
Abstract
The rRNA cistron (18S–ITS1–5.8S–ITS2–28S) is used widely for phylogenetic analyses. Recent studies show that compensatory base changes (CBC) in the secondary structure of ITS2 correlate with genetic incompatibility between organisms. Rhizoctonia solani consists of genetically incompatible strain groups (anastomosis groups, AG) distinguished by lack of anastomosis between hyphae of strains. Phylogenetic analysis of internal transcribed spacer (ITS) sequences shows a strong correlation with AG determination. In this study, ITS sequences were reannotated according to the flanking 5.8S and 28S regions which interact during ribogenesis. One or two CBCs were detected between the ITS2 secondary structure of AG-3 potato strains as compared to AG-3 tobacco strains, and between these two strains and all other AGs. When a binucleate Rhizoctonia species related to Ceratobasidiaceae was compared to the AGs of R. solani, which were multinucleate (3–21 nuclei per cell), 1–3 CBCs were detected. The CBCs in potato strains of AG-3 distinguish them from AG-3 tobacco strains and other AGs yielding further evidence that the potato strains of AG-3 originally described as R. solani are a species distinct from other AGs. The ITS1–5.8S–ITS2 sequences were analyzed by direct sequencing of PCR products from 497 strains of AG-3 isolated from potato. The same 10 and 4 positions in ITS1 and ITS2, respectively, contained variability in 425 strains (86%). Nine different unambiguous ITS sequences (haplotypes) could be detected in a single strain by sequencing cloned PCR products indicating that concerted evolution had not homogenized the rRNA cistrons in many AG-3 strains. Importantly, the sequence variability did not affect the secondary structure of ITS2 and CBCs in AG-3.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Comparison of DNA sequences encoding the internal transcribed spacers (ITS) of ribosomal RNA (rRNA) genes in the rRNA cistron (18S–ITS1–5.8S–ITS2–28S) is commonly used to study phylogenetic relationships at the species and genus levels (Buckler et al. 1997). Secondary structure predictions may be used to improve the alignment of primary sequences for comparison. With ITS sequences, there are also additional reasons because structural features of ITS1 and ITS2 may be important for positioning the transcript-processing machinery on the pre-rRNA, the precursor of the 18S, 5.8S and 28S rRNAs (Côté and Peculis 2001; Abeyrathne and Nazar 2005). Hence, the secondary structures of ITS RNA sequences provide additional evolutionary information (Coleman 2000; 2003; Coleman and Vacquier 2002; Wolf et al. 2005a) and a rather simple molecular marker, which may be particularly useful when studying closely related species (Müller et al. 2007; Selig et al. 2008).
Compensatory base changes (CBC) occur in the paired regions of a primary RNA transcript when both nucleotides of a paired site mutate while the pairing itself is maintained (e.g., G–C mutates to A–U) (Gutell et al. 1994). When over 1,300 organisms classified as different species of the same genus were analyzed, CBCs were detected between 93% of the species (Müller et al. 2007). These results indicate that a CBC in an ITS2 sequence–structure alignment provides strong evidence for distinguishing species.
Rhizoctonia solani (Kühn 1858) is an asexually growing fungal species that includes many related but genetically isolated subspecific groups. The hyphae of the closely related strains can fuse (anastomose) and, hence, form an ‘anastomosis group’ (AG), whereas the distantly related strains are unable to anastomose (Matsumoto et al. 1932; Parmeter et al. 1969; Kuninaga et al. 1978; Carling et al. 2002a; 2002b). The known strains of R. solani can be placed to at least 13 AGs, but classification based on anastomosis is not always unambiguous because some ‘bridging strains’ are able to anastomose with strains of at least two AGs (Parmeter et al. 1969; Kuninaga et al. 1978; Carling et al. 2002a; Sharon et al. 2008). Strains belonging to the AG-1, AG-2, AG-3, AG-4 and AG-8 are particularly important pathogens that cause devastating diseases such as stem canker, foliage lesions or damping off in many crops (Sneh et al. 1996; Schillinger and Paulitz 2006). On the other hand, some strains of R. solani exist merely as saprotrophs in soil (Ogoshi 1996) and AG-12 displays mycorrhizal interactions with orchids (Bernard 1909; Carling et al. 1999). The sexual (teleomorphic) stage of R. solani [Thanatephorus cucumeris (Frank) Donk; Basidiomycota, family Ceratobasidiaceae] has little significance as a plant pathogen (Sneh et al. 1996).
Rhizoctonia solani was first described as the hyphae and sclerotia (black scurf) observed on potato tubers by Kühn (1858) and later shown to cause stem canker on potato (Edson and Shapovalov 1918). The currently known anastomosis group AG-3 is specialized on potato, forms sclerotia abundantly on tubers and is likely to be the fungus originally described as R. solani. Other, less host-specific AGs may also infect potato but they show usually rather poor or no formation of sclerotia on tubers (Sneh et al. 1996; our unpublished data). Phylogenetic analysis of primary ITS sequences can be used to determine the AGs with a reasonable accuracy (Kuninaga and Yokosawa 1985; Boysen et al. 1996; Mazzola et al. 1996; Kuninaga et al. 1997; Carling et al. 2002a; Sharon et al. 2008). Also, subgroups within AG-2 have been detected (Salazar et al. 1999; 2000; Carling et al. 2002b) and the evolution of AG-1 IA analyzed using ITS sequences (Ciampi et al. 2005). The feasibility of this approach is increasing with the number of sequences determined from R. solani and other Rhizoctonia spp. However, the primary ITS sequences have not been sufficient to resolve the species complex of R. solani, e.g., the by now classical debate about the AGs and their possible establishment as species (Menzies 1970; Ogoshi 1996; González et al. 2006). The debate remains vivid because R. solani is recalcitrant to genetic studies using conventional methods (Sneh et al. 1996; Cubeta and Vilgalys 1997; Rosewich et al. 1999).
The aim of this study was to compare the secondary structures of ITS2 RNA between strains of different AGs of R. solani using the recently introduced novel method (Müller et al. 2007) and address the question about AGs and their possible establishment as species using a novel approach. There may be hundreds of tandem copies of the rRNA cistron in a eukaryotic genome and it is commonly considered that concerted evolution homogenizes them (Hillis et al. 1991) and allows ITS regions to be treated as a single gene (Coleman 2003). However, this issue has received little attention in R. solani AG-3. The molecular variability of ITS sequences was assessed using the largest number of AG-3 strains characterized so far to observe whether such variability if any would affect the CBC predictions.
Materials and Methods
Phylogenetic and Secondary Structure Analyses of Sequences
Internal transcribed spacer sequences of R. solani were obtained from Genbank and NCBI databases or determined in this study (Supplementary Table 1). Consensus sequences were obtained using the Gap-4 program (Staden Package; MRC Laboratory of Molecular Biology, Cambridge, UK). The ITS1-5.8S-ITS2 sequences used for analysis were of identical length in AG-3, but lengths slightly differed between the AGs. They were aligned with ClustalW because structural information was available for the ITS2 region only (see below). Phylogenetic analyses and building of phylogenetic trees were done using the Phylip program with default settings of the Mega 3.1 program package (Kumar et al. 2004). Bootstrap support was estimated based on 100 or 500 pseudoreplicates. Phylogenetic analyses were carried out using the neighbour-joining (Saitou and Nei 1987) and maximum parsimony (Felsenstein 1978) methods. Trees were rooted by user-specified outgroups and displayed using the TreeView program included in Mega 3.1.
ITS2 sequence–structure analysis was carried out as described (Schultz and Wolf 2009). Annotation of ITS2 sequences was according to the 5.8S–28S rDNA interaction during ribogenesis (Côté and Peculis 2001; Keller et al. 2009). Optimal and suboptimal secondary structures for AG-3 potato strain sequences (AG-3 PT) were obtained by RNAstructure (Mathews et al. 2004). One of the suboptimal structures followed the common core of eukaryotic ITS2 secondary structures (Schultz et al. 2005) and was used as a template for homology modelling. Homology modelling (Wolf et al. 2005b) of all secondary structures from sequences of AG strains used in further analyses was done using the ITS2 PAM 50 matrix with the custom modelling option (Selig et al. 2008) available at the ITS2 Database (Schultz et al. 2005; 2006; Wolf et al. 2005a; Selig et al. 2008). Secondary structures were displayed with 4SALE (Seibel et al. 2008). Using 4SALE (Seibel et al. 2006; 2008) with its ITS2 specific scoring matrix, sequences were automatically aligned with their secondary structures simultaneously. Subsequently, CBCs were counted using the CBCAnalyzer (Wolf et al. 2005a). The sequence–structure alignment is available as supplementary material (Supplementary Fig. 1).
Determination of ITS Sequences from Finnish AG-3 Strains
Strains of R. solani were collected from 281 potato fields in 53 communes distributed throughout Finland (latitude 60°–65° N) in 2002–2004 (Lehtonen et al. 2008). The numbers of strains collected from different regions were proportional to the acreage of potato production from the total potato production area in Finland. Isolation was made from canker lesions on stems, stolons and roots, from teleomorph or mycelium at the basal part of the stem, or sclerotia on tubers to 1.2% (w/v) water agar supplemented with streptomycin (50 ppm). Pure cultures of strains were obtained by excising single hyphal tips from the edge of actively growing mycelia and transferring and growing them on potato dextrose agar (PDA) (Biokar).
Of the 503 strains isolated, the ITS sequences of 106 strains and also the virulence, fungicide tolerance and growth rate of 98 strains have been reported (Lehtonen et al. 2008). The remaining strains were characterized for ITS sequences in this study. For DNA isolation, strains were grown on cellophane discs on PDA. Hyphae were homogenized with the FastPrep equipment (FP120-230; Qbiogene/BIO101 System Matrix, Thermo Savant) and DNA was isolated as described (Edwards et al. 1991). ITS1 and ITS2 regions, including the 5.8S rRNA gene, were amplified by polymerase chain reaction (PCR) using the universal ITS-1 and ITS-4 primers that anneal to the flanking 18S and 28S rRNA genes (White et al. 1990). The PCR conditions were those described by Lehtonen et al. (2008). The size and quality of PCR products were checked by electrophoresis of an aliquot (5 μl) of the reaction on a 1% agarose gel (w/v TAE electrophoresis buffer; ethidium bromide 0.1 μg/ml) and viewing the bands under UV-light. Direct sequencing of the amplification products was carried out in both directions using the ITS-1 and ITS-4 primers at the Haartman Institute and the DNA Sequencing Laboratory of the Institute of Biotechnology, University of Helsinki. Sequences were deposited to the NCBI sequence database (Supplementary Table 1).
The amplicons from three AG-3 strains differing in the number of polymorphic ITS positions (double-peaks revealed in the electropherogram generated as an output of direct sequencing) were inserted in the pGEM-T vector (Promega Corporation) according to the manufacturer’s instructions and cloned in Escherichia coli (DH5α) cells. Plasmids were purified with the QIAprep Miniprep kit (QIAGEN). The inserts were sequenced using the universal primers M13-F and M13-R with annealing sites flanking the insert in pGEM-T.
Anastomosis Tests
Anastomosis was tested with 12 strains to confirm the results from phylogenetic analysis. The method of Parmeter et al. (1969) was used with modifications introduced by Kronland and Stanghellini (1988). Reference strains of R. solani representing the anastomosis groups AG-2-1 (strain PS-4), AG-3 (strain ST 11-6), AG-4 (strain HG-II Rh 184) and AG-5 (Rh 184) were included for comparison with the strains isolated in this study. The references strains were obtained from the collection of A. Ogoshi (kindly provided by S. Naito, Graduate School of Agriculture, Hokkaido University, Sapporo, Japan). Surface-sterilized microscope slides were coated with 2% (w/v) water agar. A plug (diameter 5 mm) from the culture of the test strain and reference strain were placed on the slide 10 mm apart and incubated in a moist chamber at 25°C in the dark. Contacts of hyphae were monitored under a microscope (Laborlux S; Leitz) at ×100 and ×400 magnifications at 24-h intervals. The anastomosing hyphae were carefully traced back to their source plug in order to exclude false positives (self-anastomosis). At least nine hyphal contacts between a pair of strains were analyzed. All pairings were repeated thrice. Anastomosis reactions were classified according to Carling (1996) as C0, no interaction (not related); C2, wall contact and death of anastomosing and adjacent cells (related); and C3, fusion of cell walls and membranes (closely related).
Determination of the Number of Nuclei
Nuclei were counted as described (Martin 1987) in the cells of hyphae of the strains included in the anastomosis tests. The strains were grown on microscope slides at 20°C in the dark for 24–72 h as above, but without growth medium on the slide. Hyphae were fixed in 3% formaldehyde for 2 min and rinsed with distilled water for 1 min. Nuclei were stained with 4′-6-diamidino-2-phenylindole (DAPI; D9542, Sigma) at 1 ppm concentration in distilled water for 5 min in the dark and the slides subsequently rinsed in distilled water. Specimens were studied under a fluorescence microscope (Laborlux S; Leitz) using a DAPI filter. Nuclei were counted in the five cells closest to the hyphal tip in 10 hyphae of three colonies (150 cells in total) of each strain. Data were subjected to the Kruskal–Wallis analysis of variance by ranks, and ranks were separated with Tukey’s multiple range test (Zar 1996).
Results
Compensatory Base Changes in Secondary Structures of ITS2
The structural predictions are sensitive to possible errors in sequence annotation, e.g. possible inclusion of nucleotides from the rRNA genes in the ITS sequences analyzed. Preliminary prediction of the secondary structure for ITS2 RNA using a recently developed homology-based algorithm, as implemented in the web server of the ITS2 database (Wolf et al. 2005a; Selig et al. 2008; http://its2.bioapps.biozentrum.uni-wuerzburg.de), suggested that, indeed, the ITS2 sequences of AG-3 deposited to databases might contain bases of the 28S rRNA gene. The ITS1, 5.8S rRNA and ITS2 sequences for AG-3 were reported to contain 218, 155 and 270 bases, respectively, but the original information used to annotate was difficult to trace. Annotation of ITS2 sequences was done according to the 5.8S–28S rDNA interaction (Côté and Peculis 2001; Keller et al. 2009). The predicted secondary structure was used as criteria to refine the ITS2/28S junction in R. solani AG-3, as shown in Fig. 1. Consequently, the ITS2 sequences of AG-3 strains determined in this study contained 235 bases. Also the previously determined sequences of R. solani in databases should be refined as indicated in Fig. 1.

Annotation of the ITS2 sequences. ITS2 sequences were annotated according to the flanking regions (5.8 and 28S rDNA), which interact during ribogenesis (shown below)
The ITS2 secondary structures as predicted in Rhizoctonia in this study are in accordance with the familiar four-helix domain (Fig. 2) found in a wide range of organisms (Schultz et al. 2005). However, helix III sometimes forms a small sidearm (Fig. 2). The known ITS2 motifs like the U–U mismatch in helix II and the UGGU motif 5′ to the apex of helix III were present (Figs. 2, 3). The structures were compared among the well-characterized anastomosis tester strains representing 12 AGs and their subsets (Table 1). The ITS2 of AG-3 contained one or two CBCs in the sequence–structure alignment as compared with the ITS2 of all other AGs (Table 1). The CBCs are shown in a sequence comparison in Figs. 3 and 4. Results showed that the different subsets of AGs differed in the number of CBCs when compared to AG-3. For example, subsets AG-1-IA and AG-1-IB had two CBCs, whereas AG-1-IC had only one CBC, and subset AG-2-3 had one CBC, whereas the five other subsets of AG-2 had two CBCs (Table 2). An interesting difference between subsets was the existence of two CBCs between the potato and the tobacco strains of AG-3, as illustrated with the anastomosis tester strains PT and TB, respectively (Fig. 3, Table 1).

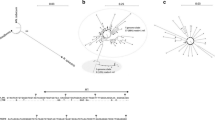
Secondary structure information for ITS2 sequences in different anastomosis groups (AGs) of Rhizoctonia solani and a binucleate Rhizoctonia sp. (strain R92). Above: representation of the complete multiple global sequence–structure alignment including gaps. The four helixes are indicated I–IV. The conserved parts are indicated with colours from the most conserved (>51%; green) to the least conserved (red, not conserved). Nucleotides that are 100% conserved throughout the alignment are highlighted in yellow. Below: the complete multiple global sequence–structure alignment without gap columns. The conserved parts are indicated as above. The common ITS2 motifs are highlighted in yellow. (Color figure online)

Two compensatory base changes in helix I of ITS2 between the AG-3 potato strain (PT) and tobacco strain (TB) derived from the multiple global sequence–structure alignment (shown without gap columns). In the uppermost sequence–structure alignment PT and TB are merged and the four helixes I–IV are indicated. The figures in the middle and at the bottom are the sequence–structure alignments of PT and TB, respectively. The compensatory base changes are highlighted in orange. (Color figure online)

The compensatory base change detected in helix III of the ITS2 secondary structure of a binucleate, unknown Rhizoctonia species (strain R92) as compared to the multinucleate AG-3 potato strain. Only the structure of Rhizoctonia sp. is shown. The CBC is highlighted in orange (electronic version online). Gaps as derived from the global multiple sequence–structure alignment are indicated by a dash. (Color figure online)
The binucleate Rhizoctonia strain R92 of an unknown species (Lehtonen et al. 2008) had a slightly different motif (CGGU) 5′ to the apex of helix III (Fig. 4). When this strain was included for comparison, it was found to have one, two or three CBCs in the sequence–structure alignment as compared to the strains representing 12 AGs of R. solani (Fig. 4).
Phylogenetic Clustering of Rhizoctonia Strains Based on ITS Sequences
Phylogenetic analysis of ITS sequences with representative sequences from the different AGs was carried out to compare the results to those obtained with the CBC analysis. The 80 different ITS sequences available from AG-3 strains in databases showed little variability, except the difference between potato and tobacco strains reported previously (Kuninaga et al. 2000; Ceresini et al. 2002a). It was considered worthwhile to analyze a larger number of potato strains to capture variability which might affect CBC predictions. The sequences of 503 Finnish potato strains of AG-3 were determined following isolation from canker lesions on potato stems (245 strains), stolons (7), roots (12), teleomorphic stage (17), vegetative mycelium (6) on stem bases, from sclerotia on tubers (215) or roots (1). Sampling was done from 31 potato cultivars, with the highest number of samples obtained from Van Gogh (36%), Nicola (9%), Fambo (8%), Saturna (6%) and Idole (5%). A total of 499 samples were collected from 268 potato fields in the mainland and 13 fields from the archipelago of Åland (Ahvenanmaa) in Finland. In addition, one strain was obtained from a stem canker lesion of a potato plant grown in Russian Carelia and three strains were obtained from sclerotia on seed potato tubers imported from The Netherlands.
Direct sequencing of the ITS1–5.8S–ITS2 region amplified from the 503 strains by PCR revealed 73 distinguishable sequences (Supplementary Table 1), of which 26 were reported in a previous study (Lehtonen et al. 2008). These predominant haplotype sequences (see below) revealed by direct sequencing of PCR products were compared with the ITS sequences of 80 strains of AG-3 from other countries, and the ITS sequences of the AG reference strains (Supplementary Table 1) by phylogenetic analysis. The neighbour-joining (Fig. 5) and maximum parsimony methods provided similar grouping of sequences (data not shown). Almost all (497 of 503) Finnish strains were clustered with the AG-3 tester strain PT ST 11-6 from potato and the AG-3 potato strains from other countries (Fig. 5).

Neighbour-joining tree of the 72 different ITS sequences of Finnish AG-3 strains (Supplementary Table 1) of Rhizoctonia solani with comparison to the different AG-3 strain ITS sequences from other countries, and the tester strains and Finnish strains of other anastomosis groups. The tree was rooted using the sequence of the Finnish binucleate, unknown Rhizoctonia sp. (R92), and the nearly identical sequence (AY634128) of a species from Ceratobasidiaceae. The horizontal distances are proportional to the genetic distances (scale indicates Kimura units; Kimura 1980), which does not allow visualization of the smallest differences. Bootstrap values greater than 90 are shown in support of the inferred branches
The AG-3 strains clustered into two host-specific subgroups that contained either the strains from potato or tobacco, respectively, clearly distinguished from each other (Fig. 5). The tobacco strains of AG-3 (Kuninaga et al. 2000; Ceresini et al. 2002a) differed from potato strains at 35–40 positions. All new strains described here were placed in the potato subgroup. The AG-3 cluster did not have groups of strains based on geographic origin although strains from all continents were included (Fig. 5).
Only a few Finnish strains were different from AG-3. Phylogenetic analysis indicated that three strains, R25, 131-L7 and 173-4, isolated from a stem canker lesion, sclerotium and teleomorphic stage on stem, respectively, were related to the previously characterized strains of AG-2-1 (Fig. 5). Strains 239-3 and R96 from stem canker lesions belonged to AG-4 and AG-5, respectively (Fig. 5). The binucleate, non-pathogenic strain R92 of an unknown Rhizoctonia species isolated from a potato stem canker lesion (Lehtonen et al. 2008) was included as an outgroup. The ITS sequence of strain R92 showed the highest identity (99%) to an ectomycorrhiza (AY634128; Bidartondo et al. 2004) (data not shown). The grouping based on differences in the ITS sequences was consistent with the results from anastomosis tests (Supplementary Table 2).
ITS Consists of Sequence Variants in AG-3 Strains
Rhizoctonia solani is known to have multinucleate hyphae but there is little data on the exact number of nuclei. Nuclear counts were carried out on hyphal cells of 11 strains representing the four AGs detected in Finland in this study. All strains were multinucleate (Fig. 6) and contained 3–21 nuclei per cell. There were significant differences in the numbers of nuclei between the strains, including the strains of AG-3 (Kruskal–Wallis test; χ2 = 176.249, df = 10, P < 0.001) (Supplementary Table 3).

Anastomosis (a) and DAPI-staining of nuclei (b) in the hyphae of Rhizoctonia solani AG-3. Arrowheads point to septa
The electropherograms obtained by direct sequencing revealed heterogeneity observed as overlapping double-peaks (two different bases) at the same 10 and 4 positions of the ITS1 and ITS2 sequences, respectively (Table 2). Identical results were obtained from sequencing both strands. The manual evaluation of the electropherograms revealed up to eight unambiguous double-peaks per strain and resulted in 42 different degenerated sequences (b1–b42; Table 2) in 425 (86%) of the 497 AG-3 strains studied. The remaining 72 sequences of the AG-3 strains showed no heterogeneity and contained only one of the nine ‘unisequences’ (a1–a9, Table 2) that were considered as haplotypes of the ITS locus. Of these, 45 sequences were identical (haplotype a1), and the rest represented eight additional haplotypes (Table 2). The majority (88.7%) of the heterogeneity observed in the sequence data could be reconstructed by combining two of the nine haplotypes (Table 2).
To understand the basis for the observed heterogeneity, amplification products were cloned for sequencing from three strains (201-3, 157-2 and 157-1). Strain 201-3 had one of the most common AG-3 ITS sequence types (b12 in Table 2; represented by R102 in Supplementary Table 1), of which the heterogeneity could not be explained by a combination of any of the nine detected haplotypes. This strain showed heterogeneity in 8 of the 14 polymorphic positions. Among the 47 clones sequenced, nine ‘haplotypes’ (H1–H9) were detected. As before, haplotypes were defined by the unique combination of bases detected at the 14 polymorphic positions (Table 3). The most abundant haplotype (H1, 18 clones) (Table 3) was identical to haplotype a4 (Table 2) and was revealed also by direct sequencing of the PCR products of this strain. In contrast, the second most abundant haplotype H2 (14 clones; Table 3) and the remaining seven rare haplotypes were not detected by direct sequencing of the PCR products. Only one of them (H4, identical to a2; Tables 2, 3) was detected in a few other AG-3 strains by direct sequencing. The second strain analyzed (no. 157-2) contained two polymorphic positions as revealed by direct sequencing of the PCR products (similar to R43 in Supplementary Table 1). Among the 21 clones sequenced, all the four possible ‘haplotypes’ were detected (Table 3). Two of them were identical to ‘haplotypes’ H1 and H4 detected in strain 210-3 and two others were new (H10 and H11) (Table 3). The third strain that was analyzed (no. 157-1) represented those 72 strains that showed no ITS sequence heterogeneity as detected by direct sequencing of the PCR products (similar to R120 in Supplementary Table 1). The 15 sequenced clones were identical to ‘haplotype’ H10 of the strain 157-2 (Table 3).
Internal transcribed spacer sequences of the previously studied AG-3 strains have also been obtained by direct sequencing of the PCR products (e.g. Kuninaga et al. 1997; 2000; González et al. 2001; Carling et al. 2002a, 2002b). The electropherograms were not available, but sequence comparisons revealed that differences in 91% of the sequences were confined to the same 14 positions as found in the AG-3 strains characterized in this study, regardless of the geographic origin (data not shown). Only seven sequences had changes in additional ITS positions (data not shown), which did not distinguish them from other AG-3 potato strains in phylogenetic analysis (Fig. 5).
Confinement of major variability to four positions of the ITS2 sequence in AG-3 strains was considered in terms of its possible influence of the ITS2 secondary structures. The analysis for detection of CBCs was repeated. The secondary structure predictions indicated that the alternate bases observed at positions 15, 45, 62 and 128 of ITS2 (Tables 2, 3) did not alter the secondary structure of ITS2. The CBCs reported above were detected in all sequence variants of AG-3 (data not shown).
Discussion
In this study, secondary structures were predicted for the ITS2 transcripts and used to elucidate the relationships between AG-3 and other AGs of R. solani on a novel basis as previously done with other eukaryotic organisms (Müller et al. 2007; Schultz and Wolf 2009). The secondary structures of ITS2 RNA provide a rather simple molecular marker, which is particularly useful when studying closely related species (Müller et al. 2007) and also organisms such as R. solani whose genetics is otherwise difficult to study. These difficulties with R. solani are associated with (i) heterokaryotization and multinucleate cells with 3–21 nuclei per cell detected in the strains examined in this study and 2–19 nuclei in a potato strain of R. solani studied by Sanford and Skoropad (1955); (ii) the lack of clamp connections in hyphae making it impossible to distinguish homokaryons from heterokaryons and to detect mating reactions (Cubeta and Vilgalys 1997) and (iii) difficulty to produce the sexual, teleomorphic stage (T. cucumeris) and basidiospores in vitro (Ceresini et al. 2002b). ITS2 secondary structures provide additional evolutionary information (Coleman 2000, 2003; Coleman and Vacquier 2002; Schultz et al. 2005, 2006; Wolf et al. 2005a, 2005b; Selig et al. 2008). When sufficient evolutionary distance has accumulated to produce one CBC in a relatively conserved pairing position of the ITS2 RNA secondary structure, the taxa differing by the CBC are observed to be sexually incompatible (Coleman and Vacquier 2002), which was found in studies including algae, plants and metazoans (Coleman 2000; 2003; Coleman and Vacquier 2002). This hypothesis has been recently re-examined with over 1,300 closely related organisms using a large collection of 25,000 ITS2 structures (Müller et al. 2007). Results show that the plants and fungi, which differ by a CBC, are different species with a 93% reliability (Müller et al. 2007).
We detected one or two CBCs in the ITS2 of AG-3 potato strains as compared to other AGs, which provided evidence that AG-3 is a distinct species. Phylogenetic analysis of the whole ITS1–5.8S–ITS2 cistron sequences provided results consistent with the CBC analysis of ITS2. The AG-3 strains from potato appeared as a highly homogenous population with no significant differences between strains from the different parts of the world. They were clearly distinguished also from the AG-3 strains from tobacco based on the two CBCs detected between the ITS2 of potato and tobacco strains. Similarly, the tobacco strains of AG-3 were distinguished from all other AGs based on one or two CBCs in ITS2. These results were consistent with phylogenetic grouping of the strains based on the ITS1–5.8S–ITS2 cistron. Hence, based on these criteria, also the tobacco strain of AG-3 can be considered a distinct species.
Historically, strains of R. solani AG-3 from potato have been considered host-specific to potato (Johnk et al. 1993) and were likely to be the fungus described as R. solani by Kühn (1858). Some of the strains infecting tobacco can anastomose with AG-3 strains from potato, but there are also systematic biochemical (Johnk et al. 1993) and genetic differences (Kuninaga et al. 2000) between potato and tobacco strains, and somatic compatibility groups within the groups of potato and tobacco strains of AG-3 (Ceresini et al. 2002a). These results are consistent with those of this study, which indicate that the potato and tobacco strains of AG-3 belong to different species. Furthermore, classification based on anastomosis is not always unambiguous because some ‘bridging strains’ are able to anastomose with strains of at least two AGs (Parmeter et al. 1969; Kuninaga et al. 1978; Carling et al. 2002a). In accordance with these previous results, the strains classified to AGs other than AG-3 based on various biological, biochemical and/or molecular criteria in this study and previous studies were not all placed to phylogenetic clusters that entirely consisted of strains of the same AG. No CBC was detected among them, with the exception of a possible CBC between a strain of AG-2-3 and AG-11 compared. This result was consistent with AG-2-3 being the most different of the six AG-2 subsets analyzed. Finally, one to three CBCs were detected between the ITS2 structures of the multinucleate strains of R. solani and the binucleate, unknown Rhizoctonia species that was isolated from potato and found to be closest related to an ectomycorrhiza (Ceratobasidiaceae) from orchids (Bidartondo et al. 2004).
Taken together, the difficulties with classification of strains of R. solani using the biological, biochemical and molecular criteria available in the past have been widely recognized (Ceresini et al. 2002a, 2002b, 2003; González et al. 2006; Sharon et al. 2008). The results of this study contribute to resolving the R. solani complex by providing novel evidence with strong support from massive analysis of ITS2 sequences, and especially from the CBC analysis. The results indicate that the potato strain of AG-3 should retain the name R. solani, whereas the tobacco strain of AG-3 and other AGs should be named as distinct species. It is, however, emphasized that the lack of a CBC in the ITS2 secondary structure alignment does not indicate that two organisms must belong to the same species. Hence, it is possible that one or several of the other AGs are distinct species. For example, González et al. (2006) utilized indels in sequence alignments as a source of phylogenetic information to reach novel resolution to the polyphyletic R. solani complex and proposed that AG-1, AG-4, AG-6 and AG-8 should be recognized as phylogenetic species. Furthermore, CBC analysis has been recently utilized to further clarify the taxonomy of puffball fungi (Lycoperdaceae; Basidiomycota) (Krüger and Gargas 2008).
This study also reports the largest survey to date of R. solani in a crop species in the field and arguably the most comprehensive analysis of the ITS sequence heterogeneity of R. solani with a special focus on AG-3. The reason for the analysis was to find out whether possible variability of ITS2 sequences would affect the CBCs, which was not the case. Genetic variability of the ITS cistron in AG-3 strains was resolved to a level not previously achieved by analysis of sequences in 497 new AG-3 strains and comparing them with 80 sequences of AG-3 strains available in GenBank. Variability was confined to ten and four positions in ITS1 and ITS2, respectively, in all strains described in this study and in the great majority (91%) of the previously described strains. This variability did not affect the CBCs as discussed above. Thirteen of these 14 variable ITS positions in potato strains were detected by Justesen et al. (2003), who determined the entire ITS1–5.8S–ITS2 cistron in 13 strains and the ITS1 sequence in 45 additional strains of AG-3 from potato in Denmark using direct sequencing similar to our study. They also detected double-peaks in the electropherogram, similar to our data, and therefore determined four individual ITS1 clones from two strains. They concluded that the strains contained two ITS1 sequences. The data of Justesen et al. (2003) support our findings that most strains of AG-3 contain one or two predominant ITS sequences designated as ITS haplotypes defined by the unique combination of bases detected in the 14 polymorphic positions. Additional haplotypes were detected in three AG-3 strains re-tested by sequencing individual clones of the PCR products. The data provide compelling evidence for an unexpectedly wide variability of ITS sequences within individual strains of AG-3. It may be hypothesized that the heterokaryotic nature of multinucleate cells (Butler and Bracker 1970; Sneh et al. 1996; Cubeta and Vilgalys 1997; Rosewich et al. 1999), up to 59 copies of the ITS1–5.8S–ITS2 cistron in the genome of R. solani (Vilgalys and Gonzales 1990) and possible exchange of nuclei during anastomosis of the somatically compatible strains may contribute to the detected heterogeneity. Apparently, concerted evolution (Hillis et al. 1991; Coleman 2003) has not yet homogenized the ITS sequences in R. solani AG-3 for reasons that are not fully understood.
References
Abeyrathne PD, Nazar RN (2005) Parallels in rRNA processing: conserved features in the processing of the internal transcribed spacer 1 in the pre-rRNA from Schizosaccharomyces pombe. Biochemistry 44:16977–16987
Bernard N (1909) L’evolution dans la symbiose des orchidéés et leur champignons commensaux. Annales des Sciences Naturelles de Paris 9 9:1–196
Bidartondo MI, Burghardt B, Gebauer G, Bruns TD, Read DJ (2004) Changing partners in the dark: isotopic and molecular evidence of ectomycorrhizal liaisons between forest orchids and trees. Proc R Soc B 271:1799–1806
Boysen M, Borja M, delMoral C, Salazar O, Rubio V (1996) Identification at strain level of Rhizoctonia solani AG4 strains by direct sequence of asymmetric PCR products of the ITS regions. Curr Genet 29:174–181
Buckler ES, Ippolito A, Holtsford TP (1997) The evolution of ribosomal DNA: divergent paralogues and phylogenetic implications. Genetics 145:821–832
Butler EE, Bracker CE (1970) Morphology and cytology of Rhizoctonia solani. In: Parmeter JR (ed) Rhizoctonia solani: biology and pathology. University of California Press, Berkeley, pp 32–51
Carling DE (1996) Grouping in Rhizoctonia solani by hyphal anastomosis. In: Sneh B, Jabaji-Hare S, Neate S, Dijst G (eds) Rhizoctonia species: taxonomy, molecular biology, ecology, pathology and disease control. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp 37–47
Carling DE, Pope EJ, Brainard KA, Carter DA (1999) Characterization of mycorrhizal strains of Rhizoctonia solani from an orchid, including AG-12, a new anastomosis group. Phytopathology 89:942–946
Carling DE, Baird RE, Gitaitis RD, Brainard KA, Kuninaga S (2002a) Characterization of AG-13, a newly reported anastomosis group of Rhizoctonia solani. Phytopathology 92:893–899
Carling DE, Kuninaga S, Brainard KA (2002b) Hyphal anastomosis reactions, rRNA-internal transcribed spacer sequences, and virulence levels among subsets of Rhizoctonia solani anastomosis group-2 (AG-2) and AG-BI. Phytopathology 92:43–50
Ceresini PC, Shew HD, Vilgalys RJ, Cubeta MA (2002a) Genetic diversity of Rhizoctonia solani AG-3 from potato and tobacco in North Carolina. Mycologia 94:437–449
Ceresini PC, Shew HD, Vilgalys RJ, Rosewich UL, Cubeta MA (2002b) Genetic structure of populations of Rhizoctonia solani AG-3 on potato in eastern North Carolina. Mycologia 94:450–460
Ceresini PC, Shew HD, Vilgalys RJ, Gale LR, Cubeta MA (2003) Detecting migrants in populations of Rhizoctonia solani anastomosis group 3 from potato in North Carolina using multilocus genotype probabilities. Phytopathology 93:610–615
Ciampi MB, Kuramae EE, Fenille RC, Meyer MC, Souza NL, Ceresini PC (2005) Intraspecific evolution of Rhizoctonia solani AG-1 IA associated with soybean and rice in Brazil based on polymorphisms at the ITS-5.8S rDNA operon. Eur J Plant Pathol 113:183–196
Coleman AW (2000) The significance of a coincidence between evolutionary landmarks found in mating affinity and a DNA sequence. Protist 151:1–9
Coleman AW (2003) ITS2 is a double-edged tool for eukaryote evolutionary comparisons. Trends Genet 19:370–375
Coleman AW, Vacquier VD (2002) Exploring the phylogenetic utility of ITS sequences for animals: a test case for abalone (Haliotis). J Mol Evol 54:246–257
Côté CA, Peculis BA (2001) Role of the ITS2-proximal stem and evidence for indirect recognition of processing sites in pre-rRNA processing in yeast. Nucl Acids Res 29:2106–2116
Cubeta MA, Vilgalys R (1997) Population biology of the Rhizoctonia solani complex. Phytopathology 87:480–484
Edson HA, Shapovalov M (1918) Potato stem lesions. J Agric Res 14:213–219
Edwards K, Johnstone C, Thompson C (1991) A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucl Acids Res 19:1349
Felsenstein J (1978) Cases in which parsimony and compatibility methods will be positively misleading. Syst Zool 27:401–410
González D, Carling DE, Kuninaga S, Vilgalys R, Cubeta MA (2001) Ribosomal DNA systematics of Ceratobasidium and Thanatephorus with Rhizoctonia anamorphs. Mycologia 93:1138–1150
González D, Cubeta MA, Vilgalys R (2006) Phylogenetic utility of indels within ribosomal DNA and β-tubulin sequences from fungi in the Rhizoctonia solani species complex. Mol Phylogenet Evol 40:459–470
Gutell RR, Larsen N, Woese CR (1994) Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol Rev 58:10–26
Hillis DM, Moritz C, Porter CA, Baker RJ (1991) Evidence for biased gene conversion in concerted evolution of ribosomal DNA. Science 251:308–310
Johnk JS, Jones RK, Shew HD, Carling DE (1993) Characterization of populations of Rhizoctonia solani AG-3 from potato and tobacco. Phytopathology 83:854–858
Justesen AF, Yohalem D, Bay A, Nicolaisen M (2003) Genetic diversity in potato field populations of Thanatephorus cucumeris AG-3, revealed by ITS polymorphism and RAPD markers. Mycol Res 107:1323–1331
Keller A, Schleicher T, Schultz J, Müller T, Dandekar T, Wolf M (2009) 5.8S–28S rRNA interaction and HMM-based ITS2 annotation. Gene 430:50–57
Kimura M (1980) A simple method of estimating evolutionary rates of base substitution through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kronland WC, Stanghellini ME (1988) Clean slide technique for the observation of anastomosis and nuclear condition of Rhizoctonia solani. Phytopathology 78:820–822
Krüger D, Gargas A (2008) Secondary structure of ITS2 rRNA provides taxonomic characters for systematic studies—a case in Lycoperdaceae (Basidiomycota). Mycol Res 112:316–330
Kühn J (1858) Die Krankheiten der Kulturwachse, ihre Ursachen und ihre Verhutung. Gustav Bosselman, Berlin, p 312
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Kuninaga S, Yokosawa R (1985) DNA base sequence homology in Rhizoctonia solani Kühn. VI. Genetic relatedness among seven anastomosis groups. Ann Phytopathol Soc Jpn 51:127–132
Kuninaga S, Yokosawa R, Ogoshi A (1978) Anastomosis grouping of Rhizoctonia solani Kühn straind from non-cultivated soils. Ann Phytopathol Soc Jpn 44:591–598
Kuninaga S, Natsuaki T, Takeuchi T, Yokosawa R (1997) Sequence variation of the rDNA ITS regions within and between anastomosis groups in Rhizoctonia solani. Curr Genet 32:237–243
Kuninaga S, Carling DE, Takeuchi T, Yokosawa R (2000) Comparison of rDNA-ITS sequences between potato and tobacco strains of Rhizoctonia solani AG-3. J Gen Plant Pathol 66:2–11
Lehtonen MJ, Ahvenniemi P, Wilson PS, German-Kinnari M, Valkonen JPT (2008) Biological diversity of Rhizoctonia solani (AG-3) in a northern potato cultivation environment in Finland. Plant Pathol 57:141–151
Martin B (1987) Rapid tentative identification of Rhizoctonia spp associated with diseased turfgrasses. Plant Dis 71:47–49
Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH (2004) Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci USA 101:7287–7292
Matsumoto T, Yamamoto W, Hirane S (1932) Physiology and parasitology of the fungi generally referred to as Hypochnus Sasakii Shirai I. Differentiation of the strains by means of hyphal fusion and culture in different media. J Soc Tropical Agric 4:370–388
Mazzola M, Smiley RW, Rovira AD, Cook RJ (1996) Characterization of Rhizoctonia strains, disease occurrence and management in cereals. In: Sneh B, Jabaji-Hare S, Neate S, Dijst G (eds) Rhizoctonia species: taxonomy, molecular biology, ecology, pathology and disease control. Kluwer Academic Publishers, Dordrecht, pp 259–268
Menzies JD (1970) Introduction: the first century of Rhizoctonia solani. In: Parmeter JR Jr (ed) Rhizoctonia solani: biology, pathology. University of California Press, Berkeley, pp 3–5
Müller T, Philippi N, Dandekar T, Schultz J, Wolf M (2007) Distinguishing species. RNA 13:1469–1472
Ogoshi A (1996) Introduction—the genus Rhizoctonia. In: Sneh B, Jabaji-Hare S, Neate S, Dijst G (eds) Rhizoctonia species: taxonomy, molecular biology, ecology, pathology and disease control. Kluwer Academic Publishers, Dordrecht, pp 1–9
Parmeter JR Jr, Sherwood RT, Platt WD (1969) Anastomosis grouping among strains of Thanatephorus cucumeris. Phytopathology 59:1270–1278
Rosewich UL, Pettway RE, McDonald BA, Kistler HC (1999) High levels of gene flow and heterozygote excess characterize Rhizoctonia solani AG-1 IA (Thanatephorus cucumeris) from Texas. Fungal Genet Biol 28:148–159
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Evol Biol 4:406–425
Salazar O, Schneider JHM, Julian MC, Keijer J, Rubio V (1999) Phylogenetic subgrouping of Rhizoctonia solani AG 2 strains based on ribosomal ITS sequences. Mycologia 91:459–467
Salazar O, Julian MC, Hyakumachi M, Rubio V (2000) Phylogenetic grouping of cultural types of Rhizoctonia solani AG 2–2 based on ribosomal ITS sequences. Mycologia 92:505–509
Sanford GB, Skoropad WP (1955) Distribution of nuclei in hyphal cells of Rhizoctonia solani. Can J Microbiol 1:412–415
Schillinger WF, Paulitz TC (2006) Reduction of Rhizoctonia bare patch in wheat with barley rotations. Plant Dis 90:302–306
Schultz J, Wolf M (2009) ITS2 sequence-structure analysis in phylogenetics: a how-to manual for molecular systematics. Mol Phylogenet Evol 52:520–523
Schultz J, Maisel S, Gerlach D, Wolf M (2005) A common core of secondary structure of the internal transcribed spacer 2 (ITS2) throughout the Eukaryota. RNA 11:361–364
Schultz J, Müller T, Achtziger M, Seibel PN, Dandekar T, Wolf M (2006) The internal transcribed spacer 2 database—a web server for (not only) low level phylogenetic analyses. Nucl Acids Res 34:W704–W707
Seibel P, Müller T, Dandekar T, Schultz J, Wolf M (2006) 4SALE—a tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinform 7:498
Seibel P, Müller T, Dandekar T, Wolf M (2008) Synchronous visual analysis and editing of RNA sequence and secondary structure alignments using 4SALE. BMC Res Notes 1:91
Selig C, Wolf M, Mueller T, Dandekar T, Schultz J (2008) The ITS2 database II: homology modelling RNA structure for molecular systematics. Nucl Acids Res 36:D377–D380
Sharon M, Kuninaga S, Haykumachi M, Naito S, Sneh B (2008) Classification of Rhizoctonia spp. using rDNA-ITS sequence analysis supports the genetic basis of the classical anastomosis grouping. Mycoscience 49:93–114
Sneh B, Jabaji-Hare S, Neate S, Dijst G (eds) (1996) Rhizoctonia species: taxonomy, molecular biology, ecology, pathology and disease control. Kluwer Academic Publishers, Dordrecht, p 578
Vilgalys R, Gonzales D (1990) Ribosomal DNA restriction fragment length polymorphism in Rhizoctonia solani. Phytopathology 80:151–158
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols—a guide to methods and applications. Academic Press, San Diego, CA, pp 315–322
Wolf M, Friedrich J, Dandekar T, Müller T (2005a) CBCAnalyzer: inferring phylogenies based on compensatory base changes in RNA secondary structures. In Silico Biol 5:291–294
Wolf M, Achtziger M, Schultz J, Dandekar T, Müller T (2005b) Homology modeling revealed more than 20,000 rRNA internal transcribed spacer 2 (ITS2) secondary structures. RNA 11:1616–1623
Zar JH (1996) Biostatistical analysis, 3rd edn. Prentice-Hall, New Jersey
Acknowledgements
We are grateful to M. Rännäli for skillful technical assistance, L. Paulin and P. Kristo for sequence analysis services, and A. Ogoshi and S. Naito for the AG reference strains. This work was funded by the Ministry of Agriculture and Forestry, Finland (grant 4655/501/2003), University of Helsinki, and the following Finnish companies and organizations: Berner Ltd., Chips Ltd., Finnamyl Ltd., Finnish Horticultural Products Society, Finnish Seed Potato Centre Ltd., HG Vilper Ltd., Jepuan Peruna Ltd., Järviseudun Peruna Ltd., Kesko, Kemira GrowHow, Krafts Food Ltd., MTT AgriFood Finland North Ostrobothnian Research Station, ProAgria Oulun maaseutukeskus, Pohjoisen Kantaperuna Ltd., Potwell Ltd., ProAgria Association of Rural Advisory Centers, Ravintoraisio Ltd., Ruokaperuna, Ruokakesko Ltd., Saarioinen Ltd. and Solanum Ltd.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
239_2009_9260_MOESM1_ESM.jpg
{kind=link}
Multiple global sequence-structure alignment (ITS2) of the strains of different anastomosis groups (AGs) of Rhizoctonia solani and a binucleate Rhizoctonia sp. (R92). The sequence length is given after the sequence name (JPG 2,846 kb)
Rights and permissions
About this article
Cite this article
Ahvenniemi, P., Wolf, M., Lehtonen, M.J. et al. Evolutionary Diversification Indicated by Compensatory Base Changes in ITS2 Secondary Structures in a Complex Fungal Species, Rhizoctonia solani . J Mol Evol 69, 150–163 (2009). https://doi.org/10.1007/s00239-009-9260-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-009-9260-3