunderland
Takamatsu H, Mellor PS, Mertens PP, Kirkham PA, Burroughs JN, Parkhouse RM (2003) A possible overwintering mechanism for bluetongue virus in the absence of the insect vector. J Gen Virol 84:227–235
Wang TH, Donaldson YK, Brettle RP, Bell JE, Simmonds P (2001) Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J Virol 75:11686–11699
Wilson A, Mellor P (2008) Bluetongue in Europe: vectors, epidemiology and climate change. Parasitol Res 103:S69–S77
Wilson A, Darpel K, Mellor PSS (2008) Where does bluetongue virus sleep in the winter? PLoS Biol 6:e210
Woelk CH, Holmes EC (2002) Reduced positive selection in vector-borne RNA viruses. Mol Biol Evol 19:2333–2336
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Carpi, G., Holmes, E.C. & Kitchen, A. The Evolutionary Dynamics of Bluetongue Virus. J Mol Evol 70, 583–592 (2010). https://doi.org/10.1007/s00239-010-9354-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-010-9354-y