
Abstract
The evolution of a multigene family (MGF) is affected by the structure and function of its regulatory elements, specifically by the link between recombination and DNA transcription and/or replication. The ribosomal DNA (rDNA) MGF is often hierarchically repetitive, combining function with repetition in a single genic system. Its tandemly repeated operons contain the transcription unit of the 45S ribosomal RNA precursor alternating with an intergenic spacer (IGS) that commonly includes repeated transcription regulatory elements. To study the evolution of repeated sequences and the influence of repeat characteristics on their sequence divergence, we sequenced and characterized a single complete IGS from 11 daphniid species and analyzed their repeat arrays along with those from an additional 21 species of arthropods. We tested the hypotheses that sequence similarity is higher among tandemly arrayed repeats than among interleaved or dispersed repeats, and that the homogeneity of repeat arrays is affected by the number and the length of repeats, as well as by the presence of putative regulatory elements. We found that both tandem repeat organization and the presence of a TATA motif are significantly correlated with increased sequence similarity among homologous IGS repeats. We also observed that some repeat types are only found in a single species, while others appear to have persisted for >100 MY, with evidence for homologous repeat types in sister species. Taken together, these data suggest that both drift and natural selection influence repeat evolution within the IGS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The distribution of sequence variation among the members of a multigene family (MGF) is determined by the relative rates of homogenization due to gene conversion and unequal crossing over, and divergence due to mutation, natural selection, and genetic drift. Differences in the relative rates of these processes lead to very different patterns of genetic variation among the members of a MGF. Divergent evolution refers to the pattern in which a new function is acquired by a duplicate copy and interspecific orthologs are more similar to one another than they are to intraspecific paraglogs (Ota and Nei 1994); birth-and-death evolution is the pattern in which some duplicated copies are eliminated, some become inactivated (pseudogenes), while others persist in the genome (Ota and Nei 1994; Nei and Rooney 2005); and concerted evolution (CE) is the pattern in which intraspecific paralogs are more similar to one another than they are to interspecific orthologs (Arnheim 1983).
Eukaryotic ribosomal DNA (rDNA) is a large MGF whose copies all retain the same function: encoding ribosomal RNA (rRNA). It consists of tandem repeats of the ribosomal RNA (rRNA) precursor sequence separated by an intergenic spacer (IGS) (Fig. 1). Although purifying selection on the rRNA-coding sequences results in remarkable interspecific sequence conservation (Nei and Rooney 2005), the primary sequence of the cis-regulatory elements in the IGS is less conserved. Even so, intraindividual and intraspecific sequence divergence in this region is low relative to interspecific sequence divergence, which suggests that the former is maintained by mechanisms of CE rather than by stringent selection (Arnheim 1983; Nei and Rooney 2005). Furthermore, cis-regulatory elements in the IGS, the very elements that are believed to affect rates of recombination (Kobayashi et al. 2001; Mancera et al. 2008), may also be tandemly repeated.

Organization of eukaryotic ribosomal DNA arrays. The intergenic spacer (IGS) alternates with the 45S rRNA precursor (dotted line with arrow), which begins at the transcription start point (tsp) and includes the external transcribed spacer (ETS), the internal transcribed spacers (ITS1 and ITS2) and the rRNA-coding sequences (18S, 5.8S, 28S). Repeats within the IGS can be tandem (three consecutive open rectangles), interleaved (black ovals alternating with open rectangles) or dispersed (striped hexagons)
Although it is generally accepted that repeated sequences in the IGS evolve in a concerted fashion (Liao 1999), previous results for North American Daphnia pulex suggest that not all IGS repeat arrays conform to this pattern (Crease 1995). To investigate the predominant pattern of MGF evolution for repeated sequences in the IGS, we examined the repeat arrays in IGS sequences from 32 arthropod species. Divergence times within the subgenera or genera represented in our study range between ~2.2 MY to greater than ~60 MY.
We looked for variation in the homogeneity of different repeat types in a single IGS and also tested whether properties of these repeats, such as array organization (tandem, interspersed, or dispersed; Fig. 1) and array length, influence rates of homogenization. Because molecular interactions like gene conversion and unequal crossing over between copies of MGF are more probable when copies are tandemly arrayed, we tested the hypothesis that the mean sequence divergence is lower among tandemly arrayed repeats than among repeats whose copies are dispersed or interleaved with other repeated sequences. As well, the first and last repeats of an array are reportedly more divergent from their paralogs than internal repeats are, presumably because their peripheral position reduces the opportunity for molecular exchange by unequal crossing over (Markos and Baldwin 2002). Thus, the mean sequence divergence among repeats in a long array is expected to be lower because these divergent repeats will have less impact on the mean, compared to a short array, where they may represent most of the total sequence divergence. Therefore, we tested the hypothesis that the number of repeats affects overall mean sequence divergence. Similarly, we determined whether the length of repeats within an array affects the mean sequence divergence among them. Shorter repeats may undergo higher rates of molecular exchange by mechanisms such as replication slippage, while gene conversion and unequal crossing over are likely to be the major homogenizing mechanisms for longer repeats.
Promoter-like sequences have been documented in IGS repeats (frog: Caudy and Pikaard 2002; mosquito: Baldridge and Fallon 1992). The relationship between recombination hotspots and enhancers and/or promoters suggests that something intrinsic to these sequences may predispose them to double-strand break/repair (Lin and Keil 1991; Mancera et al. 2008). Furthermore, purifying selection is believed to play a role in the evolution of rDNA-coding sequences. Therefore, we tested the effect of elements that are putatively involved in transcription regulation on sequence divergence among paralogous IGS repeats. Because sequence conservation may not be a functional requirement for these elements (Dermitzakis and Clark 2002), we also used promoter-prediction software to locate promoter-like sequences in IGS repeats and compared the homogeneity of repeats with and without these motifs.
Methods
Cloning and Sequencing the rDNA IGS of Daphniids
We extracted total genomic DNA from 15 to 20 pooled parthenogenetic offspring of a single female from each of 11 daphniid species using the IsoQuick Nucleic Acid Extraction Kit (MicroProbe, Inc., Carlsbad, CA). The IGS fragment was PCR amplified with the Expand Long Template PCR System (Roche Diagnostics, Laval, QC) according to manufacturer’s specifications using primers complimentary to highly conserved regions at the 3′-end of the 28S rRNA gene (5′-GTTTAGACCGTCGTGAGACAGGTTAG) and the 5′-end of the 18S rRNA gene (5′-TCAGGCTCCCTCTCCGG). Depending on the species, the major fragment generated by these reactions ranged between 4 and 7 kbp, which agrees with previous characterizations of the D. pulex IGS (Crease 1995). Gel purified PCR products from this band were cloned using the TOPO XL PCR cloning Kit (Invitrogen Corp., Carlsbad, CA) according to manufacturer’s recommendations. Plasmid clones were pre-screened for the presence of appropriately sized inserts and the presence of 18S and 28S rRNA gene termini were confirmed by sequencing plasmid DNA with M13 Forward and Reverse primers and the ABI Prism TaqFS dye terminator kit (Applied Biosystems, Foster City, CA). Sequences were resolved on an ABI 377 automated sequencer (Applied Biosystems, Foster City, CA). The Erase-a-base system (Promega, Madison, WI) was used to generate nested deletion subclones from a single insert of each species. Overlapping deletion subclones were sequenced and a contig sequence of each insert from each species was created in Sequencher (Gene Codes, Ann Arbor, MI). These daphniid IGS sequences are deposited in GenBank under the accession numbers (EU595546–EU595565, Table 1). Nucleotide sequences for the remainder of the IGS in this analysis were obtained from GenBank (Table 1). The 32 sequences represent five insect genera (Acyrthosiphon, Aedes, Chiromonus, Drosophila, and Simulidae) and three branchiopod crustacean genera (Artemia, Daphnia, and Simocephalus).
IGS Sequence Analysis
We located the 5′-end of the 18S rRNA gene and the 3′-end of the 28S rRNA gene in each IGS sequence (Table 1) by consensus alignment with the homologous sequence from Daphnia pulex (AF014011) and Daphnia longicephala (AF346516) (Omilian and Taylor 2001), respectively. Repetitive regions in individual IGS sequences were located visually in Dotmatcher (window size 25, threshold 40, available at http://bioweb.pasteur.fr/seqanal/interfaces/dotmatcher.html; Rice et al. 2000). We estimated IGS repeat boundaries by performing 25 local self-alignments using SIM (default settings, available at http://genome.cs.mtu.edu/align/align.html, Huang and Miller 1991; Huang et al. 1990). Finally, we refined putative IGS repeat boundaries by visual inspection of the aligned IGS repeat sequences (Multiple Sequence Alignment Program [MAP], default settings, available at http://genome.cs.mtu.edu/map/map.html, Huang and Miller 1991; Huang et al. 1990). The MAP alignment algorithm computes a multiple global alignment of sequences using an iterative pairwise method; it does not heavily penalize long internal gaps, and terminal gaps are not penalized at all (Huang and Miller 1991; Huang et al. 1990).
To test the hypothesis that the presence of promoter-like sequences increases IGS repeat homogeneity, we used the neural network promoter-prediction algorithm of Reese (2001, http://www.fruitfly.org/seq_tools/promoter.html) to locate putative transcription promoters within the repeat sequences of each IGS. Because these motifs did not always fall within repeat boundaries, we counted repeats with complete or partial motifs as promoter-positive in our statistical analyses. We also searched visually for sequences similar to a previously identified putative promoter/enhancer motif (TATATACG) in D. pulex (Crease 1993) and Drosophila (Tautz et al. 1987).
To test whether repeats within the IGS exhibit patterns more typical of concerted, divergent (previously reported by Crease [1995] in D. pulex), or birth-and-death evolution (not previously reported for the IGS), we identified putatively homologous repeat types in different individuals by aligning all repeat copies of similar arrays in individuals from sister species. We assigned the same label (i.e., A, B, etc.) to repeat copies that were reliably alignable within each subgenus. For example, we attempted to align all of the A-repeat copies from all six of the individuals in the subgenus Daphnia (Table S1). The A-repeats from Daphnia ambigua could not be reliably aligned with those of the five remaining Daphnia individuals, and were omitted from interspecific analyses.
Because of their high frequency in non-coding (nc) DNA, we incorporated indels (insertions and deletions) as characters in subsequent analyses. After generating a multiple alignment for each IGS repeat type in MAP (default parameters), we adjusted these interindividual repeat alignments by eye. We replaced the first position in each deletion with a unique nucleotide (i.e., a nt not present at the same position in any other sequence), which allowed the deletion to be treated as a single mutational event in estimates of sequence divergence.
We used the nucleotide p-distance algorithm in MEGA v3.1 (Kumar et al. 2004) to calculate the pairwise sequence divergence among all putatively homologous repeat copies (i.e., an intra and interindividual comparison among all A-repeats from five of the six individuals in the subgenus Daphnia). The mean intraindividual pairwise sequence divergence, for each repeat type in individuals from each pair of sister species, was calculated from these values (Table S2). Indels were included as described above. To visualize the matrices of sequence divergence between repeats, we constructed neighbour-joining dendrograms of repeat copies using the nucleotide p-distance matrix, assuming homogeneous patterns of nucleotide substitution among lineages and uniform mutation rates among sites (MEGA v3.1, Kumar et al. 2004). Support for the nodes in these trees was estimated by calculating bootstrap values based on 500 replicates.
We performed an Analysis of Molecular Variance (AMOVA) using Arlequin 3.0 (Excoffier et al. 2005) to hierarchically partition the genetic variance of alignable repeats within individuals and among individuals from the same genus. Repeat copies were analyzed after grouping them by individual as well as by their position in the array. The genetic variability for each repeat type within and between individuals (F ST) was also calculated between pairs of individuals in the genera Daphnia, Drosophila, and Chironomus.
Statistical Analyses
To avoid the non-independence of pairwise values of sequence divergence among a group of homologous repeat copies in subsequent statistical tests, we created a consensus sequence of each repeat type within each IGS sequence of each individual and calculated the p-distance between it and each of the contributing repeat copies using MEGA v3.1 (see Glass et al. 2008). Each consensus sequence was based on an nt frequency greater than 50% at each position. If 2 nt were equally frequent at the same position, the nt represented by the letter nearest the beginning of the alphabet was used in the consensus. If a gap was present in 50% or more of the sequences, the position was not included in the consensus sequence.
An estimate of consensus sequence divergence (CSD) was calculated for each repeat type in each IGS sequence by taking the mean of the p-distances between each repeat copy and its consensus sequence. For example, we created a consensus sequence of the A-repeats from D. ambigua and calculated the p-distance between it and each of the D. ambigua A-repeats, and took the mean of these values. The correlation between the CSD (Table S1) and the pairwise sequence divergence within all individuals is 0.961 (Spearman’s rho, 2-tailed, α = 0.01, p < 0.001), suggesting that use of CSD in our statistical analyses will yield similar results to those obtained with pairwise sequence divergence (which is typically used), but without violating the independence of data points.
The mean CSD for each repeat type in each individual was used for all statistical analyses performed with SPSS (Chicago, Illinois; version 12.0 for Windows). Results were considered to be statistically significant if p < 0.05. The CSD was ln−1 transformed to minimize distribution skewness and allow parametric analyses when necessary.
To determine whether all repeat types within a single IGS evolve at the same relative rate, we compared the CSD among all the repeat types of any individual with multiple repeat types in its IGS using a Kruskal–Wallis nonparametric one-way analysis of variance (ANOVA).
We analyzed the effect of six independent characteristics (parameters) on CSD. (1) The effect of taxonomy (i.e., family) is of interest because some lineages may have acquired mutations that affect rates of sequence divergence among repeat copies following speciation. We used Tamhane’s post hoc multiple comparisons to compare CSD between taxonomic families. (2) We tested for significant effects of the organization of IGS repeat types (i.e., tandem, interleaved with other repeat types, and dispersed, Fig. 1) on CSD using Tukey’s HSD post hoc multiple comparisons. Correlations between CSD and (3) array length (the number of repeats of each type in the IGS), and between CSD and (4) mean length of a repeat type were identified using Spearman’s rho. Lastly, we compared CSD between repeat types (5) with and without promoter-like motifs and/or (6) the presence of a TATA-like motif using a nonparametric pairwise comparison (Mann–Whitney U test).
We tested for significant main effects of each of these six parameters on CSD with a univariate ANOVA. We tested for independence of each parameter by searching for correlations between them (e.g., between taxonomy and number of repeats of each type) with Spearman’s rho. And finally, we tested for interactions between parameters with statistically significant main effects on CSD using univariate two-way ANOVA, followed by a partial correlation analysis if an interaction was identified.
Results
The Organization and Relationships Among Repeated Sequences in the IGS
The organization of repeat arrays in the IGS of the individuals from the family Daphniidae, and the genera Drosophila and Chironomus are illustrated in Figs. 2a, 3a, and 4a. The IGS of the remaining arthropod individuals have been described elsewhere (Aedes albopictus, Baldridge and Fallon 1992; Aedes aegypti, Wu and Fallon 1998; Acyrthosiphon pisum, Kwon and Ishikawa 1992). A summary of the structure of the IGS and the %GC of repeat types for all 32 sequences is given in Table S1.

IGS variation in individuals representing 11 species in the family Daphniidae. a Species phylogeny and IGS structure. Gray ovals represent repeats containing putative enhancer motifs (TATAGGG) similar in sequence to the putative core promoter in Daphnia pulex. The black rectangle in Daphnia obtusa represents 11 tandem copies of the 28 nt E-repeat. The black rectangle in Simocephalus vetulus represents 14 tandem copies of the 16 nt B-repeat. Repeat types are labelled according to Table S1. The letter code assigned to each repeat type does not necessarily reflect homology across species, except within the subgenus Daphnia. Branch lengths in the topology (Colbourne and Hebert 1996) are not to scale. Divergence time estimates are from Colbourne and Hebert (1996). AMB = Daphnia ambigua, CUR = D. curvirostris, GAL = D. galeata, LCP = D. longicephalus, LGR = D. longiremis, OBT = D. obtusa, PAR = D. parvula, PCA = D. pulicaria, PX1 = D. pulexNA, PX2 = D. pulexE, VET = Simocephalus vetulus. b Unrooted Neighbor-joining dendrogram of p-distances between A-repeat sequences from species in the subgenus Daphnia. c Unrooted Neighbor-joining dendrogram of p-distances between B-repeat sequences. d Unrooted Neighbor-joining dendrogram of p-distances between C-repeat sequences. e Unrooted Neighbor-joining dendrogram of p-distances between D-repeat sequences. Indels were coded as single mutational events in each analysis. Bootstrap values >70, based on 500 replicates, are indicated at the nodes. The p-distance scale is given below each dendrogram

IGS variation in five species in the genus Drosophila. a Species phylogeny and IGS structure. Gray ovals represent repeats containing putative enhancer motifs (TATAGGG) similar in sequence to the putative core promoter identified by Tautz et al. (1987). Branch lengths in the topology (Tautz et al. 1987) are not to scale. Divergence time estimates are from Russo et al. (1995). HYD = Dr. hydei, VIR = Dr. virilis, FUN = Dr. funebris, ORE = Dr. orena, MEL = Dr. melanogaster. b Unrooted Neighbor-joining dendrogram of p-distances between B-repeat sequences from species in the subgenus Drosophila. c Unrooted Neighbor-joining dendrogram of p-distances between D-repeat sequences from species in the subgenus Sophophora. d Unrooted Neighbor-joining dendrogram of p-distances between E- and F-repeat sequences from species in the subgenus Sophophora. Indels were coded as single mutational events in each analysis. Bootstrap values >70, based on 500 replicates, are indicated at the nodes. The p-distance scale is given below each dendrogram

IGS variation in 12 species of the superfamily Chironomoidea. a Species phylogeny and IGS structure. Repeats within arrays are shown as ovals. Gray ovals (A-repeat) represent the only repeat type with sequence similarity across chironomid species. The black rectangle in Chironomus annularius represents six tandem copies of a 26-nt repeat. Branch lengths in the topology, from Guryev et al. (2001), are not to scale. Divergence time estimates are from Kao et al. (1994). The phylogenetic relationship of Chironomus halophilus to the other chironomid species has been estimated from 28S-rRNA gene sequences obtained from Genbank. ANN = Chironomus annularius, CIN = C. cingulatus, DOR = C. dorsalis, HAL = C. halophilus, LUR = C. luridus, PAL = C. pallidivittatus, PLU = C. plumosus, PSE = C. pseudothummi, TEN = C. tentans, THT = C. thummi thummi, THP = C. thummi piger, SAN = Simulium sanctipauli. b Unrooted Neighbor-joining dendrogram of p-distances between A-repeat sequences. Indels were coded as single mutational events as described in the text. Bootstrap values >70, based on 500 replicates, are indicated at the nodes. The p-distance scale is given below the dendrogram
Subgenus Daphnia
We identified four repeat types in the IGS of individuals from the subgenus Daphnia, and the mean pairwise sequence divergence (PSD) within types is summarized in Table 2. The A-repeats of Daphnia individuals (Fig. 2a, Table S1) contain a sequence similar to the core promoter previously identified in the D. pulexNA IGS (Crease 1993). Although the A-repeats of D. ambigua contain fragments similar to the core promoter and to other motifs in the A-repeats of other subgenus Daphnia individuals, they have diverged to the extent that we could not confidently incorporate them into an alignment with the other individuals. Thus, we have omitted them from interindividual analyses.
Almost 53% of the sequence variation among A-repeats is partitioned among individuals (Table 2) and for the most part, they cluster by individual (Fig. 2b). B-repeats are limited to the three individuals in the D. pulex group (D. pulexE, D. pulicaria, and D. pulexNA). Although their mean interindividual PSD is higher than their mean intraindividual PSD, less than half of the sequence variation occurs between individuals (Table 2). Pairwise F ST values for both A- and B-repeats are large (A: 0.295–0.778, B: 0.598–0.625) between all pairs of individuals except between D. pulicaria and D. pulexNA, where they are negative (Table S4). This is illustrated by the dendrograms in which both A-repeats (Fig. 2b) and B-repeats (Fig. 2c) cluster by individual except for repeats from D. pulicaria and D. pulexNA, which group by position in the array.
The C-repeat is also limited to the three individuals in the D. pulex group (Fig. 2d). It occurs as two tandem copies in the IGS of D. pulexNA and D. pulicaria, but there is only a single copy in the IGS of D. pulexE. All the variation in C-repeats is within individuals, although neither the AMOVA nor the negative pairwise F ST values are statistically significant (Table 2, Table S4). D-repeats, found only in the D. obtusa and D. pulexE IGS (Fig. 2a), are highly differentiated between individuals, with over 81% of the variation among D-repeats occurring between individuals (Table 2; Fig. 2e). With the exception of the C-repeat, all mean interindividual PSD are higher than mean intraindividual PSD, and range from a 1.2-fold difference for B-repeats to nearly a 5-fold difference for D-repeats (Table 2).
The only similarity we found among the various repeat types in the IGS of the other Daphnia individuals is the presence of a TATA motif similar to the putative core promoter in the D. pulexNA IGS (Fig. 2a). Despite its presence in the majority of daphniid individuals and the fact that a similar motif was reported in the IGS repeat arrays of Dr. melanogaster (Coen and Dover 1982), the TATA motif was not found in any of the repeats from the D. longiremis or S. vetulus IGS.
Subgenus Drosophila
We identified three repeat types in the IGS of the three subgenus Drosophila individuals (Fig. 3a). Arrays of an A-repeat type appear in the IGS of both Dr. hydei and Dr. virilis, but interindividual alignments are not reliable and this repeat type is absent from the IGS of Dr. funebris. Only the B-repeat, which contains an enhancer motif similar to the core promoter (Tautz et al. 1987), has orthologs in all three individuals. Over 51% of the sequence variation among B-repeats is partitioned between individuals (Table 3) with pairwise F ST values ranging from 0.447 to 0.542 (Table S7). These repeats cluster by individual (Fig. 3b).
Subgenus Sophophora
The subgenus Sophophora is represented here by individuals from Dr. melanogaster and Dr. orena. Their IGS contain tandem arrays of two main repeat types, D and E–F (Fig. 3a). In Dr. melanogaster, E-repeats are present in an array separate from F-repeats, however, they are alignable with the F-repeats of both individuals. Because the IGS sequence for Dr. orena is incomplete, the presence of an array of E-repeats cannot be determined. The intraindividual PSD among D-repeats is low relative to interindividual PSD (Table 3), and although levels of interindividual variation among D-repeats is only 33% (Table 3), they still cluster by individual (Fig. 3c).
The interindividual PSD among F-repeats in both Dr. melanogaster and Dr. orena is surprisingly high when compared to their intraindividual PSD (Table 3 and Table S5) in light of the fact that these repeats are reported to contain an enhancer sequence similar to the core promoter (Tautz et al. 1987), as well as being involved in X–Y pairing in Dr. melanogaster (Ren et al. 1997). The intraindividual PSD of E-repeats (present only in Dr. melanogaster) is more than an order of magnitude smaller than the PSD of either Dr. orena or Dr. melanogaster F-repeats, with over 60% of the sequence variation partitioned between individuals and/or repeat types (Table 3 and Table S5). Pairwise F ST values range from 0.263 between the F repeats in Dr. melanogaster and Dr. orena, to 0.679 between the E and F repeats in Dr. melanogaster (Table S7), which justifies distinguishing each type with different letter codes. E–F repeats cluster by individual and repeat type with the exception of the 3′ F-repeats in both individuals, which cluster together (Fig. 3d).
Genus Chironomus
The structure of the Chironomus IGS is as variable as that in Daphnia but the structural rearrangements in the Daphnia IGS represent more than 200 MY divergence (Fig. 2a), while differences in the Chironomus IGS have occurred in ~60 MY (Fig. 4a). Only the A-repeat is found in the IGS of all eleven Chironomus individuals (Fig. 4a) and it clusters by position (A1 or A2) in the IGS (Fig. 4b). While this suggests that the duplication predates the expansion of this lineage, the topology of the repeat dendrogram is not consistent with that of the species phylogeny (Fig. 4a, b). When the A-repeats are grouped by individual, less than 25% of the variation occurs between individuals. However, grouping them by position yields similar results (Table 4). This phenomenon may be due to the fact that there are only two copies of the repeat in each IGS. No similarities were identified between other IGS repeat types in Chironomus.
No repeat types are shared between the two Aedes individuals. Each of the other two families (Simulidae and Artemiidae) in this study is represented by a single species.
Impact of Phylogeny and IGS Structure on Consensus Sequence Divergence (CSD) Between Repeats
The mean CSD values reported in Table S1 indicate either that distinct repeat types within individuals are evolving at different rates, or that they have existed for different lengths of time. However, these differences are only significant in Ae. albopictus, D. longiremis, Dr. virilis, and D. curvirostris (Table S10). The first three individuals have only two repeat types and the differences are clear. In D. curvirostris, there are no significant differences in CSD between pairs of the seven repeat types, although mean CSD is <0.01 for C- and E-repeats, 0.02 for G-repeats, and >0.06 for the other four types. The C-, A- and G-repeats are tandemly arrayed, while the other repeat-types are dispersed (Fig. 1a).
On finding a significant between-family difference in the mean CSD of repeat copies (univariate ANOVA, F = 4.791, p = 0.000), we used Tamhane’s T2 post hoc multiple comparisons test to identify specific differences (the assumption of equal variances is violated, Table S11). The mean CSD is significantly lower among repeats in the family Aphididae when compared to Chironomidae, Simulidae, Culicidae, Drosophilidae, and Daphniidae, but not Artemiidae (Table S11). While there is no significant difference in mean CSD among repeats between the families Daphniidae and Drosophilidae, both are significantly lower than the mean CSD among repeats in the families Chironomidae and Simulidae. Finally, the mean CSD among repeats is lower in the family Culicidae than in Simulidae (Table S11).
When we looked for main effects of repeat organization (tandem, interleaved, or dispersed), the number of repeats, their mean length, the presence of a putative promoter-like motif, and the presence of a TATA-like motif on CSD of repeats types within an IGS, only organization was significant. Only 35% of the variation is explained by the corrected model (Table S12) and there are more tandem repeat types (N = 43) than either interleaved (N = 20) or dispersed repeat types (N = 11) in this data set.
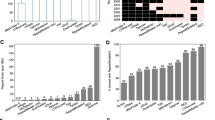
Neither the repeat length, nor the number of repeats (which ranges between 2 and 15), has a significant affect on CSD, however, we found a significant interaction between them (Table S13). In general, CSD decreases as the number and the length of repeats in an IGS increases. After identifying significant between-family differences in the magnitude of CSD (Table S11), we controlled for the effects of family with a partial correlation, and found that repeat organization and mean repeat length each have a significant affect on CSD (Table 5). Tandem and interleaved repeats are significantly more homogeneous than dispersed repeats (Table S14), and longer repeats are significantly more homogeneous than shorter ones. The length of a repeat is a nearly continuous variable ranging from ~16 to 865 nt, with a mean of 158 nt and a standard deviation of 127. When we grouped repeats into 50 nt categories, we found significant differences in mean CSD between some categories. Repeats with a mean length of 200–249 nt are significantly more homogeneous (mean CSD = 0.029) than those with a mean length of 50–99 nt (0.089) or 100–149 nt (mean CSD = 0.077) and repeats with a mean length of 150–199 nt are significantly more homogeneous (mean CSD = 0.053) than those with a mean length of 50–99 nt (mean CSD = 0.089) (Fig. 5; Tables S15 and S16).

Mean consensus sequence divergence in repeats of different lengths (nt) from 32 species of arthropods. Bars with the same letter are significantly different from one another (p < 0.05, Table S16). N number of repeats in the category
The partial correlation analysis also revealed a significant interaction between repeat organization and number of repeats (Table 5). In general, repeat homogeneity increases in tandem and interleaved repeats as the number of repeats increases. In addition, there is a significant interaction between mean repeat length and the presence of both promoter-like and TATA motifs (Table 5). These motifs are more likely to be found in longer repeat types than in shorter ones. A post hoc Mann–Whitney U test on the effect of these motifs revealed no significant difference in mean CSD between repeat types with and without promoter-like sequences, however, repeats containing a TATA motif are significantly more homogeneous than those without [mean CSD (±SD) = 0.043 (±0.023) versus 0.073 (±0.040), U = 266, p = 0.006]. The TATA motif is present in the IGS of the Drosophila individuals and the majority of the Daphnia individuals, however, it is absent from the IGS of the Chironomus individuals.
Discussion
For the most part, identifying homologous IGS repeat arrays between congeneric taxa was unambiguous because differences in repeat sequences are composed of point mutations and short indels. The majority of IGS repeats are tandemly arrayed, however, some are interleaved or dispersed. Varying combinations of similar sequences made defining the repeat boundaries subjective, so we relied on interindividual rather than intraindividual alignments when delimiting repeats. For example, Crease (1993) treated the A–B unit in D. pulex as a single repeat type, however, D. obtusa and D. parvula IGS lack the ~125-nt B-repeat, so we split it into A- and B-repeats to facilitate interspecific comparisons. The result is that while A-repeats in the subgenus Daphnia are interleaved in some species and tandem in others, they are ~200 nt in length in most species. The relatively long A-repeat in D. ambigua is treated as a single unit of 356 nt. However, ~145 nt at the 3′-end of the third copy is missing, suggesting that this may be a compound repeat, similar to the A- and B-repeats in the D. pulex group, or the interleaved A- and C-repeats in D. obtusa. Unfortunately, without the IGS sequence of a sister species, the ancestral length of the A-repeat in D. ambigua cannot be determined.
Even at short evolutionary distances, the chimeric nature of the IGS is evident. For example, the IGS sequences of Aedes and Chironomus individuals are characterized by the rapid expansion or elimination of repeat arrays. The challenge is to determine the sequence characteristics that predispose the IGS to such dynamic behavior. Previous evolutionary studies of the IGS within and between individuals and populations of D. pulexNA demonstrated that gene conversion and unequal crossing over in the IGS operate at the level of the repeat array as well as the entire rDNA operon (Crease 1995). Even so, the distinct repeat consensus sequence at each of the four positions in the IGS array suggests that the rate of unequal intrachromosomal exchange within the IGS is low relative to the rate of exchange at the level of the entire rDNA operon (Crease 1995). Luchetti et al. (2006) obtained a similar result in their analysis of four IGS sequences from the branchiopod, Triops cancriformis. However, unlike D. pulexNA, the IGS repeats in this species do not contain promoter-like sequences.
In Arabidopsis, where the estimated divergence time between A. thaliana and A. halleri is 5 MY (Haubold and Wiehe 2001), the rate of homogenization within the IGS exceeds the rate of mutation, resulting in sufficient phylogenetic signal to permit the resolution of major species groups but not subspecies or historically isolated populations (Hayworth 2000). It appears that the disparity in the degree of homogeneity between IGS repeats in Arabidopsis and Daphnia is a function of evolutionary distance between species because we found that the pattern of repeat evolution in the IGS of all but two of the 32 arthropod sequences we analyzed have evolved in concert. The exceptions are the IGS repeats of D. pulexNA and D. pulicaria, which are more similar to their orthologs than to their paralogs. This pattern is typical of divergent or birth and death evolutions, rather than concerted evolution. Nonetheless, the occurrence of unequal crossing over and gene conversion between the repeats in this array has been documented, and it has spread to all the IGS within D. pulexNA, which is consistent with CE (Crease 1995). Thus, a more parsimonious explanation for the lack of reciprocal repeat monophyly between these two species is the fact that they hybridize with one another (Colbourne et al. 1998).
Orthologous copies of the 3′ F-repeats in the IGS of Dr. orena and Dr. melanogaster (subgenus Sophophora) are more similar than conspecific paralogous copies (Fig. 3d). This is consistent with previous findings that repeats positioned at the end of an array are less often involved in the homogenization process relative to their upstream counterparts (Lassner and Dvorak 1986; Dvorak et al. 1987; Da Rocha and Bertrand 1995). In Lessingia spp. (Compositae, Astereae, Markos and Baldwin 2002) and in the genus Calycadenia (Asteraceae, Baldwin and Markos 1998), interior repeats appear to evolve in concert, yet the sequence similarity of flanking repeats is higher between orthologs than paralogs. It is tempting to suggest that these repeats are evolving according to the model of divergent or birth-and-death evolution. However, when homologous repeats in closely related species primarily cluster according to species despite the occasional misplaced family member, sequence homogenization must still be occurring (Liao 1999).
In some cases, repeat homogeneity may be the result of recent amplification events rather than long-term homogenization of an existing array (Liao 1999). The Ac. pisum IGS repeat array, in which sequence divergence among repeats is zero, may be an example of such an array. Again, without a comparison to sequences from closely related species, it is impossible to determine whether this is a newly amplified array or an old array in which homogenization is particularly efficient. Similarly, the differing degrees of sequence homogeneity exhibited within repeat arrays in this study may be due to the relative age of the arrays within each species, with newer arrays being more homogeneous and older arrays exhibiting patterns of divergent or birth and death evolution. Alternatively, it may be a reflection of differences in selective constraints or in rates of homogenization caused by different array characteristics. Similar observations have been reported in other studies whereby some MGF members or parts of genes conform to patterns of CE, while others do not (Polanco et al. 1998).
It has been demonstrated that rates of recombination alone are sufficient to account for the homogeneity or divergence of repeat copies in MGF (Dover and Tautz 1986). Here, we speculated that parameters such as array length, repeat organization and repeat length, as well as the presence of putative enhancer motifs, affect the homogeneity of repeats by influencing rates of recombination. We did not find a significant correlation between repeat array length and homogeneity among repeats. However, the occurrence of intraspecific IGS length variation due to differences in the number of IGS repeats is a common feature of eukaryotic rDNA (Weider et al. 2005 and references within). We only analyzed a single IGS sequence from each species, so it is possible that some of the sequences are not representative of the most common length variant in that species. Even so, we did find that repeat organization has a significant effect on homogeneity. Tandem and interleaved repeats are more homogeneous than dispersed repeats, which is consistent with the expectation that recombination occurs more often among sequences that are in close proximity to one another. Aside from taxonomy, the only other significant effect is the presence of the putative enhancer/promoter TATA motifs. Thus, it could be argued that CE is enhanced by selective constraints favouring repeats that contain this motif.
Although Nei and Rooney (2005) argue that examining the relative contribution of purifying selection and random genetic drift in the evolution of MGF is important, selection is difficult to demonstrate in ncDNA where it is not possible to compare ratios of synonymous to nonsynonymous mutations. Still, natural selection may contribute to sequence homogeneity in these presumably functional arrays by preserving sequence motifs that are predisposed to frequent DNA repair of double- or single-strand breaks in actively transcribed sequences. Gonzales and Sylvester (2001) suggested that rates of homogenization in the IGS may be affected by the hypomethylation of regulatory regions thought to affect recombination rates in rDNA (Jiang and Liao 1999) as well as by RNA polymerase-I-associated topoisomerase I in actively transcribed rDNA (Zhang et al. 1988; Rose et al. 1988). While the IGS cannot be considered a coding region, “active” copies are frequent binding sites for transcription factors and RNA polymerase I. González-Barrera et al. (2002) reported transcription-mediated recombination in yeast, though the molecular mechanisms are still unclear. Takeuchi et al. (2003) reported that recombination might be caused by a collision between transcription and replication forks. Furthermore, cis-acting elements can influence whether duplicated genes undergo concerted or divergent evolution. One example can be found in the silk moth Bombyx mori, which has two families of early chorionic genes, ErA and ErB that are closely linked on one chromosome (Hibner et al. 1991). The genes of the ErA family exhibit 96% sequence identity, whereas those in the ErB family exhibit only 63% sequence identity. Sequence analysis has suggested that microsatellite-like simple repeats that are present in the ErA family but not in the ErB family may account for the difference in homogenization, because simple sequence repeats can be the site for initiation of gene conversion (Hibner et al. 1991). Microsatellite sequences in the human RNU2 locus may also play a role in CE (Liao and Weiner 1995).
The presence of putative enhancer motifs in homologous IGS repeat-arrays from Daphnia, Drosophila, and Sophophora suggests that selection does play a role in the evolution of repeats in the IGS. Indeed, some studies have suggested that IGS repeat copy number variation can influence growth rate and tolerance to different environmental conditions via its effect on rates of rRNA transcription (see Weider et al. 2005 for a review). But what of those species whose IGS repeat arrays do not contain TATA motifs? Grummt (1999) and Reeder (1999) suggested that the repetitive nature of IGS repeats themselves might enhance transcription. On the other hand, it has been argued that it is not essential for enhancers to occur in multiple copies (Wang et al. 2003; Burton et al. 2005). Thus, species whose IGS contain repeat arrays that appear and disappear in short order may have evolved other means of rDNA transcription regulation.
References
Arnheim N (1983) Concerted evolution of multigene families. In: Nei M, Koehn RK (eds) Evolution of genes and proteins. Sinauer Associates, Sunderland, pp 38–61
Baldridge GD, Fallon AM (1992) Primary structure of the ribosomal DNA intergenic spacer from the Mosquito, Aedes albopictus. DNA Cell Biol 11:51–59
Baldwin BG, Markos S (1998) Phylogenetic utility of the external transcribed spacer (ETS) of 18S–26S rDNA: congruence of ETS and ITS trees of Calycadenia (Compositae). Mol Phylogenet Evol 10:449–463
Burton RS, Metz EC, Flowers JM, Willett CS (2005) Unusual structure of ribosomal DNA in the copepod Tigriopus californicus: intergenic spacer sequences lack internal subrepeats. Gene 344:105–113
Caudy AA, Pikaard CS (2002) Xenopus ribosomal RNA gene intergenic spacer elements conferring transcriptional enhancement and nucleolar dominance-like competition in oocytes. J Biol Chem 277:31577–31584
Coen ES, Dover GA (1982) Multiple Pol I initiation sequences in rDNA spacers of Drosophila melanogaster. Nucleic Acids Res 10:7017–7026
Colbourne JK, Hebert PDN (1996) The systematics of North American Daphnia (Crustacea: Anomopoda): a molecular phylogenetic approach. Philos Trans R Soc B 351:349–360
Colbourne JK, Crease TJ, Weider LJ, Hebert PDN, Dufresne F, Hobaek A (1998) Phylogenetics and evolution of a circumarctic species complex (Clardocera: Daphnia pulex). Biol J Linn Soc 65:347–365
Crease TJ (1993) Sequence of the intergenic spacer between the 28S and 18S rRNA encoding genes of the crustacean, Daphnia pulex. Gene 134:245–249
Crease TJ (1995) Ribosomal DNA evolution at the population level: nucleotide variation in intergenic spacer arrays of Daphnia pulex. Genetics 141:1327–1337
Da Rocha PS, Bertrand H (1995) Structure and comparative analysis of the rDNA intergenic spacer of Brassica rapa. Implications for the function and evolution of the Cruciferae spacer. Eur J Biochem 229:550–557
Dermitzakis ET, Clark AG (2002) Evolution of transcription factor binding sites in Mammalian gene regulatory regions: conservation and turnover. Mol Biol Evol 19:1114–1121
Dover GA, Tautz D (1986) Conservation and divergence in multigene families: alternatives to selection and drift. Philos Trans Roy Soc B 312:275–289
Dvorak J, Jue D, Lassner M (1987) Homogenization of tandemly repeated nucleotide sequences by distance-dependent nucleotide sequence conversion. Genetics 116:487–498
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinformatics 1:47–50
Glass SK, Moszczynska A, Crease TJ (2008) The effect of transposon Pokey insertions on sequence variation in the 28S rRNA gene of Daphnia pulex. Genome 51:988–1000
Gonzales IL, Sylvester JE (2001) Human rDNA: evolutionary patterns within the genes and tandem arrays derived from multiple chromosomes. Genomics 73:255–263
González-Barrera S, Garcia-Rubio M, Aguilera A (2002) Transcription and double-strand breaks induce similar mitotic recombination events in Saccharomyces cerevisiae. Genetics 162:603–614
Grummt I (1999) Regulation of mammalian ribosomal gene transcription by RNA polymerase I. Prog Nucl Acid Res Mol Biol 62:109–154
Guryev V, Makarevitch I, Blinov A, Martin J (2001) Phylogeny of the genus Chironomus (Diptera) inferred from DNA sequences of mitochondrial cytochrome b and cytochrome oxidase I. Mol Phylogenet Evol 19:9–21
Haubold B, Wiehe T (2001) Statistics of divergence times. Mol Biol Evol 18:1157–1160
Hayworth DA (2000) Hierarchical domains in concerted evolution of ribosomal DNA intergenic spacers in Arabidopsis (Brassicaceae). PhD Thesis, Washington University
Hibner BL, Burke WD, Eickbush TH (1991) Sequence identity in an early chorion multigene family is the result of localized gene conversion. Genetics 128:595–606
Huang X, Miller W (1991) A time-efficient, linear-space local similarity algorithm. Adv Appl Math 12:337–357
Huang X, Hardison RC, Miller W (1990) A space-efficient algorithm for local similarities. Comput Appl Biosci 6:373–381
Jiang C, Liao D (1999) Striking bimodal methylation of the repeat unit of the tandem array encoding human U2 snRNA (the RNU2 locus). Genomics 62:508–518
Kao WY, Trewitt PM, Bergtrom G (1994) Intron-containing globin genes in the insect Chironomus thummi. J Mol Evol 38:241–249
Kobayashi T, Nomura M, Horiuchi T (2001) Identification of DNA cis elements essential for expansion of ribosomal DNA repeats in Saccharomyces cerevisiae. Mol Cell Biol 21:136–147
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinformatics 5:150–163
Kwon OY, Ishikawa H (1992) Unique structure in the intergenic and 5′ external transcribed spacer of the ribosomal RNA gene from the pea aphid Acyrthosiphon pisum. Eur J Biochem 206:935–940
Lassner M, Dvorak J (1986) Preferential homogenization between adjacent and alternate subrepeats in wheat rDNA. Nucleic Acids Res 14:5499–5512
Liao D (1999) Concerted evolution: molecular mechanism and biological implications. Am J Hum Genet 64:24–30
Liao D, Weiner AM (1995) Concerted evolution of the tandemly repeated genes encoding primate U2 small nuclear RNA (the RNU2 locus) does not prevent rapid diversification of the (CT)n. (GA)n microsatellite embedded within the U2 repeat unit. Genomics 30:583–593
Lin YH, Keil RL (1991) Mutations affecting RNA polymerase I-stimulated exchange and rDNA recombination in yeast. Genetics 127:31–38
Luchetti A, Scanabissi F, Mantovani B (2006) Molecular characterization of ribosomal intergenic spacer in the tadpole shrimp Triops cancriformis (Crustacea, Branchipoda, Notostraca). Genome 49:888–893
Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM (2008) High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 454:479–485
Markos S, Baldwin BG (2002) Structure, molecular evolution, and phylogenetic utility of the 5(‘) region of the external transcribed spacer of 18S–26S rDNA in Lessingia (Compositae, Astereae). Mol Phylogenet Evol 23:214–228
Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Ann Rev Genet 39:121–152
Omilian AR, Taylor DJ (2001) Rate acceleration and long-branch attraction in a conserved gene of cryptic daphniid (Crustacea) species. Mol Biol Evol 18:2201–2212
Ota T, Nei M (1994) Divergent evolution and evolution by the birth-and-death process in the immunoglobulin VH gene family. Mol Biol Evol 11:469–482
Polanco C, Gonzalez AI, de la Fuente A, Dover GA (1998) Multigene family of ribosomal DNA in Drosophila melanogaster reveals contrasting patterns of homogenization for IGS and ITS spacer regions. A possible mechanism to resolve this paradox. Genetics 149:243–256
Reeder RH (1999) Regulation of RNA polymerase I transcription in yeast and vertebrates. Prog Nucl Acid Res Mol Biol 62:293–327
Reese MG (2001) Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem 26:51–56
Ren X-J, Eisenhour L, Hong C-S, Lee Y, McKee BD (1997) Roles of rDNA spacer and transcription unit-sequences in X-Y meiotic chromosome pairing in Drosophila melanogaster males. Chromosoma 106:29–36
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277
Rose KM, Szopa J, Han FS, Cheng YC, Richter A, Scheer U (1988) Association of DNA topoisomerase I and RNA polymerase I: a possible role for topoisomerase I in ribosomal gene transcription. Chromosoma 96:411–416
Russo CA, Takezaki N, Nei M (1995) Molecular phylogeny and divergence times of drosophilid species. Mol Biol Evol 12:391–404
Takeuchi Y, Horiuchi T, Kobayashi T (2003) Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev 17:1497–1506
Tautz D, Tautz C, Webb D, Dover GA (1987) Evolutionary divergence of promoters and spacers in the rDNA family of four drosophila species. J Mol Biol 195:525–542
Wang S, Zhao M, Li T (2003) Complete sequence of the 10.3 kb silkworm Attacus ricini rDNA repeat, determination of the transcriptional initiation site and functional analysis of the intergenic spacer. DNA Seq 14:95–101
Weider LJ, Elser J, Crease T, Mateos M, Cotner J, Markow T (2005) The functional significance of ribosomal(r)DNA variation: impacts on the evolutionary ecology of organisms. Ann Rev Eco Evol Syst 36:219–242
Wu CCN, Fallon AM (1998) Analysis of ribosomal DNA Intergenic spacer region from the yellow fever mosquito, Aedes aegypti. Insect Mol Biol 7:19–29
Zhang H, Wang JC, Liu LF (1988) Involvement of DNA topoisomerase I in transcription of human ribosomal RNA genes. Proc Natl Acad Sci USA 85:1060–1064
Acknowledgments
This work was supported by a grant from the Natural Sciences and Engineering Research Council of Canada to TJC. CDA was supported by Ontario Graduate Scholarships. We thank A. Holliss for assistance with the DNA sequencing. D. Irwin, R. Danzmann, L. Weider, S. McTaggart, and two anonymous reviewers provided helpful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ambrose, C.D., Crease, T.J. Evolution of Repeated Sequences in the Ribosomal DNA Intergenic Spacer of 32 Arthropod Species. J Mol Evol 70, 247–259 (2010). https://doi.org/10.1007/s00239-010-9324-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-010-9324-4