
Abstract
Multiple independent recruitments of the S-pollen component (always an F-box gene) during RNase-based gametophytic self-incompatibility evolution have recently been suggested. Therefore, different mechanisms could be used to achieve the rejection of incompatible pollen in different plant families. This hypothesis is, however, mainly based on the interpretation of phylogenetic analyses, using a small number of divergent nucleotide sequences. In this work we show, based on a large collection of F-box S-like sequences, that the inferred relationship of F-box S-pollen and F-box S-like sequences is dependent on the sequence alignment software and phylogenetic method used. Thus, at present, it is not possible to address the phylogenetic relationship of F-box S-pollen and S-like sequences from different plant families. In Petunia and Malus/Pyrus the putative S-pollen gene(s) show(s) variability patterns different than expected for an S-pollen gene, raising the question of false identification. Here we show that in Petunia, the unexpected features of the putative S-pollen gene are not incompatible with this gene’s being the S-pollen gene. On the other hand, it is very unlikely that the Pyrus SFBB-gamma gene is involved in specificity determination.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
RNase-based gametophytic self-incompatibility (GSI) has been studied, at the molecular level, in species of the Solanaceae, Plantaginaceae, and Rosaceae families. A single S-locus, containing separate pistil and pollen components, determines specificities differences. To maintain GSI, the two genes should coevolve. For diploid plant species, when the S-haplotype present in the haploid pollen grain matches one of the two S-haplotypes present in the pistil, then the pollen is recognized as “self” and rejected by the pistil (de Nettancourt 1977). In polyploid Prunus species, a single match is also enough to cause pollen rejection (Hauck et al. 2006).
The pistil component in all three families is known to be the S-RNase gene (Roalson and McCubbin 2003). This gene shows the expected features of an S-pistil component, namely, high levels of synonymous and nonsynonymous divergence, as well as enough positively selected amino acid sites to account for the many specificities known to be present in natural populations [between 10% and 20% of all amino acid sites, spread over the protein; for Antirrhinum, however, no positively selected amino acid sites have been identified; J. Vieira et al. 2007]. Evidence for historical recombination has been detected at the S-RNase gene in species of the Solanaceae and Rosaceae families, which is unexpected (Schierup et al. 2001; Wang et al. 2001; Vieira et al. 2003; Ortega et al. 2006). Nevertheless, most recombination events are inferred to be intragenic gene conversion events that only affect small regions of the gene and, thus, do not cause specificity breakdown (Vieira et al. 2003, 2007; Tsukamoto et al. 2008). Using the S-RNase gene, phylogenetic analyses suggest that RNase-based GSI has evolved only once, before the split of the Asteridae and Rosidae, about 120 million years ago (Igic and Kohn 2001; Steinbachs and Holsinger 2002; Vieira et al. 2008a).
The pollen component has been identified as one (in the case of Petunia [SLF], Antirrhinum [SLF], and Prunus [SFB]) or several (in the case of Malus/Pyrus [SFBB] and Nicotiana [DD]) F-box protein coding genes (Lai et al. 2002; Qiao et al. 2004; Kao and Tsukamoto 2004; Sijacic et al. 2004; Entani et al. 2003; Ushijima et al. 2003, 2004; J. Vieira et al. 2008b; Sassa et al. 2007; Cheng et al. 2006). It should be noted that Petunia, Nicotiana, and Antirrhinum belong to the Asteridae, while Prunus and Malus/Pyrus belong to the Rosidae. The lack of correlation between the pollen component being one or several genes and known phylogenetic relationships is unexpected.
The suggestion that the pollen component is composed of several genes in Malus, Pyrus, and Nicotiana is based on the observation that, in the vicinity of the S-RNase gene, there are two or more F-box genes showing pollen-specific expression, linkage disequilibrium with the S-RNase, and low variability levels (Sassa et al. 2007; Wheeler and Newbigin 2007). Indeed, synonymous and nonsynonymous variability levels are similar at the pollen and pistil component genes only in Prunus (Ikeda et al. 2004; Nunes et al. 2006; Vieira et al. 2008b, c; Tsukamoto et al. 2008). Furthermore, so far, only at Prunus SFB have enough positively selected amino acid sites been detected to account for the large numbers of specificities known to be present in natural populations (Nunes et al. 2006; Vieira et al. 2008b, c). Although the low variability levels reported in Malus/Pyrus could somehow be related to the suggestion that there are several pollen genes, in Petunia and Antirrhinum a single pollen gene has been reported, and variability levels at the pollen gene are similarly low.
The reported differences between RNase-based GSI systems motivated the studies of the origin (single vs multiple) of the S-pollen component. Phylogenetic studies are, nevertheless, faced with many difficulties. For instance, the sequences being compared have been independently evolving for, at least, the last 120 million years. Furthermore, F-box genes belong to a large gene family (L. Wang et al. 2004).
The phylogenetic relationship of the S-pollen genes has been addressed using neighbor-joining analyses and mostly small data sets (N = 144, Wang et al. 2004; N = 14, Ushijima et al. 2004; N = 31, Cheng et al. 2006) and maximum parsimony (N = 36, Wheeler and Newbigin 2007). In these studies the pollen S-genes from Antirrhimum (SLFs), Petunia (SLFs), and Malus (SFBBs) grouped together with SLF-like Nicotiana sequences and Prunus SLFLs genes (genes that are not involved in determining pollen specificity; Matsumoto et al. 2008; Vieira et al. 2008d). Prunus SFB alleles fall into one clade that appeared to be more closely related to A. thaliana (a self-compatible species) F-box sequences than to other S-pollen genes. Therefore, Prunus SFB gene has been postulated to diverge early during RNase-based GSI evolution (Wang et al. 2004; Wheeler and Newbigin 2007). Assuming that SLF and SFB genes are involved in GSI, this observation suggests that S-pollen genes may have been independently recruited in RNase-based GSI (Wheeler and Newbigin 2007). Therefore, different mechanisms may be used to achieve the rejection of incompatible pollen in each plant family (Wheeler and Newbigin 2007).
To make the inferences discussed above, nucleotide sequences from very divergent species are being aligned. It is thus conceivable that the alignment algorithm used may affect some of the conclusions. Here we explore this possibility using a nonredundant set of 210 complete nonpseudogene nucleotide F-box S-pollen and S-like sequences and three different alignment schemes. To determine the possible effect of different phylogenetic reconstruction methods, we use both a maximum likelihood and a Bayesian approach. Here we show that both the alignment algorithm used and the phylogenetic reconstruction method greatly influence the inferred relationship of very divergent groups of sequences. In the phylogenies obtained here the sequences reported to be the S-pollen gene in Solanaceae, Plantaginaceae, and Malus/Pyrus cluster together with sequences not involved in specificity determination, as previously reported (Wheeler and Newbigin 2007). Nevertheless, here we argue that this observation does not imply independent recruitments of the S-pollen gene during RNase-based GSI evolution. We also show that, for the Petunia SLF gene, although more sequence data are needed, the findings so far are compatible with this gene being the S-pollen gene. Moreover, we show that the Pyrus SFBB-gamma gene is a very unlikely candidate for an S-pollen gene.
Material and Methods
Datasets and Sequence Alignment
The dicot NCBI plant database was queried using the protein sequence that corresponds to the entry gi29420812 (Prunus mume SLFL3) and TBLASTN, to get the nonredundant set of 209 nucleotide nonpseudogene complete sequences used in this study (Supplementary Table S1). One SLF-like Oryza sativa nucleotide sequence (gi115487495) was added to this set in order to have a putative root when performing phylogenetic analyses. The translated amino acid sequences were aligned using the accurate CLUSTALW algorithm as implemented in DAMBE (Xia and Xie 2001). This amino acid alignment was used as a guide to obtain the corresponding nucleotide alignment. The same set of sequences was aligned using the software T-Coffee (Notredame et al. 2000). Two alignments were created using T-Coffee’s accurate mode and expresso mode. In both modes, the default options of the program were used.
For the purpose of phylogenetic reconstruction, distance-based methods are avoided here since they use information on the distance between the sequences and not the nucleotide states themselves. Thus, to infer the relationship of the 210 complete nucleotide sequences, a fast maximum likelihood method of tree reconstruction, as implemented in GARLI (Zwickl 2006), was used with the default options. The model implemented is the generalized time-reversible (GTR) model of sequence evolution, allowing for among-site rate variation and a proportion of invariable sites. For large data sets containing very divergent sequences this is almost always the best fit model of sequence evolution (Zwickl 2006). Due to computational burden, when using a Bayesian approach (the software MrBayes; Huelsenbeck and Ronquist 2001), a subset of 84 complete nucleotide sequences that represents the most divergent lineages (underlined sequences in Supplementary Table S1) was used. Once more, we use the GTR model of sequence evolution, thus allowing for among-site rate variation and a proportion of invariable sites. Third codon positions may have a gamma distribution shape parameter that is different from that of first and second codon positions. Two simultaneous and completely independent analyses, starting from different random trees, were run for 500,000 generations (each with one cold and three heated chains). Samples were taken every 100th generation and the first 1250 samples were discarded (burn-in).
For the single gene analyses four data sets were used, namely, five Petunia S-RNase sequences, the cognate Petunia S-RNase sequences (Supplementary Table S2), a set of 24 Pyrus SFBB-gamma sequences (stars in Supplementary Table S1), and seven complete Pyrus S-RNase sequences from the same cultivars and annotated as the same specificity as the corresponding SFBB-gamma sequences (Supplementary Table S2). SLF, SFB, and S-RNase amino acid sequences were aligned using ClustalX version 1.64b (Thompson et al. 1997). This alignment was used as a guide to align the corresponding nucleotide sequences. Only two and six alignment gaps of size 3 are present in the Petunia SLF and S-RNase alignment, respectively. All 24 Pyrus SFBB-gamma sequences are of identical size, thus the alignment of these sequences is not ambiguous. In the Pyrus S-RNases data set there are seven alignment gaps.
Population Genetics Estimates
Population genetics estimates (synonymous and nonsynonymous divergence values, linkage disequilibrium, minimum number of recombination events, and Tajima’s D values) were calculated using DnaSP 4.1 (Rozas et al. 2003) software. When calculating divergence values, the Jukes-Cantor correction for multiple hits was used.
Inferences on Positively Selected Amino Acid Sites
For the identification of sites under positive selection we used both the codeml software implemented in PAML 3.13 (Yang 1997) and the method of Wilson and McVean (2006) as implemented in the omegaMap v 0.5 software (www.danielwilson.me.uk) that uses a population genetics approximation.
Using Yang’s (1997) method, maximum-likelihood trees were generated with PAUP* (Swofford 2002) after using Modeltest (Posada and Crandall 1998) to find the simplest model of nucleotide sequence evolution that best fits the data (using the AIC). At each step 100 replicates were used. Starting branch lengths were obtained using the Rogers-Swofford approximation method, and branch-length optimization was performed using the one-dimensional Newton-Raphson method with a pass limit equal to 20. Starting trees were obtained via stepwise addition and tree-bisection-reconnection (TBR) was used as the branch-swapping algorithm. Addition of sequences was random. A molecular clock was not enforced.
For the Petunia SLF sequences the best model is the TVM + I + G model, with base frequencies A = 0.3578, C = 0.2530, G = 0.2027, and T = 0.1865, the rate matrix A ↔ C = 0.5419, A ↔ G = 1.0766, A ↔ T = 0.5614, C ↔ G = 0.8286, C ↔ T = 1.0766, and G ↔ T = 1.0000, a proportion of invariable sites equal to 0.0945, and a gamma distribution shape parameter equal to 0.7677 for variable sites. For Petunia S-RNase sequences the best model is the TVM + G model, with base frequencies A = 0.3504, C = 0.1863, G = 0.2018, and T = 0.2615, the rate matrix A ↔ C = 3.1706, A ↔ G = 6.7285, A ↔ T = 2.9849, C ↔ G = 3.7660, C ↔ T = 6.7285, and G ↔ T = 1.0000, no invariable sites, and a gamma distribution shape parameter equal to 1.2853.
For the Pyrus SFBB-gamma sequences (24) the best model is the HKY85 + G + I model, with base frequencies A = 0.28840, C = 0.17310, G = 0.20960, and T = 0.32890, a transition/transversion ratio of 4.8388, a proportion of invariable sites equal to 0.8039, and a gamma distribution shape parameter equal to 1.0575 for variable sites. For Pyrus S-RNase sequences the best model is GTR + I, with base frequencies A = 0.3195, C = 0.2196, G = 0.2011, and T = 0.2599, the rate matrix A ↔ C = 2.1104, A ↔ G = 3.9279, A ↔ T = 1.7817, C ↔ G = 2.3518, C ↔ T = 2.5177, and G ↔ T = 1.0000, and a proportion of invariable sites equal to 0.4077.
When using the method of Wilson and McVean (2006) we use the same settings as by Nunes et al. (2006) and Vieira et al. (2008b). Briefly, 250,000 iterations were performed and a burn-in of 25,000 was used. Both an objective and a subjective prior specification approach was used and the results were checked for convergence. In the subjective approach the values used as priors reflect the prior belief that there is little recombination in the data set, as by Nunes et al. (2006).
Looking for Correlated Evolutionary Histories
Linear correlations were calculated using K a and K s values and the SPSS Statistics 16.0 software (SPSS Inc., Chicago, IL). The partition homogeneity test was performed, as implemented in PAUP* (heuristic search using as optimality criterion parsimony; 1 million replicates).
Results
Phylogenetic Analyses of SLF-Like Sequences
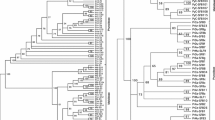
RNase-based GSI is thought to have arisen about 120 million years ago, before the Asteridae-Rosidae split. Thus, to make inferences on the evolution of this system, nucleotide sequences from very divergent species must be compared. Since divergent nucleotide sequences are being compared, the alignment and phylogenetic methods used may affect the conclusions that are made. Therefore, we have used three sequence alignment approaches, as well as two phylogenetic inference approaches (Materials and Methods) for each of the three resulting alignments. The phylogenetic trees obtained are shown in Supplementary Figs. S1 and S2. Based on these phylogenies we define eight sequence groups (Fig. 1; see also Supplementary Table S1). Two hundred five of 210 sequences are always classified in the same group, irrespective of the alignment and phylogenetic method used. Nevertheless, as shown in Fig. 1, the relationships between the different sequence groups differ depending on the alignment and phylogenetic method used. When only amino acid aligned positions with a score >3 are used, as suggested by Notredame et al. (2000), different phylogenetic trees are still obtained when different alignment algorithms are used (data not shown).

Schematic representations of the phylogenetic trees shown in Supplementary Figs. S1 and S2. Panel 1: using 210 complete F-box S-pollen and S-like nucleotide sequences and a fast maximum likelihood method, as implemented in GARLI (Zwickl 2006). Numbers are the percentage of times a given cluster is observed. Panel 2: using 84 complete F-box S-pollen and S-like nucleotide sequences (that represent all most divergent lineages) and a Bayesian approach (Huelsenbeck and Ronquist 2001). Numbers are posterior credibility values >0.60. The sequences were aligned using: (a) T-Coffee’s accurate alignment mode (Notredame et al. 2000); (b) T-Coffee’s express alignment mode (Notredame et al. 2000); and (c) the CLUSTALW algorithm implemented in DAMBE (Xia and Xie 2001). Based on these phylogenies we define eight sequence groups, called A to H
All phylogenies obtained here suggest that putative S-pollen sequences from Petunia, Antirrhinum, and Pyrus/Malus cluster together with sequences not involved in specificity determination, an observation previously made by Wheeler and Newbigin (2007).
Recently, Newbigin et al. (2008) pointed out that, in at least some species, the putative S-pollen gene shows variability patterns and genealogical histories far different than expected for an S-pollen gene (Newbigin et al. 2008). Those authors conclude that either some S-pollen genes have been falsely identified or there is a major problem with theoretical expectations. The possible wrong identification of the S-pollen gene in a given plant family can obviously affect the conclusions about whether this gene has been independently recruited several times during RNase GSI evolution. So far, most expected features have been observed for the Prunus SFB gene only (Ikeda et al. 2004; Nunes et al. 2006; Vieira et al. 2008b). Most importantly, only in Prunus have enough positively selected amino acid sites been reported at both the S-RNase and the SFB gene to account for the many specificities found in natural populations. At present, enough nucleotide sequences are available to perform the needed analyses for the Petunia SLF and Pyrus SFBB-gamma genes only. Those results are presented in the following Results sections.
Variability Levels at the Petunia SLF and S-RNase Genes
Using the set of five Petunia haplotypes available (Supplementary Table S2), synonymous (K s ) and nonsynonymous (K a ) values were calculated for the S-RNase and SLF genes. The average K s value is 0.736 and 0.145 for the S-RNase and SLF genes, respectively. The average K a value is 0.360 and 0.050 for the S-RNase and SLF genes, respectively. The low SLF K s and K a values are unexpected given an estimated proportion of invariable sites of 9.5% for this gene (Materials and Methods). A Tajima’s D value of –0.98 and –0.81 is obtained for the S-RNase and SLF gene, respectively, indicating an excess of rare variants. Nevertheless, for both cases, the Tajima’s D value is not statistically different from that expected under a neutral scenario, under the conservative assumption of no recombination.
The low variability levels at the SLF gene compared with those at the S-RNase gene are suggestive of nonnegligible levels of recombination. Indeed, 158 (1.2%) of the 13,336 possible pairwise comparisons show all four gametic types. Nevertheless, the five S-RNase gene sequences show a similar pattern (680 [4.6%] of 14,878 possible comparisons). The minimum number of recombination events (Hudson and Kaplan 1985) implied by the small data set of five SLF and S-RNase sequences is 12 and 27, respectively. For both the SLF and the S-RNase gene there are no pairs of sites showing significant linkage disequilibrium after Bonferroni correction, although this could be, in principle, accounted for by the small sample size. The average level of linkage disequilibrium is lower for SLF (Kelly’s 1997; Zns = 0.273) than for the S-RNase gene (Zns = 0.350).
Testing Whether the Petunia S-RNase and SLF Evolutionary Histories Are Compatible
The difference in average K s values between the SLF and the S-RNase genes is unexpected under the assumption that the two genes are linked, i.e., that they share a common evolutionary history (see Variability Levels at the Petunia SLF and S-RNase Genes, above). Thus, to test whether the Petunia S-RNase and SLF evolutionary histories are compatible, using the set of five available Petunia haplotypes, synonymous (K s ) and nonsynonymous (K a ) divergence values were calculated for all S-RNase pairwise comparisons and for the corresponding SLF pairwise comparisons, as performed by Nunes et al. (2006). For K s the Pearson correlation coefficient is –0.303 (n = 10; p > 0.05). Similar results were obtained for K a (the Pearson correlation coefficient is –0.372; n = 10; p > 0.05). Therefore, the relative allele ages of the two genes are not correlated (Fig. 2). When the partition homogeneity test is performed a highly significant value is obtained (p < 0.000001), indicating that the S-RNase and SLF evolutionary histories are incompatible.

Synonymous divergence values for the Petunia SLF and corresponding S-RNase gene
Evidence for Positively Selected Amino Acid Sites at the Petunia SLF and S-RNase S-Haplotype Genes
Under the assumption that the maximum-likelihood tree obtained here reflects the true evolutionary relationship of the five SLF sequences, the simpler model that best fits the data is model M2 (three classes, one of them with K a /K s >1; Supplementary Table S3). This assumption may be unrealistic, since the analyses presented under Variability Levels at the Petunia SLF and S-RNase Genes and Testing Whether the Petunia S-RNase and SLF Evolutionary Histories Are Compatible (above) suggest nonnegligible levels of recombination. Eighty-one and eight-tenths percent of the codons fall into a class with K a /K s <1 (purifying selection), 17.9% fall into a class with K a /K s = 1 (neutrally evolving) and 0.3% of the codons (site 293) fall into a class with K a /K s = 12.4 (strong positive selection). For the latter amino acid site the posterior probability of selection is >95% (Table 1). At this site there are five different amino acids, all charged (E, D, R, K, and H). When using the 95% criterion and the Wilson and McVean (2006) method (using both the subjective and the objective prior specification approaches), no amino acid sites are detected as being positively selected. Nevertheless, this method assumes, for instance, that the alleles sampled are a random sample of the alleles present in the population, and this may not be true. Violating this assumption may affect the conclusions that are made. Site 293 has a posterior probability of selection of 79% and 76% using the objective and subjective prior specification approach, respectively. Overall there is weak evidence for one positively selected amino acid site at the reported Petunia pollen gene, but this can be attributed to the small sample size (five sequences only). It is therefore imperative to obtain further sequences for this gene.
When Yang’s (1997) method is applied to the five S-RNase sequences of the corresponding SLF- S-haplotypes, the best model that fits the data is M2 (three classes, one of them with K a /K s >1; Supplementary Table S3). Only two amino acid sites show a posterior probability of selection >95% (Table 1). When using the 95% criterion and the Wilson and McVean (2006) method (using both the subjective and the objective prior specification approach), no amino acid sites are detected as being positively selected. This is in contrast with 13 sites previously identified using the same methodology and 64 Solanaceae S-RNase sequences (Vieira et al. 2007).
Variability and Recombination Levels at the Pyrus SFBB-gamma Gene
The Pyrus SFBB-gamma nucleotide sequences (14, 8, and 2 from P. pyrifolia, P. bretschneideri, and P. sinkiangensis, respectively) are of identical size, thus the alignment is not ambiguous. Although there is no information about these sequences other than that in GenBank, it is known that in all cases but two, they are from different cultivars. In the phylogenetic analyses they always cluster together with high support. The average K s is 0.024 and the average K a is 0.008, therefore we assumed that they are alleles of the same gene. There is an excess of rare variants as indicated by a negative Tajima’s D (1989) value of –1.54, although this value is not statistically different from that expected under a neutral scenario under the conservative assumption of no recombination.
The low K s and K a values are compatible with an estimated proportion of invariable sites of 0.8039 (Materials and Methods). Although there are no Pyrus polymorphism reference values, such low levels of variation suggest that the Pyrus SFBB-gamma gene experiences nonnegligible levels of recombination. Indeed, 118 (3.9%) of the 3003 possible pairwise comparisons show all four gametic types. The minimum number of recombination events (Hudson and Kaplan 1985) implied by the sequence data is six. There are only five pairs of sites showing significant linkage disequilibrium after Bonferroni correction (sites 6 and 29, 864 and 885, 864 and 918, 885 and 918, and 1135 and 1153), and the average level of linkage disequilibrium is low (Kelly’s 1997; Zns = 0.063).
Evidence for Positively Selected Sites at the Pyrus SFBB-gamma and a Subsample of S-RNase Genes from the Same S-Haplotypes
Assuming that the maximum-likelihood tree obtained here reflects the true evolutionary relationship of the 24 Pyrus SFBB-gamma sequences, the simpler model that best fits the data is model M2 (three classes, one of them with K a /K s >1; Supplementary Table S3). Ninety-six and six-tenths percent of the codons fall into a class with K a /K s <1 (purifying selection), 0% fall into a class with K a /K s = 1 (neutrally evolving), and 3.4% of the codons fall into a class with an average K a /K s = 3.99 (positive selection). For amino acid sites 7, 10, and 349 the posterior probability of selection is >95%. There are three (S, G, and I), two (L and P), and three (R, C and H) different amino acids at sites 7, 10, and 349, respectively.
Using the 95% criterion and the Wilson and McVean (2006) method (using both the subjective and the objective prior specification approach), no amino acid sites are detected as being positively selected. Nevertheless, as stated before, this method assumes that the sampled alleles are a random sample of the alleles present in the population, and this is likely not true for domesticated species such as Pyrus. Violating this assumption may affect the conclusions that are made. Sites 7, 10, and 349 have a posterior probability of selection of 94% and 83%, 89% and 74%, and 19% and 28% under the objective and subjective prior specification approach, respectively. Overall, there is weak evidence for two positively selected amino acid sites (sites 7 and 10) at the Pyrus SFBB-gamma gene.
When Yang’s (1997) method is applied to seven S-RNase sequences of the corresponding SLF- S-haplotypes (S-RNases from the same cultivar, annotated as the same specificity), the model that best fits the data is M2 (three classes, one of them with K a /K s >1; Supplementary Table S3). Twenty-eight amino acid sites show a posterior probability of selection >95% (Table 1). Using the 95% criterion and the Wilson and McVean (2006) method, 21 of those amino acid sites identified by Yang’s (1997) method as being positively selected are also identified using both the subjective and the objective prior specification approach (Table 1).
Discussion
The phylogenetic analyses performed here suggest that few conclusions can be drawn regarding the relationship of F-box S-pollen and S-like sequences from different plant families. Only three sequence groups include sequences from both Asteridae and Rosidae species (Fig. 1), and none of the groups containing S-pollen gene sequences includes sequences from both the Asteridae and the Rosidae species. The Petunia/Nicotiana SLF sequences are not a sister group to the Pyrus/Malus sequences (Fig. 1), as previously suggested (Cheng et al. 2006). Depending on the settings and alignment algorithm used, Prunus SFB sequences are shown as a very divergent group or, alternatively, as a sister group to a group of sequences that include the Petunia, Antirrhinum, and Pyrus/Malus S-pollen genes (with a posterior probability of 77% in Bayesian analyses; Supplemental Fig. S2B). Moreover, depending on the alignment used, the most closely related sequence to Prunus SFB sequences could be either none, a sequence from Vitis vinifera, or a sequence from Populus trichocarpa. The inference of very old lineages that are only observed in a single plant family, despite the larger sample size (namely, the Prunus SFB sequences, a group composed exclusively of A. thaliana sequences, a group composed exclusively of M. truncatula sequences, and a group composed of Malus/Pyrus/Prunus sequences), suggests that it may be impossible to infer the relationship of divergent F-box S-like sequences with certainty. All such groups, however, include Rosidae species only. Despite the availability of large amounts of genome sequencing data for several Rosidae species, such as, A. thaliana, M. truncatula, L. japonicus, V. vinifera, and P. trichocarpa, the known relationships between Rosidae families is rarely retrieved in the phylogenies. Nevertheless, for Asteridae, the known relationships are recovered (Plantaginaceae and Solanaceae sequences). It is, however, difficult to establish the generality of this finding, since fewer data are available for Asteridae species. Given these results and the finding that the S-RNase gene (based on phylogenetic analyses of T2 RNases [Igic and Kohn 2001; Steinbachs and Holsinger 2002; Vieira et al. 2008a]) evolved only once before the Rosidae/Asteridae split, it is not possible to conclude, based on a phylogenetic argument alone, that S-pollen genes have been independently recruited during RNase GSI evolution.
The sequences that cluster together with the putative S-pollen gene in Solanaceae and Malus/Pyrus are sequences not involved in specificity determination. Assuming that these genes are determining pollen GSI, this result could suggest multiple independent recruitment of the S-pollen gene (Wheeler and Newbigin 2007). Nevertheless, it is conceivable that, during evolution, the S-pollen gene has been duplicated. Although the most likely fate of such duplication is pseudogeneization, in some cases the duplicate gene may acquire a new function, thus explaining the observed pattern. It should be noted that the closest neighbors of S-pollen genes are very often genes that are in the immediate vicinity of the S-pollen gene and that they show pollen specific expression, as expected according to the proposed evolutionary scenario. Duplication and acquisition of a new function have been reported for genes that belong to the S-RNase lineage (Igic and Kohn 2001; Vieira et al. 2008a), although the pistil gene is more rarely duplicated than the S-pollen gene. In conclusion, in the absence of further evidence, the default scenario for RNase-based GSI evolution should be that both the pistil and the pollen components evolved only once.
The Petunia S-pollen SLF gene presents low synonymous divergence (0.145; see Results), an observation previously made by Wheeler and Newbigin (2007) and Newbigin et al. (2008). Although there are no good estimates for synonymous diversity levels in Petunia neutrally evolving genes, we used for comparison the average level of synonymous diversity for the Nicotiana alata 48A gene (0.0266; data not shown). It is likely that the value for the SLF gene is indeed high. The pistil and pollen component histories are also apparently uncorrelated. If this is true, in Petunia, historical recombination was enough to uncouple the evolutionary history of the SLF and S-RNase genes. Nevertheless, the SLF variability patterns are still likely affected by the presence of the S-locus in its vicinity due to incomplete linkage. It is conceivable that genetic exchange within the S-RNase or SLF genes rather than between the two genes is the explanation for such a finding. Evidence for intragenic recombination at the Petunia S-RNase gene has been reported before by several authors (Wang et al. 2001; Schierup et al. 2001; Vieira et al. 2003), and here we show evidence for recombination at the Petunia SLF gene. Recombination analysis of 1205 segregating plants showed no recombination between the SLF and the S-RNase gene, although the two genes are about 161 kb apart (Wang et al. 2003). Even very low recombination rates within or between the two genes over a long evolutionary time can, however, have an impact on the inferred allele relationships. A similar scenario has recently been proposed for the Prunus SLF1 gene that is the neighbor gene of the Prunus S-RNase gene (Vieira et al. 2008d) and is not involved in determining S-pollen specificity (Matsumoto et al. 2008).
For the five Petunia SLF and S-RNase S-haplotype genes, there is evidence for a few positively selected amino acid sites, using Yang’s (1997) method only. Nevertheless, when similar analyses were performed using 64 Solanaceae S-RNase sequences, 13 positively selected amino acid sites were identified (Vieira et al. 2007). Therefore, many more Petunia S-pollen SLF gene sequences are needed to determine the number of positively selected amino acid sites with a high degree of confidence.
The other unexpected feature of Petunia SLF is that it is expressed in pollen but not at the expected time (Sijacic et al. 2004). The expression of an S-pollen gene is expected to be maximal during pollen tube growth, and not at anthers stage 4, as observed for Petunia SLF gene. The transcript abundance may not, however, reflect protein levels, and in vitro germination may not reflect the situation in vivo (Sijacic et al. 2004). Nevertheless, it is clear that this is the gene responsible for competitive interaction in Petunia (Sijacic et al. 2004; Tsukamoto et al. 2005) and it has been assumed, although never decisively proven, that the gene responsible for competitive interaction is the same as the S-pollen gene. It should be noted that polyploidy in Prunus, and therefore duplication of the S-pollen gene, does not lead to self-compatibility. Hence, competitive interaction is not a universal feature of S-RNase-based GSI (Hauck et al. 2006; Nunes et al. 2006).
At the Pyrus SFBB-gamma gene there is weak evidence for two positively selected amino acid sites (sites 7 and 10). This is in contrast with the results obtained with fewer S-RNase sequences from the same S-haplotypes. Although, only seven sequences were analyzed, 21 amino acid sites show a high (>95%) posterior probability of being positively selected. Furthermore, the two sites at the Pyrus SFBB-gamma gene are located in the F-box motif region of the protein (Cheng et al. 2006), an a priori unlikely region for specificity determination. Since these were detected with Yang’s method only, they could represent false positives. It is known that violation of the assumptions of this method, such as recombination in the data set, can result in the detection of false-positive sites (Wilson and McVean 2006). For comparison, the F-box region in Prunus shows strong hydrophobicity and no amino acid sites under positive selection (Ikeda et al. 2004). Levels of synonymous and nonsynonymous variability are low at the Pyrus SFBB-gamma gene (the average K s and K a per site is 0.024 and 0.008, respectively). Furthermore, the average level of linkage disequilibrium is also low (Kelly’s 1997; Zns = 0.063). Therefore, there is little evidence for an effect of the S-locus on the patterns of evolution of the Pyrus SFBB-gamma gene. Overall, it seems unlikely that the Pyrus SFBB-gamma gene plays a role in specificity determination, in contrast with what was suggested by Sassa et al. (2007). This conclusion is not dependent on whether the S-pollen component is monogenic or multigenic. Indeed, if the system is monogenic, then enough positively selected amino acid sites must be found in a given gene to account for the number of different specificities found in natural populations, and this is clearly not the case for Pyrus SFBB-gamma gene. If the system is multigenic, then each gene involved in specificity determination will show a fraction of the total number of positively selected amino acid sites needed to account for the number of different specificities found in natural populations. This seems also not to be the case for the Pyrus SFBB-gamma gene.
Overall, three main conclusions are made here regarding the evolution of the S-pollen gene: (i) there is no convincing evidence to suggest that the S-pollen gene was independently recruited multiple times during evolution since it is not possible to address the phylogenetic relationship of F-box S-pollen and S-like sequences from different plant families; (ii) the gene identified as being the Petunia S-pollen shows several unexpected features, although, at present, there is no major reason to suspect that it has been wrongly identified; and (iii) it is very unlikely that the Pyrus SFBB-gamma gene is involved in specificity determination.
References
Castric V, Vekemans X (2004) Plant self-incompatibility in natural populations:a critical assessment of recent theoretical and empirical advances. Mol Ecol 13:2873–2889
Cheng J, Han Z, Xu X, Li X (2006) Isolation and identification of the pollen-expressed polymorphic F-box genes linked to the S-locus in apple (Malus x· domestica). Sex Plant Reprod 19:175–183
de Nettancourt D (1977) Incompatibility in angiosperms. Springer-Verlag, Berlin
Entani T, Iwano M, Shiba H, Che FS, Isogai A, Takayama S (2003) Comparative analysis of the self-incompatibility (S-) locus region of Prunus mume: identification of a pollen-expressed F-box gene with allelic diversity. Genes Cells 8:203–213
Golz JF, Oh HY, Su V, Kusaba M, Newbigin E (2001) Genetic analysis of Nicotiana pollen-part mutants is consistent with the presence of an S-ribonuclease inhibitor at the S locus. Proc Natl Acad Sci USA 98:15372 1536
Hauck NR, Yamane H, Tao R, Iezzoni AF (2006) Accumulation of non-functional S-haplotypes results in the breakdown of gametophytic self-incompatibility in tetraploid Prunus. Genetics 172:1191–1198
Hudson RR, Kaplan NL (1985) Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111:147–164
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Igic B, Kohn JR (2001) Evolutionary relationships among self-incompatibility RNases. Proc Natl Acad Sci USA 98:13167–13171
Ikeda K, Igic B, Ushijima K, Yamane H, Hauck NR, Nakano R, Sassa H, Iezzoni AF, Kohn JR, Tao R (2004) Primary structural features of the S haplotype-specific F-box protein, SFB, in Prunus. Sex Plant Reprod 16:235–243
Kao TH, Tsukamoto T (2004) The molecular and genetic bases of S-RNase-based self-incompatibility. Plant Cell 16:S72–S83
Kelly JK (1997) A test of neutrality based on interlocus associations. Genetics 146:1197–1206
Lai Z, Ma W, Han B, Liang L, Zhang Y, Hong G, Xue Y (2002) An F-box gene linked to the self-incompatibility (S) locus of Antirrhinum is expressed specifically in pollen and tapetum. Plant Mol Biol 50:29–42
Matsumoto D, Yamane H, Tao R (2008) Characterization of SLFL1, a pollen-expressed F-box gene located in the Prunus S locus. Sex Plant Reprod 21:113–121
Newbigin E, Paape T, Kohn JR (2008) RNase-based self-incompatibility: puzzled by pollen S. Plant Cell 20:2286–2292
Notredame C, Higgins DG, Heringa J (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217
Nunes MDS, Santos RAM, Ferreira SM, Vieira J, Vieira CP (2006) Variability patterns and positively selected sites at the gametophytic self-incompatibility pollen SFB gene in a wild self-incompatible Prunus spinosa (Rosaceae) population. New Phytol 172:577–587
Ortega E, Boskovic RI, Sargent DJ, Tobutt KT (2006) Analysis of S-RNase alleles of almond (Prunus dulcis): characterization of new sequences, resolution of synonyms and evidence of intragenic recombination. Mol Genet Genomics 276:413–426
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Qiao H, Wang F, Zhao L, Zhou J, Lai Z, Zhang Y, Robbins TP, Xue Y (2004) The F-box protein AhSLF-S2 controls the pollen function of S-RNase-based self-incompatibility. Plant Cell 16:2307–2322
Roalson EH, McCubbin AG (2003) S-RNases and sexual incompatibility:structure, functions, and evolutionary perspectives. Mol Phylogenet Evol 29:490–506
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Sassa H, Kakui H, Miyamoto M, Suzuki Y, Hanada T, Ushijima K, Kusaba M, Hirano H, Koba T (2007) S locus F-box brothers:multiple and pollen-specific F-box genes with S haplotype-specific polymorphisms in apple and Japanese pear. Genetics 175:1869–1881
Schierup MH, Mikkelsen AM, Hein J (2001) Recombination, balancing selection and phylogenies in MHC and self-incompatibility genes. Genetics 159:1833–1844
Sijacic P, Wang X, Skirpan AL, Wang Y, Dowd PE, McCubbin AG, Huang S, Kao TH (2004) Identification of the pollen determinant of S-RNase-mediated self-incompatibility. Nature 429:302–305
Sonneveld T, Tobutt KR, Vaughan SP, Robbins TP (2005) Loss of pollen-S function in two self-compatible selections of Prunus avium is associated with deletion/mutation of an S haplotype-specific F-box gene. Plant Cell 17:37–51
Steinbachs JE, Holsinger KE (2002) S-RNase-mediated gametophytic self-incompatibility is ancestral in eudicots. Mol Biol Evol 19:825–829
Swofford DL (2002) PAUP*: phylogenetic analysis using Parsimony (*and other methods), version 4.0b10. Sinauer, Sunderland, MA
Thompson J, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX window interface: flexible stategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tsukamoto T, Ando T, Watanabe H, Marchesi E, Kao TH (2005) Duplication of the S-locus F-box gene is associated with breakdown of pollen function in an S-haplotype identified in a natural population of self-incompatible Petunia axillaris. Plant Mol Biol 57:141–153
Tsukamoto T, Potter D, Tao R, Vieira CP, Vieira J, Iezzoni AF (2008) Genetic and molecular characterization of three novel S-haplotypes in sour cherry (Prunus cerasus L.). J Exp Bot 59:3169–3185
Ushijima K, Sassa H, Dandekar AM, Gradziel TM, Tao R, Hirano H (2003) Structural and transcriptional analysis of the self-incompatibility locus of almond:identification of a pollen-expressed F-box gene with haplotype-specific polymorphism. Plant Cell 15:771–781
Ushijima K, Yamane H, Watari A, Kakehi E, Ikeda K, Hauck NR, Iezzoni AF, Tao R (2004) The S haplotype-specific F-box protein gene, SFB, is defective in self-compatible haplotypes of Prunus avium and P. mume. Plant J 39:573–586
Vieira CP, Charlesworth D, Vieira J (2003) Evidence for rare recombination at the gametophytic self-incompatibility locus. Heredity 91:262–267
Vieira J, Morales-Hojas R, Santos RAM, Vieira CP (2007) Different positively selected sites at the gametophytic self-incompatibility pistil S-RNase gene in the Solanaceae and Rosaceae (Prunus, Pyrus and Malus). J Mol Evol 65:175–185
Vieira J, Fonseca NA, Vieira CP (2008a) An S-RNase-based gametophytic self-incompatibility system evolved only once in eudicots. J Mol Evol 67:179–190
Vieira J, Santos RAM, Ferreira SM, Vieira CP (2008b) Inferences on the number and frequency of S-pollen gene (SFB) specificities in the polyploid Prunus spinosa. Heredity 101:351–358
Vieira J, Fonseca NA, Santos RAM, Habu T, Tao R, Vieira CP (2008c) The number, age, sharing and relatedness of S-locus specificities in Prunus. Genet Res 90:17–26
Vieira J, Teles E, Santos RAM, Vieira CP (2008d) Recombination at Prunus S-locus region SLFL1 gene. Genetics 180:483–491
Wang L, Dong L, Zhang Y, Zhang Y, Wu W, Deng X, Xue Y (2004) Genome-wide analysis of S-locus F-box-like genes in Arabidopsis thaliana. Plant Mol Biol 56:929–945
Wang X, Hughes AL, Tsukamoto T, Ando T, Kao T (2001) Evidence that intragenic recombination contributes to allelic diversity of the S-RNase gene at the self-incompatibility (S) locus in Petunia inflata. Plant Physiol 125:1012–1022
Wang Y, Wang X, McCubbin AG, Kao TH (2003) Genetic mapping and molecular characterization of the self-incompatibility (S) locus in Petunia inflata. Plant Mol Biol 53:565–580
Wheeler D, Newbigin E (2007) Expression of 10 S-class SLF-like genes in Nicotiana alata pollen and its implications for understanding the pollen factor of the S locus. Genetics 177:2171–2180
Wilson DJ, McVean G (2006) Estimating diversifying selection and functional constraint in the presence of recombination. Genetics 172:1411–1425
Xia X, Xie Z (2001) DAMBE: software package for data analysis in molecular biology and evolution. J Hered 92:371–373
Yang Z (1997) PAML a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Zwickl DJ (2006) Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Ph.D. dissertation. The University of Texas at Austin, Austin
Acknowledgments
This work was partially funded by Fundação para a Ciência e Tecnologia (FCT; research project POCI/BIA-BDE/59887/2004, funded by POCI 2010 and cofunded by FEDER funds). Nuno A. Fonseca is the recipient of postdoctoral grant SFRH/BPD/26737/2006 from the FCT.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Vieira, J., Fonseca, N.A. & Vieira, C.P. RNase-Based Gametophytic Self-Incompatibility Evolution: Questioning the Hypothesis of Multiple Independent Recruitments of the S-Pollen Gene. J Mol Evol 69, 32–41 (2009). https://doi.org/10.1007/s00239-009-9249-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-009-9249-y