
Abstract
NfeD-like proteins are widely distributed throughout prokaryotes and are frequently associated with genes encoding stomatin-like proteins (slipins). Here, we reveal that the NfeD family is ancient and comprises three major groups: NfeD1a, NfeD1b and truncated NfeD1b. Members of each group are associated with one of four conserved gene partners, three of which have eukaryotic homologues that are membrane raft associated, namely stomatin, paraslipin (previously SLP-2) and flotillin. The first NfeD group (NfeD1b), comprises proteins of approximately 460-aa long that have three functional domains: an N-terminal protease, a middle membrane-spanning region and a soluble C-terminal region rich in β-strands. The nfeD1b gene is adjacent to eoslipin in prokaryotic genomes except in Firmicutes and Deinococci, where yqfA replaces eoslipin. Proteins in the second major group (NfeD1a) are homologous to the C-terminus of NfeD1b which forms a β-barrel-like domain, and their genes are associated with paraslipin. Using OrthoMCL clustering, we show that nfeD1b genes have become truncated on many independent occasions giving rise to the third major group. These short NfeD homologues frequently remain associated with their ancestral gene neighbour, resembling NfeD1a in structure, yet are much more related to full-length NfeD1b; we term these “truncated NfeD1b”. These conserved associations suggest that NfeD proteins are dependent on gene partners for their function and that the site of interaction may lie within the C-terminal portion that is common to all NfeD homologues. Although NfeD homologues are confined to prokaryotes, this conserved association could represent an excellent system to study slipin and flotillin proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
NfeD homologues are widely distributed throughout prokaryotic genomes, being present in both archaeal and bacterial species, although at present no eukaryotic NfeD homologue has been found (Green et al. 2004). Simple sequence analyses reveal the family to be comprised of both long (~460 aa) and short (~145 aa) forms. Long NfeD forms contain an N-terminal serine protease domain (Yokoyama and Matsui 2004) that shows structural similarity to the ClpP protease of Escherichia coli (Yokoyama et al. 2006), a predicted multi-spanning membrane domain and a soluble C-terminal NfeD domain (PFAM: PF01957). Short members of this family contain an N-terminal hydrophobic region followed by a soluble five-stranded β-barrel domain (Kuwahara et al. 2008; Walker et al. 2008), which is homologous to the C-terminal PF01957 domain of long NfeD forms.
Although NfeD homologues are widely distributed amongst prokaryotes, their functions remain largely unknown. Borthakur and Gao (1996) demonstrated that a Tn5-induced nfeD mutant of Rhizobium etli was defective in nodulation competition. More recently, a Bacillus subtilis nfeD homologue, yqeZ, located in the yqeZ–yqfAB operon, was shown to provide resistance to the SPβ prophage-encoded bacteriocin sublancin (Butcher and Helmann 2006), although this region is not sufficient to confer immunity to the producing strain.
nfeD homologues are frequently found co-localised with genes encoding stomatin-like proteins (slipins) (Green and Young 2008) (Green et al. 2004; Kuwahara et al. 2008). Slipins are an ancient group of highly conserved proteins whose members can be found in all three domains of life. Human slipins are associated with a variety of medical conditions, including cancer, haemolytic anaemia and kidney failure, although precise functions remain unclear (Boute et al. 2000; Cui et al. 2007; Stewart et al. 1992). Our recent phylogenetic analysis of the stomatin protein family (Green and Young 2008) revealed two distinct groups of prokaryotic slipins, which we termed eoslipins (previously p-stomatin, Tavernarakis et al. 1999) and paraslipins (previously SLP-2, Wang and Morrow 2000). Much of the experimental work into NfeD function has endeavoured to explain the nfeD–slipin association, as protein conservation and phylogenetic analyses suggest that functional conclusions drawn from prokaryotic slipins may be relevant to eukaryotic slipin function (Green and Young 2008). Yokoyama and Matsui (2004) demonstrated cleavage of a slipin by the N-terminal portion of NfeD from Pyrococcus horikoshii. In a different experiment, a short C-terminal NfeD homologue was shown to function, along with the slipin QmcA encoded by the neighbouring gene, as a multicopy suppressor that alleviated the growth defect of the ftsH/htpX double mutant at 42°C in E. coli (Chiba et al. 2006). Recently, Hinderhofer et al. (2009) have extended this enquiry to show more generally that, within prokaryotes, nfeD homologues are frequently associated with genes encoding other members of the SPFH superfamily. The SPFH concept was first proposed by Tavernarakis et al. (1999) to describe a domain shared among stomatin, prohibitin, flotillin and HflC/K proteins, although poor alignments and variable domain positions have led some to question the monophyly of this group (Rivera-Milla et al. 2006).
It is important to emphasise that the NfeD proteins discussed in this article are totally unrelated to the original 320 aa NfeD (nodulation formation efficiency) sequence, first described by Soto et al. (1994) and later by Garcia-Rodriguez and Toro (2000), that showed similarity to an ornithine cyclodeaminase. An incorrect annotation of the protein domain PF01957, followed by automated gene classification, has led to a large number of sequences being inappropriately termed NfeD in the databases. Despite an attempt to rectify this (Green et al. 2004), the terminology is now so well established that we reluctantly accept the name NfeD. On the other hand, the gene mutated by Borthakur and Gao (1996), which affected nodulation competitiveness, is a member of this wider nfeD family, although the authors did not formally name it. Hinderhofer et al. (2009) have recently proposed a nomenclature for NfeD-like proteins: full-length NfeD is termed NfeD1b and short-proteins homologous to the C-terminus of NfeD1b are termed NfeD1a.
So far, the NfeD protein family has not been subject to any rigorous phylogenetic analysis, and statements regarding homology have been based on amino acid similarity and protein length. We have conducted a wide-ranging study of nfeD homologues in prokaryotic genomes and, using a variety of bioinformatic and phylogenetic methods, shed light on the origin, evolution and function of both nfeD homologues and their conserved gene neighbours.
Materials and Methods
Gene Neighbour Searching
For the gene neighbour analysis, NfeD homologues classified as belonging to the PFAM NfeD family (PF01957) were downloaded from the PFAM website (http://pfam.sanger.ac.uk/). Of these, 500 proteins, from 394 taxa, were from sufficiently assembled genomes that provided gene positional information in the NCBI records. Using a python script, we searched the GenBank records for genes upstream (−1) and downstream (+1) of our nfeD queries. Protein products of the gene neighbours were then retrieved and aligned using MAFFT (Katoh et al. 2002) to identify homologous groups. Proteins found in groups represented by more than five genera and encoded on the same strand as the query sequence were considered to represent a conserved gene pair, and these were used to create a spreadsheet of nfeD gene neighbours. This analysis was then repeated for lone genes to ensure no conserved gene pairs had been missed. Although flotillin sequences were only found to be conserved amongst three genera, they were kept in the analysis because of their perceived relatedness to slipins. To obtain a comprehensive understanding of NfeD1b distribution and evolution, a BLASTP search, using Sinorhizobium meliloti 1021 Smb20990 as the query sequence, was performed against completed prokaryotic genomes (using archaeal or firmicute NfeD1b sequences as the query recovered essentially the same NfeD1b sequences). Sequences of 350–500-aa long that aligned with the query using Muscle (Edgar 2004) and contained the SAG protease motif were considered NfeD1b homologues.
Multiple Sequence Alignment
NfeD homologues were aligned using Muscle; all other alignments were performed using MAFFT. Duplicates were removed from alignments using ElimDupes at the HCV (http://hcv.lanl.gov/content/sequence/ELIMDUPES/elimdupes.html) and the long-branched NfeD from Bdellovibrio bacteriovorus (GI:42523756) was removed to prevent the problem of long branch attraction (Felsenstein 1978; Johannes 2005). All full-length alignments were edited using the Gblocks server (Castresana 2000) before phylogenetic reconstruction, whilst domain-specific phylogenies were edited by eye using SeaView (Galtier et al. 1996).
Phylogenetic Analyses
Maximum likelihood (ML) trees were constructed using PhyML 3.0 (Guindon and Gascuel 2003) using Nearest-Neighbour Interchange for tree improvement and MrBayes 3.1.1 for Bayesian Inference (Ronquist and Huelsenbeck 2003). For ML analyses, empirical matrices of protein evolution and their parameters were determined separately for each alignment using ProtTest (Abascal et al. 2005) and goodness-of-fit estimated using the Akaike Information Criterion. In all cases, the LG model (Le and Gascuel 2008) with +I, +G (four rate categories) and +F parameters was selected as the optimal model. For MrBayes analyses, fixed rate models were estimated (prset aamodel = mixed) using the Markov Chain Monte Carlo (MCMC) sampler: WAG for NfeD (Whelan and Goldman 2001), RtRev for slipin (Dimmic et al. 2002), VT for NfeD1b-2 (Müller and Vingron 2000) and Cprev for YqfA (Adachi et al. 2000) were chosen. A discrete-gamma distribution with four rate categories was combined with the proportion of invariable sites model using lset rates = invgamma for all phylogenies. Support for the ML tree topologies was determined using 100 bootstrap repetitions whilst Bayesian posterior probabilities were used in MrBayes. The MCMC analysis comprised two runs, each with one cold and three hot chains for 1,000,000 generations (NfeD), 3,000,000 generations (slipin), 500,000 generations (NfeD1b-2 and YqfA) and sampled every 100 generations, with a burnin of 25% of the samples. Sufficient convergence of runs was checked by ensuring random fluctuation of log likelihood values of the cold chain and that the potential scale reduction factor was 1.0 for all parameters. Trees were viewed using FigTree v.1.2.1 (Rambaut 2007).
Clustering Analysis of the C-Terminal Domains
All 500 NfeD homologues were clustered using the OrthoMCL algorithm (Li et al. 2003). A Muscle alignment of all short and long NfeD homologues was manually truncated at the point where full-length and C-terminal NfeD proteins no longer aligned, leaving only the C-terminal domain. Gaps were removed from this alignment and clustering was performed on ~150 aa fragments using an all-against-all BLASTP analysis. BLASTP hits were passed to OrthoMCL for Markov Chain Clustering.
Structural Prediction
Membrane topology was predicted by PolyPhobius (Käll et al. 2005) from alignments that were edited minimally by eye using the displayed sequence as the template. To prevent bias, only one species was selected from each genus.
Results
nfeD Homologues Are Associated With One of Four Conserved Gene Neighbours
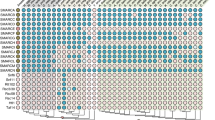
To gain an overview of the NfeD protein family, a simple BLASTP search was performed using the S. meliloti NfeD1b sequence as the query (Fig. 1). Full-length homologues (NfeD1b) have a widespread yet patchy distribution, being absent from major bacterial groups such as the Chlamydiae, Cyanobacteria, Actinobacteria, Spirochetes, Epsilonproteobacteria and Mollicutes. Indeed this patchiness occurs even at the strain level: for example, Rhizobium leguminosarum bv. trifolii WSM2304 encodes NfeD1b whilst R. leguminosarum bv. viciae 3841 does not. The BLASTP search was essentially comprehensive for NfeD1b sequences, because the N-terminal protease domain is well conserved, but also recovered some short sequences (approximately 150 aa) that had only the C-terminal domain. However, most NfeD1a sequences were not recovered because this domain is highly divergent. We then assembled all 500 proteins that are annotated as possessing the Pfam PF01957 domain, which includes both NfeD1b and NfeD1a. Just over 85% of these nfeD homologues had, as immediate neighbour, one of four conserved gene partners: eoslipin, paraslipin, yqfA or flotillin (Fig. 2; see Supplementary Material). Full-length sequences of yqeZ (the nfeD1b homologue found in Firmicutes and Deinococci) were located upstream of yqfA. Other nfeD1b genes were always found to be associated with eoslipin, in either an upstream (bacteria and Thermococci) or a downstream (archaea only) orientation, except in the case of Pyrobaculum spp., where nfeD1b is associated with paraslipin. Short nfeD1a genes were found to be either upstream or downstream of paraslipin, yqfA or flotillin genes.

Distribution of NfeD1b within the major prokaryotic groups. NfeD1b homologues were identified through a BLAST search against completed prokaryotic genomes using Sinorhizobium meliloti 1021 NfeD1b as the query. Only proteins of length 350–600 aa, containing the three functional domains, were accepted. n the number of genomes searched for each group

Schematic diagram of the main nfeD conserved gene clusters and truncated forms. The nfeD numbers correspond to the four phylogenetic clades in Fig. 3. Solid boxes show the four major conserved gene partners: yqfA, flotillin, eoslipin and paraslipin, whereas dashed boxes show truncated forms. Percentages correspond to the proportion of all nfeD homologues found to exist in that specific gene pair. 13.8% of nfeD homologues were not found to exist in any conserved cluster, whereas 1.4% of nfeD homologues were found in other arrangements from those shown above. Arrows are proportional to length of the genes, whereas intergenic regions are not to scale
NfeD1b Family Phylogeny
A phylogeny of those long NfeD1b sequences (>400 aa) that have conserved gene neighbours reveals four distinct groups, which we term NfeD1b-1,2,3 and 4 (Fig. 3). ML and Bayesian analyses produced congruent topologies with mostly well-resolved internal branches. NfeD1b-4 is the major archaeal group, with monophyletic clades of euryarchaeotes and crenarchaeotes. However, certain archaea have NfeD1b-2 or NfeD1b-3 in place of, or as well as, NfeD1b-4. Within bacteria, we see two main groups, comprising NfeD1b-1 and NfeD1b-2 (YqeZ). NfeD1b-2 is restricted to Firmicutes and Deinococci and the long-branched Pyrobaculum homologues fall at the base of this group. The major groups identified in Fig. 3 correlate with the type and arrangement of gene neighbour, as illustrated in Fig. 2. All nfeD1b-1 genes are upstream of eoslipin, whereas the nfeD1b-2 sequences are upstream of a different partner, yqfA. Within archaea, nfeD1b-4 is associated with eoslipin but is downstream of it, unlike the arrangement in bacteria. In contrast, the eoslipin–nfeD1b-3 clusters of Pyrococcus and Thermococcus have the ‘bacterial’ arrangement, the significance of which will be dealt with later.

Unrooted phylogeny of NfeD proteins using ML. ML and Bayesian trees were constructed from a 239 aa alignment. The ML tree is shown with branch lengths proportional to the number of amino acid substitutions per site, as indicated by the scale bar. ML bootstraps (based on 100 replicates) and Bayesian posterior probabilities are displayed using branch thickness according to the key (top left). Gene neighbour arrangements are shown to the right of each major group. Arrows indicate positions of the two most plausible root placements mentioned in the section “Discussion”. The clade marked with an asterisk was strongly supported by Bayesian analysis but, in contrast to ML, included Legionella pneumophila, whereas the clade marked with a filled circle additionally included Desulfovibrio desulfuricans in the Bayesian analysis
Phylogeny of Slipin Neighbours Suggest an Ancient Origin for the NfeD–Slipin Cluster
The majority (76%) of NfeD homologues were associated with a slipin gene. To investigate further the coevolution of the nfeD–slipin gene clusters, a phylogeny of all nfeD-associated slipin sequences was constructed (Fig. 4). There are two major groups separated by a long, well-supported, internal branch. The upper portion of the tree contains eoslipin sequences encoded by genes adjacent to nfeD1b-1, nfeD1b-3 or nfeD1b-4. The only exceptions are sequences from Dehalococcoides spp., where eoslipin is a downstream of a C-terminal nfeD-1b homologue. The lower group comprises paraslipin sequences from bacteria and euryarchaeota that are associated with NfeD1a. Paraslipin phylogeny does not totally reflect organism phylogeny; for example, the paraslipin sequences of Cyanobacteria, Clostridia and Deinococci species all fall within the major proteobacterial clade, suggesting horizontal transfer of genes between these groups. Archaea are paraphyletic in both portions of the tree, with Thermococci species branching away from the main archaeal clade. In the paraslipin portion of the tree, there is strong support for a clade uniting both bacterial and archaeal hyperthermophiles from the Thermotogae and Thermococci, suggesting horizontal transfer between these two groups. Long-branched Pyrobaculum slipins associated with nfeD1b-2 and the Methanopyrus kandleri slipin associated with truncated nfeD1b-4, fall within the paraslipin and eoslipin portion of the tree, respectively, but were omitted from the final tree in Fig. 4 (with no change in topology) to increase branch resolution.

Phylogeny of nfeD-associated slipin sequences using ML. ML and Bayesian trees were constructed from a 201-aa alignment. Branches are drawn proportional to the number of amino acid substitutions per site as indicated by the scale bar. ML bootstraps (based on 100 replicates) and Bayesian posterior probabilities are displayed for important internal branches using branch thickness according to the key (top left). Arrows indicate positions of the two most plausible roots (root 1, branch between eoslipins and paraslipins; root 2, branch between crenarchaeota and euryarchaeota eoslipins). The upper groups contain eoslipins associated with nfeD1b-1,2,3,4, whereas the lower group comprises paraslipin sequences associated with nfeD1a
Some C-Terminal Homologues Cluster With Full-Length NfeD1b Proteins
It was clear from the BLAST searches that not all short NfeD homologues were equally related, as some short fragments produced a better BLAST score to full-length NfeD1b than to NfeD1a proteins. Unfortunately, determining relatedness of short and long NfeD forms using phylogenetic analysis is problematic due to the high level of sequence divergence among short C-terminal forms. However, OrthoMCL was able to cluster all NfeD homologues based on amino acid similarity of the C-terminal domain. Twenty-five clusters were recovered in total and 20 of these contained members with conserved gene pairs (see Supplementary Material); 4 of these clusters include both long and short NfeD forms (Table 1). Cluster 1 is the largest mixed cluster and consists mostly of long bacterial NfeD1b-1 sequences (>400 aa) that are eoslipin-associated, as well as some short C-terminal NfeD sequences from Dehalococcoides spp. and Pseudomonas entomophila (96 and 175 aa, respectively) that are also associated with eoslipin. Pyrococcus, Thermococcus and Thermoplasma NfeD1b-3 sequences also fall within this group. Cluster 2, reflecting NfeD1b-4, contains only archaeal species and comprises long NfeDs (plus a single short form from M. kandleri); all are associated with eoslipin. Cluster 3 groups both long (NfeD1b-2) and short NfeD homologues, all are associated with yqfA except one which has no conserved neighbour. The fourth cluster comprises flotillin-associated C-terminal NfeD forms. Remaining clusters are paraslipin-associated NfeD1a forms, with one of these clusters containing a single long NfeD from the archaeon, Archaeoglobus fulgidus. Five clusters of C-terminal forms have no conserved neighbour.
Truncation and Fusion in NfeD Evolution
The above observation suggests that some of the short C-terminal NfeD homologues are in fact truncations of full-length NfeD1b proteins. To test this hypothesis, we constructed a phylogeny from a C-terminal alignment of short and long yqfA-associated NfeD1b-2 homologues from cluster 3 in Table 1 (Fig. 5a). Short truncated forms do not form a single distinct group on the tree; rather, they are dispersed amongst long NfeD1b-2 forms, suggesting multiple independent origins. The congruent phylogeny of the gene neighbour yqfA (Fig. 5b) together with their co-localisation on the genome suggests that truncated forms have evolved repeatedly from full-length NfeD1b-2 and have remained associated with their ancestral gene neighbour, yqfA.

ML phylogeny of NfeD1b-2 homologues and their associated YqfA. ML and Bayesian trees were constructed from a an alignment of C-terminal domains (152 aa) from both full-length and truncated NfeD1b-2 homologues associated with yqfA and b an alignment of associated YqfA (329 aa). Branch lengths are proportional to the number of amino acid substitutions per site, as indicated by the scale bars. One hundred replicates were performed for the ML analyses. Bootstraps/posterior probabilities are shown for the major internal branches if >75/0.95, respectively. Filled circles indicate truncated NfeD1b-2 C-terminal fragments
Domain Structure of the NfeD Family and Its Partner Proteins
NfeD proteins are predicted to be membrane proteins, although the exact number of membrane-spanning helices remains unclear. Both Phobius and TMHMM predict four TMDs for some NfeD1b proteins, and five for other NfeD1b proteins. Prediction of TMDs can be improved using an alignment of homologues, since they are likely to share sequence features (Käll et al. 2005). Separate alignments were constructed of 5 NfeD1b-2 proteins, 54 NfeD1b-1,3,4 proteins, and 132 NfeD1a proteins and submitted to the PolyPhobius server. Essentially, congruent profiles were predicted for NfeD1b-1,3,4 and NfeD1b-2 proteins (Fig. 6), with an N-terminal signal peptide sequence (posterior label probability = 0.8) and five TMDs towards the C-terminus. Truncated NfeD forms possess an N-terminal hydrophobic domain followed by the C-terminal soluble region, although these were based on single submissions to Phobius rather than global alignments. NfeD1a proteins possess a hydrophobic N-terminus followed by a soluble C-terminal region.

Hydrophobic profiles of NfeD1b, NfeD1a and truncated NfeD1b sequences. a(i) Polyphobius prediction of TMD based on an alignment of 54 NfeD1b-1,3,4 sequences and a(ii) single Phobius prediction of the truncated NfeD1b-4 of M. kandleri. b(i) Polyphobius prediction of TMD based on an alignment of five NfeD1b-2 sequences and b(ii) single Phobius prediction of the truncated NfeD1b-2 of Clostridium thermocellum. c Polyphobius prediction based on an alignment of 132 NfeD1a sequences. Hollow bars above each plot indicate protein length. Grey transmembrane regions, black signal peptide prediction
Discussion
The NfeD Family Is Ancient
The NfeD protein family can be considered to be composed of three major groups: NfeD1a, NfeD1b and truncated NfeD1b (Fig. 7). The largest group, NfeD1a, comprises proteins of approximately 145 aa in length that are exclusively associated with paraslipin (Fig. 4). These proteins possess an N-terminal hydrophobic domain, followed by a soluble C-terminal domain rich in beta sheets (Kuwahara et al. 2008), which is structurally very similar to the OB-fold (oligosaccharide/oligonucleotide-binding fold) domain. The second major group, NfeD1b, comprises proteins of approximately 460 aa with three functional domains after a predicted N-terminal signal peptide: (i) an N-terminal serine protease domain, (ii) a middle membrane-spanning domain and (iii) a C-terminal domain that is homologous to the soluble C-terminal region of NfeD1a. The majority (78%) of these nfeD genes are associated with eoslipin in archaeal and bacterial genomes, whilst within Firmicutes and Deinococci the partner gene is yqfA. The final group is not a ‘natural group’ in that it unites independently evolved truncated forms of NfeD1b. These truncated forms are of a similar length and domain structure to NfeD1a (Fig. 6), yet evolutionarily they are more related to NfeD1b, as demonstrated by our clustering and phylogenetic analyses (Table 1 and Fig. 5). Many of these truncated forms have remained associated with their ancestral gene partner, either yqfA or eoslipin. It is not clear whether these truncated forms are functioning in a similar way to NfeD1a–paraslipin pairings or instead are performing some novel role. It does however raise the intriguing question of whether eoslipin can, under certain circumstances, functionally replace paraslipin; an important consideration for any knockout studies. Although flotillin did not meet our criteria for being considered a conserved gene neighbour (it is restricted to Bacillus and Mycobacterium spp.), we included it in the analysis due to its SPFH domain. Our clustering analysis did not allow us to assign the flotillin-associated C-terminal NfeD homologue, YuaF, to any group, therefore we cannot discern whether these represent convergently evolved NfeD domains or instead represent divergent copies of either NfeD1a or truncated NfeD1b. Attempts to determine relatedness of these C-terminal forms using phylogenetic methods are fraught with difficulties associated with aligning and modelling the evolution of highly diverged sequences and are probably best avoided.

Model of NfeD family evolution. A slipin/nfeD protocluster duplicated to give rise to NfeD1a and NfeD1b subfamilies, which became associated with paraslipin and eoslipin, respectively. Within Firmicutes, nfeD1b-2 became associated with yqfA, which may represent a highly diverged form of eoslipin. Throughout NfeD1b evolution, multiple, independent truncation events occurred which gave rise to truncated C-terminal portions which frequently remained associated with their ancestral gene neighbour. Grey boxed clusters represent ancestral forms
The origin of the NfeD1b–eoslipin and NfeD1a–paraslipin clusters is not clear because of low branch resolution and evidence for horizontal transfer between archaea and bacteria in both portions of the slipin tree. The monophyly of crenarchaeota and euryarchaeota in Fig. 3 suggests that the common ancestor of archaea had an NfeD-1b–eoslipin cluster. What is not clear is whether this cluster was present in the common ancestor all life (root 1, Fig. 3) or instead was transferred later into bacteria through an ancient horizontal transfer from a Thermococci-like ancestor (root 2, Fig. 3). In contrast, NfeD1a–paraslipin is present in both bacteria and euryarchaeotes but is missing from crenarchaeota. Again, we cannot be certain whether the NfeD1a–paralsipin cluster was present in the last universal common ancestor or instead arose in archaea and later was transferred into bacteria.
Gene duplications can be used to root a tree that includes ancient paralogues. If we accept eoslipin and paraslipin as paralogous clades, the root of the slipin phylogeny (root 1, Fig. 4) occurs prior to the divergence of archaea and bacteria, in line with the traditional rooting of the prokaryotic tree (Gogarten et al. 1989; Iwabe et al. 1989). This suggests that both subfamilies are extremely ancient and provides evidence that nfeD1a–paraslipin and nfeD1b–eoslipin gene clusters arose from a duplication of an ancestral nfeD–slipin protocluster. Because NfeD1a and NfeD1b are exclusively associated with paraslipin and eoslipin, respectively, we are able to use the slipin root and map this onto our NfeD phylogeny (Fig. 3, root 1). If we do this, we recover archaea and bacteria as each essentially monophyletic, although strict monophyly is distorted by horizontal transfer between bacteria and Thermococci (Fig. 3). An alternative hypothesis would be to accept that eoslipin and paraslipin proteins are not equally ancient and instead propose that the NfeD1a–paraslipin cluster arose from the duplication of an NfeD1b–eoslipin ancestral cluster within archaea; this would correspond to root two in Figs. 3 and 4. Following this duplication, both NfeD1a–paraslipin and NfeD1b–eoslipin clusters where transferred into bacteria by horizontal transfer before many of the major bacterial groups had diverged. We find this second hypothesis considerably more complex and therefore less likely.
Within Firmicutes and Deinococci, NfeD1b diverged to form YqeZ (NfeD1b-2). The long branch leading to the group (Fig. 3) may reflect rapid evolution coinciding with the acquisition of a new gene partner yqfA. It is important to note that the same taxa are not found in both NfeD1b-2 and NfeD1b-1 clades, suggesting that YqeZ is an orthologous rather than paralogous NfeD group and therefore does not warrant a separate name. YqfA has been termed a stomatin homologue and recently included as a member of the SPFH superfamily (Hinderhofer et al. 2009). However, we can provide no sequence evidence to support either of these hypotheses. BLASTP searches using YqfA reveal extremely weak hits (e-value of 1.0) to the so-called prohibitin-like and SPFH proteins: this is not good evidence of homology. Indeed, regions of alignment occur in areas predicted to form coiled coils in both the query and the hit sequence, suggesting any similarity could equally be a result of convergence. It is true, however, that the genomic context suggests that YqfA may represent a highly diverged form of eoslipin that evolved within the Firmicute lineage, as suggested by Hinderhofer et al. (2009) and probably was acquired by Deinococci through HGT. What drove this change in gene partner is not clear, although it may have coincided with the change in cell envelope structure associated with Firmicutes. The distribution of NfeD1b orthologues within bacteria is intriguing and not fully understood (Figs. 1 and 2). It appears that NfeD1b has been lost from many major groups, such as Actinobacteria, Cyanobacteria and Chlamydiae and even within related groups we find a patchy distribution. These results are consistent with the idea that NfeD1b proteins have a non-essential, accessory role in prokaryotic genomes; intriguingly, this ‘role’ is of use to an extremely diverse set of bacteria and archaea and is not restricted to a narrow taxonomic range.
Functional Implications
NfeD homologues are clearly reliant on a conserved gene neighbour which we assume is necessary for function, either through direct physical interaction or by functioning in the same pathway. All conserved gene partners identified here share similar structural properties, with broadly comparable hydrophobic profiles and predicted coiled-coil regions (Hinderhofer et al. 2009). Exactly what is the role of these partner proteins? Paraslipin, eoslipin and flotillin all have clear eukaryotic homologues that contain the SPFH domain and reside in cholesterol-rich regions of the cell membrane known as lipid rafts (Browman et al. 2007). Increasingly, prokaryotic SPFH proteins are being found to exhibit similar characteristic to their eukaryotic relatives, being found in detergent-resistant membranes (Zhang et al. 2005) where they form discrete foci (Donovan and Bramkamp 2009).
Our results suggest that it is the C-terminal domain of NfeD1b (transmembrane region or β-barrel) that interacts with the partner protein, as even truncated nfeD1b genes remain associated with the original gene partner (Table 1). Indeed, Fig. 5 implies that NfeD1b proteins have a propensity to become truncated, as this has occurred a number of times. Whether truncation results in a novel function, or merely attenuates the original function, is not clear. We can speculate that the protease domain may be performing some regulatory role, possibly by cleaving the partner protein (Yokoyama and Matsui 2004) or some other protein, but is not required for the interaction with the partner. At present there is considerable ambiguity as to whether there are four or five transmembrane domains (TMD) within the middle portion of NfeD1b. Using a global alignment of NfeD1b-1,2,3 proteins, PolyPhobius predicts five TMDs (Fig. 6). Determining the exact topology is vital if we are to make an informed prediction of function, as with four TMD both the protease and C-terminal NfeD domains would be on the same side of the membrane, whereas with five they would reside in different compartments of the cell. This topology also affects whether the C-terminal domain is accessible to the partner protein. A considerable amount of experimental research into NfeD function has been performed using the archaeon P. horikoshii as a model; this has both the nfeD1b-eoslipin and nfeD1a-paraslipin gene clusters. Using surface plasmon resonance, Yokoyama et al. showed that the C-terminus of PH1510 (NfeD1b) interacts with the C-terminus of PH0470 (paraslipin). On this evidence, the authors suggest that NfeD1b/eoslipin proteins form a functioning complex with paraslipin/NfeD1a proteins. However, this is unlikely to be a general mechanism, since only 13% of genomes examined in this study were found to contain both clusters. It may be that the observed interaction is merely a reflection of the fact that eoslipin and paraslipin are homologous proteins with similar structures. Cross binding of paraslipin with NfeD1b would not occur in vivo if the proteins resided in different compartments within the cell, but cellular localisation of NfeD1b, eoslipin and NfeD1a still needs to be determined.
We have provided a number of discrete hypotheses that can now be tested experimentally to unravel NfeD1a and NfeD1b functions. Such studies will undoubtedly enhance our understanding of NfeD gene partners and their raft-associated eukaryotic relatives such as stomatin, flotillin and paraslipin.
References
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105
Adachi J, Waddell PJ, Martin W, Hasegawa M (2000) Plastid genome phylogeny and a model of amino acid substitution for proteins encoded by chloroplast DNA. J Mol Evol 50:348–358
Borthakur D, Gao X (1996) A 150-megadalton plasmid in Rhizobium etli strain TAL182 contains genes for nodulation competitiveness on Phaseolus vulgaris L. Can J Microbiol 42:903–910
Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C (2000) NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24:349–354
Browman DT, Hoegg MB, Robbins SM (2007) The SPFH domain-containing proteins: more than lipid raft markers. Trends Cell Biol 17:394–402
Butcher BG, Helmann JD (2006) Identification of Bacillus subtilis sigma-dependent genes that provide intrinsic resistance to antimicrobial compounds produced by Bacilli. Mol Microbiol 60:765–782
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Chiba S, Ito K, Akiyama Y (2006) The Escherichia coli plasma membrane contains two PHB (prohibitin homology) domain protein complexes of opposite orientations. Mol Microbiol 60:448–457
Cui Z, Zhang L, Hua Z, Cao W, Feng W, Liu Z (2007) Stomatin-like protein 2 is overexpressed and related to cell growth in human endometrial adenocarcinoma. Oncol Rep 17:829–833
Dimmic MW, Rest JS, Mindell DP, Goldstein RA (2002) rtREV: an amino acid substitution matrix for inference of retrovirus and reverse transcriptase phylogeny. J Mol Evol 55:65–73
Donovan C, Bramkamp M (2009) Characterization and subcellular localization of a bacterial flotillin homologue. Microbiology 155:1786–1799
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Felsenstein J (1978) Cases in which parsimony or compatibility methods will be positively misleading. Syst Zool 27:401–410
Galtier N, Gouy M, Gautier C (1996) SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Bioinformatics 12:543–548
García-Rodríguez FM, Toro N (2000) Sinorhizobium meliloti nfe (nodulation formation efficiency) genes exhibit temporal and spatial expression patterns similar to those of genes involved in symbiotic nitrogen fixation. Mol Plant Microbe Interact 13:583–591
Gogarten JP, Kibak H, Dittrich P, Taiz L, Bowman EJ, Bowman BJ, Manolson MF, Poole RJ, Date T, Oshima T (1989) Evolution of the vacuolar H+-ATPase: implications for the origin of eukaryotes. Proc Natl Acad Sci USA 86:6661–6665
Green JB, Young JP (2008) Slipins: ancient origin, duplication and diversification of the stomatin protein family. BMC Evol Biol 8:44
Green JB, Fricke B, Chetty MC, von Düring M, Preston GF, Stewart GW (2004) Eukaryotic and prokaryotic stomatins: the proteolytic link. Blood Cells Mol Dis 32:411–422
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hinderhofer M, Walker CA, Friemel A, Sturmer CA, Moller HM, Reuter A (2009) Evolution of prokaryotic SPFH proteins. BMC Evol Biol 9:10
Iwabe N, Kuma K, Hasegawa M, Osawa S, Miyata T (1989) Evolutionary relationship of archaebacteria, eubacteria, and eukaryotes inferred from phylogenetic trees of duplicated genes. Proc Natl Acad Sci USA 86:9355–9359
Johannes B (2005) A review of long-branch attraction. Cladistics 21:163–193
Käll L, Krogh A, Sonnhammer EL (2005) An HMM posterior decoder for sequence feature prediction that includes homology information. Bioinformatics 21(Suppl 1):i251–i257
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Kuwahara Y, Ohno A, Morii T, Yokoyama HY, Matsui I, Tochio H, Shirakawa M, Hiroaki H (2008) The solution structure of the C-terminal domain of NfeD reveals a novel membrane-anchored OB-fold. Protein Sci 17:1915–1924
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320
Li L, Stoeckert CJ, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189
Müller T, Vingron M (2000) Modeling amino acid replacement. J Comput Biol 7:761–776
Rambaut A (2007) FigTree, a graphical viewer of phylogenetic trees. http://tree.bio.ed.ac.uk/software/figtree/
Rivera-Milla E, Stuermer CA, Málaga-Trillo E (2006) Ancient origin of reggie (flotillin), reggie-like, and other lipid-raft proteins: convergent evolution of the SPFH domain. Cell Mol Life Sci 63:343–357
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Soto MJ, Zorzano A, García-Rodríguez FM, Mercado-Blanco J, López-Lara IM, Olivares J, Toro N (1994) Identification of a novel Rhizobium meliloti nodulation efficiency nfe gene homolog of Agrobacterium ornithine cyclodeaminase. Mol Plant Microbe Interact 7:703–707
Stewart GW, Hepworth-Jones BE, Keen JN, Dash BC, Argent AC, Casimir CM (1992) Isolation of cDNA coding for an ubiquitous membrane protein deficient in high Na+, low K+ stomatocytic erythrocytes. Blood 79:1593–1601
Tavernarakis N, Driscoll M, Kyrpides NC (1999) The SPFH domain: implicated in regulating targeted protein turnover in stomatins and other membrane-associated proteins. Trends Biochem Sci 24:425–427
Walker CA, Hinderhofer M, Witte DJ, Boos W, Möller HM (2008) Solution structure of the soluble domain of the NfeD protein YuaF from Bacillus subtilis. J Biomol NMR 42:69–76
Wang Y, Morrow JS (2000) Identification and characterization of human SLP-2, a novel homologue of stomatin (band 7.2b) present in erythrocytes and other tissues. J Biol Chem 275:8062–8071
Whelan S, Goldman N (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18:691–699
Yokoyama H, Matsui I (2004) A novel thermostable membrane protease forming an operon with a stomatin homolog from the hyperthermophilic archaebacterium Pyrococcus horikoshii. J Biol Chem 280:6588–6594
Yokoyama H, Matsui E, Akiba T, Harata K, Matsui I (2006) Molecular structure of a novel membrane protease specific for a stomatin homolog from the hyperthermophilic archaeon Pyrococcus horikoshii. J Mol Biol 358:1152–1164
Zhang HM, Li Z, Tsudome M, Ito S, Takami H, Horikoshi K (2005) An alkali-inducible flotillin-like protein from Bacillus halodurans C-125. Protein J 24:125–131
Acknowledgements
This study was supported by a BBSRC PhD studentship to J.B.G. R.P.J.L. is funded by NERC Grant NE/D011485/1 awarded to J.P.W.Y. We thank Dr Gemma Atkinson for the use of her python script to identify conserved gene partners and for help with MrBayes.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
239_2009_9304_MOESM1_ESM.xls
(i) NfeD and conserved gene neighbours and cluster, (ii) NfeD distribution in completed prokaryotic genomes (XLS 398 kb)
Rights and permissions
About this article
Cite this article
Green, J.B., Lower, R.P.J. & Young, J.P.W. The NfeD Protein Family and Its Conserved Gene Neighbours Throughout Prokaryotes: Functional Implications for Stomatin-Like Proteins. J Mol Evol 69, 657–667 (2009). https://doi.org/10.1007/s00239-009-9304-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-009-9304-8