
Abstract
Phenazines are secondary metabolites with broad-spectrum antibiotic activity against bacteria, fungi, and eukaryotes. In pseudomonad species, a conserved seven-gene phenazine operon (phzABCDEFG) is required for the conversion of chorismic acid to the broad-spectrum antibiotic phenazine-1-carboxylate. Previous analyses of genes involved in phenazine production from nonpseudomonad species uncovered a high degree of sequence similarity to pseudomonad homologues. The analyses undertaken in this study wished to eluciadate the evolutionary history of genes involved in the production of phenazines. Furthermore, I wanted to determine if the phenazine operon has been transferred through horizontal gene transfer. Analyses of GC content, codon usage patterns, frequency of 3:1 dinucleotides, sequence similarities, and phylogenetic reconstructions were undertaken to map the evolutionary history of phenazine genes from multiple bacterial species. Patchy phyletic distribution, high sequence similarities, and phylogenetic evidence infer that pseudomonad, Streptomyces cinnamonensis, Pantoea agglomerans, Burkholderia cepacia, Pectobacterium atrosepticum, Brevibacterium linens, and Mycobacterium abscessus species all contain a phenazine operon which has most likely been transferred among these species through horizontal gene transfer. The acquisition of an antibiotic-associated operon is significant, as it may increase the relative fitness of the recipient species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Horizontal gene transfer (HGT) can be defined as the nonvertical transmission of genes between different strains or species. HGT between different bacterial species can act as an evolutionary factor driving genetic diversity (Jain et al. 2003; Ochman et al. 2000). Gene transfer can alter the biochemical properties of a recipient organism and has the potential to create functional novelties allowing for the exploitation of unfamiliar environments (Ochman et al. 2000). HGT has been associated with the gain of genes that confer the ability to catabolize certain amino acids that are important virulence factors (Martin et al. 1998) and, also, the acquisition of drug resistance by benign bacteria (Woo et al. 2003). Large-scale genomic sequencing of prokaryotes has revealed that gene transfer is an important evolutionary mechanism for these organisms (Eisen 2000; Jain et al. 2003). It has been suggested that HGT is so rampant, that the prokaryote tree of life is more net-like than tree-like (Doolittle 1999). However, there is much debate as to whether lateral gene transfer is an ubiquitous influence throughout prokaryotic genome evolution. Some authors suggest that alternative explanations such as gene loss, gene duplication, and the segregation of paralogues are incorrectly described as HGT events (Kurland et al. 2003).
This analysis investigated the evolutionary history of an operon normally associated with the production of secondary metabolites (phenazines) in pseudomonad species. Phenazines are biologically active, water-soluble chorismic acid derivatives (Giddens et al. 2002; Mavrodi et al. 1998). They are produced by diverse eubacteria, including members of the Streptomyces, Pseudomonas, Pelagiobacter, Vibrio, Erwinia, and Burkholderia genera (Imamura et al. 1997; Sato et al. 1995; Turner and Messenger 1986), and are often excreted at high levels during bacterial growth in vitro (Mavrodi et al. 2006). Phenazines have a characteristic tricyclic ring system and have been shown to confer a selective growth advantage to organisms which secrete them, as they possess broad-spectrum antibiotic activity toward bacteria, fungi, and eukaryotes (Blankenfeldt et al. 2004). Phenazine antibiotic activity is linked to their ability to undergo oxidation-reduction transformations leading to the accumulation of toxic superoxide radicals in target cells (Hassett et al. 1993).
In pseudomonad species, the best-studied phenazine producers, a conserved seven-gene phenazine (phz) operon (phzABCDEFG) is required for the conversion of chorismic acid to the broad-spectrum antibiotic phenazine-1-carboxylate (PCA) (Mavrodi et al. 1998, 2001; Parsons et al. 2004b). Genes belonging to this operon are referred to as core phenazine genes in this analysis. In P. aeruginosa PAO1, the phz operon is present in duplicate, each copy possessing the ability to produce PCA (Mavrodi et al. 2001). Initial Southern hybridization studies showed that the phz operon was present in 21 phenazine producing pseudomonads, but not in 7 phenazine producing isolates of Burkholderia and Brevibacterium (Mavrodi et al. 2001). This led to the supposition that the phz operon is conserved in pseudomonads and may be unique to them (Mavrodi et al. 2001). However, subsequent analyses have located phz-like operons in phenazine producing species such as Streptomyces cinnamonensis, Pantoea agglomerans, Burkholderia cepacia, and Pectobacterium atrosepticum species (Bell et al. 2004; Giddens et al. 2002; Haagen et al. 2006; Mavrodi et al. 2006).
This analysis set out to elucidate the evolutionary history of phz operons from a diverse range of bacteria. I also wished to determine if there is evidence that any of the sequenced bacteria in GenBank contain the phz operon.
The overall patchy phyletic distribution, aberrant phylogenies, evidence of gene linkage, and high sequence similarities provide evidence that multiple components of a core phenazine biosynthesis operon found in pseudomonad, Streptomyces cinnamonensis, Pantoea agglomerans, Burkholderia cepacia, Pectobacterium atrosepticum, Brevibacterium linens, and Mycobacterium abscessus species shares a similar evolutionary history. Furthermore, it is possible that the phz operon disseminated from one of these species to the others via HGT.
Materials and Methods
Sequence Data and Database Searches
Amino acid sequences from all completely sequenced prokaryotic genomes were obtained from the NCBI ftp site (release 165). The genomes utilized and their taxonomic affiliations are listed in Supplemental Table 1. Complete bacterial genomes were utilized so as to ensure potential phenazine operons could be located. Draft protein-coding genes for Brevibacterium linens BL2 were downloaded from the DOE Joint Genome Institute. Phenazine gene clusters for Pseudomonas aeruginosa PAO1 (AF005404), Pantoea agglomerans Eh1087 (AF451953), and Streptomyces cinnamonensis (AM384985) were obtained from GenBank. All sequence data were merged to produce a local database.
Seven phenazine genes (phzABCDEFG) from a Pseudomonas aeruginosa PAO1 phz operon were used as database query sequences. Taking one phz protein at a time, gene families were located using the BlastP algorithm (Altschul et al. 1997) with a cutoff expectation (E) value of 10−20.
Accession numbers for all sequences used in this analysis are given by Supplemental Table 2.
Phylogenetic Methods
Gene families were aligned using MUSCLE (v3.6) (Edgar 2004), with the default settings.
Obvious alignment ambiguities were manually corrected. Phylogenetic relationships were inferred for full and representative datasets using maximum likelihood methods. Appropriate protein models of substitution were selected for each gene family using ModelGenerator (Keane et al. 2006). Optimum models and associated parameters for all protein families are summarized in Supplemental Table 3.
One hundred bootstrap replicates were then carried out with the appropriate protein model using the software program PHYML (Guindon and Gascuel 2003) and summarized using the majority-rule consensus method.
Bayesian and LogDet phylogenies based on reduced datasets were also reconstructed for selected gene families (phzA/B and phzE). Bayesian phylogenies were reconstructed with the software PhyloBayes version 2.3, which incorporates the heterogeneous CAT site model (Lartillot and Philippe 2004). Markov chains were run for 40,000 cycles, discarding the first 10,000 points and then saving a point every 5 cycles. The “bpcomp” command from PhyloBayes was used to ensure that Bayesian trees had converged. A neighbor-joining tree based on amino acid LogDet distances (Lockhart et al. 1994) was reconstructed using LDDIST (Thollesson 2004), the fraction of invariant sites was estimated by the method of Sidow et al. (1992), and these were excluded. Support for groups on trees were determined using the bootstrap resampling technique.
The approximately unbiased test of phylogenetic tree selection (Shimodaira 2002) was performed, to assess whether differences in topology between constrained and unconstrained gene trees are no greater than expected by chance.
Dinucleotide Frequencies
The distribution of 3:1 dinucleotide frequencies in the whole genomes of Burkholderia cepacia R18194, Pectobacterium atrosepticum SCRI1043, Mycobacterium abscessus, Brevibacterium linens BL2, and a representative pseudomonad (P. aeruginosa PA01) were determined with CODONW (http://codonw.sourceforge.net). The Spearman rank correlation coefficient (Snedecor and Cochran 1995) was used to assess the pairwise covariation of the 3:1 dinucleotide frequencies.
Codon Usage
To determine if phz genes had a different codon usage pattern to their host genome, an analysis of variation in synonymous codon usage was undertaken using the GCUA software (McInerney 1998). Individual correspondence analyses of raw codon counts for the genomes of B. cepacia, P. atrosepticum, B. linens, M. abscessus, and a representative pseudomonad (P. aeruginosa PA01) were performed, with the first four principal axes being used to evaluate synonymous codon usage patterns. Similar analyses were also carried out on the phz gene families displayed in Supplemental Figs. 3–7.
Results
Blast Analysis
Using the seven genes (phzA-G) from a Pseudomonas aeruginosa PAO1 phz operon as database query sequences, phz gene families were located in the bacterial database used in this analysis. Examination of result files showed that many species contain various phz homologues. The complete genomes of B. cepacia, P. atrosepticum, B. linens, and M. abscessus were found to contain at least four of the seven phz genes (Table 1). Furthermore, these genes were localized to specific chromosomal regions, suggesting that they may form operons. A second round of database searches showed that the phz genes within these organisms share relatively high sequence similarity to pseudomonad species (Table 1). Phenazine operons have previously been characterized in P. agglomerans and S. cinnamonensis (Giddens et al. 2002; Haagen et al. 2006). Phenazine genes from these also share a high sequence similarity to pseudomonad species (Table 1).
Therefore, an initial Blast analysis suggests that core phz genes in Pseudomonad, S. cinnamonensis, P. agglomerans, B. cepacia, P. atrosepticum, B. linens, and M. abscessus species are homologous and, due to their relatively high sequence similarity (Table 1), may share a common evolutionary history.
Chromosomal Organization of Phenazine Genes
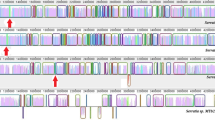
The chromosomal locations of phz genes from S. cinnamonensis, P. agglomerans, B. cepacia, P. atrosepticum, B. linens, and M. abscessus were determined and their organization was compared to that of a characterized P. aeruginosa phz operon (Mavrodi et al. 2001).
S. cinnamonensis contains six of the seven-core phenazine biosynthesis genes found in P. aeruginosa. A phzF homologue is absent in S. cinnamonensis. Ignoring this and the position of one of the phzA/B homologues, the order of the remaining phz genes is identical to that in P. aeruginosa (Fig. 1).

Comparison of phenazine loci from multiple bacterial species with P. aeruginosa PAO1. Gene names are shown within gene boxes. Homologues are also color-coded. B. linens and M. abscessus have additional homologues (not shown) but they are not linked to the putative phenazine operon
P. agglomerans contains 16 genes required for the production of a phenazine compound (d-alanylgriseoluteic acid) (Giddens et al. 2002). Homologues for five of the core pseudomonad phenazine producing genes are present within this cluster. P. agglomerans does not contain a homologue of phzC and has a single copy of phzA/B. The organization of the remaining phz genes is analogous to that observed in P. aeruginosa (Fig. 1).
B. cepacia contains six of the seven phz genes found in P. aeruginosa. It is missing one of the phzA/B homologues (Fig. 1). Overall the organization of the phz homologues in B. cepacia resembles that of P. aeruginosa (Fig. 1). The notable exception is the chromosomal position of phzC, which is separated from the remaining phenazine genes by two open reading frames (Fig. 1).
P. atrosepticum contains five core phenazine biosynthesis genes. It contains one phzA/B copy and is missing phzC. The organization of the phenazine genes also resembles that of P. aeruginosa (Fig. 1). There is a predicted open reading frame (ECA2700) separating phzE and phzF. However, this is most probably a false gene call, as it is short (116 amino acids) and has no significant database hits in GenBank.
B. linens contains seven phenazine homologues. Two copies of phzD were located. Three of the phenazine genes (phzA/B, phzD, and phzE) are found in close proximity to one another. However, the organization of this putative phz operon differs to what is observed in P. aeruginosa (Fig. 1). The remaining phenazine homologues (phzC, phzF, phzG, and the second copy of phzD) are not linked and are dispersed in the B. linens genome (not shown).
M. abscessus has six phz biosynthesis homologues. It has two copies of phzC and phzD. Three of the phenazine homologues (phzC, phzD, and phzE) are linked (Fig. 1). An open reading frame (MAB_0296) splits phzC and phzD; otherwise their organization is similar to that of P. aeruginosa (Fig. 1). The remaining homologues (phzG and the second copies of phzC and phzD) show no evidence of linkage (not shown).
GC Content
The complete genomes of B. cepacia, P. atrosepticum, B. linens, and M. abscessus are available. GC content analysis for individual phenazine genes from these species and a representative pseudomonad (P. aeruginosa PA01) shows that in general they are in the range observed for the global GC content of their corresponding genome (Table 2). This suggests that if the phenazine biosynthesis genes in these species did originate from an alien source, their GC content has ameliorated (so that it now resembles the host genomes) over time (Lawrence and Ochman 1997). A small number of genes are exceptions, however. For example, phzA/B from P. atrosepticum has a GC content ~10% lower than the average of their host genome (Table 2).
The phz core operon from P. agglomerans exhibits a GC content of 44.6%. P. agglomerans is yet to be sequenced, however, its GC is thought to be approximately 56% (Giddens et al. 2002). Such a deviation in GC may be the result of a successful HGT that has not yet ameliorated to their hosts genomes.
The genome of S. cinnamonensis has not been sequenced either. A number of Streptomyces species have been sequenced and their GC contents were found to be ~70% (Bentley et al. 2002; Ikeda et al. 2003; Ohnishi et al. 2008). Based on this, the GC content of the S. cinnamonensis phenazine genes appear to be in the range of other Streptomyces genomes (Table 2).
3:1 Dinucleotide Frequencies
The covariation of 3:1 (the third base of a codon followed by the first base of the proceeding codon) dinucleotide frequencies of the phz genes in B. cepacia, P. atrosepticum, B. linens, M. abscessus, and P. aeruginosa PA01 relative to their corresponding genomes were evaluated using the Spearman rank correlation coefficient (Supplemental Table 4). These nucleotides are subject to the weakest selective constraints, therefore mutational events are tolerated to a greater degree (Rosas-Magallanes et al. 2006). p-values < 0.0001 were observed for the majority of phenazine genes in all organisms (Supplemental Table 4). This infers that the 3:1 dinucleotide frequencies of most phenazine genes display strong covariation with their corresponding genome; this does not suggest evidence of HGT. There are a small number of exceptions, however. p-values of 0.921 and 0.1037 indicate that the 3:1 dinucleotide frequencies of phzA/B and phzD from P. atrosepticum do not exhibit covariation with the 3:1 dinucleotide frequencies of the P. atrosepticum genome (Supplemental Table 4). Interestingly, these three genes also display a deviant GC content relative to the P. atrosepticum genome (Table 2). Similarly, phzA/B from B. linens displays a p-value of 0.1074 for 3:1 dinucleotide frequencies.
Codon Usage
A correspondence analysis of codon usage in the sequenced genomes of B. cepacia, P. atrosepticum, B. linens, and M. abscessus and a representative pseudomonad (P. aeruginosa PA01) was undertaken (Supplementary Fig. 1). This was performed so the codon usage of phz genes could be compared to the global codon usage of their corresponding genomes. Greater distances from the origin correspond to greater differences in codon usage from the mean values. Therefore vertically descended genes such as essential housekeeping genes are expected to cluster around the origin. Conversely recently acquired genes often display an atypical codon preference compared to other genes in the genome and are expected to be found as outliers (Medigue et al. 1991).
The majority of phenazine genes were found to have a codon usage consistent with the rest of their genomes (Supplementary Fig. 1). Two exceptions are the phzA/B and phzD genes from P. atrosepticum (Supplementary Fig. 1). These two genes also show deviant GC (Table 2) and 3:1 dinnucleotide frequencies (Supplementary Table 4) relative to their host genome.
Individual analyses of the variation in synonymous codon usage for all phz gene families were also undertaken (Supplementary Fig. 2). However, there is no evidence of a shared codon usage pattern for core phenazine genes in pseudomonad, S. cinnamonensis, B. cepacia, P. atrosepticum, M. abscessus, or B. linens species (Supplementary Fig. 2).
Phylogenetic Analyses
PhzA/B Phylogeny
The PhzA/B amino acid sequence from P. aeruginosa PAO1 was used as a query to locate bacterial homologues from the database. The exact roles played by PhzA/B in phenazine production are unknown. However, they may help prevent unproductive decomposition of PCA. Furthermore, the overall yield of PCA has been shown to be dependent on PhzA in a dose-dependent manner (McDonald et al. 2001).
Previous analysis has been shown that pseudomonad phzB sequences share a high sequence similarity to phzA homologues and are the result of a recent duplication event (Mavrodi et al. 1998). Therefore, phzB homologues were also included in this phylogenetic analysis. In total 23 phzA/B homologues were located within 11 organisms. Most originated from pseudomonad species. However, homologues were located within the Actinobacteria (S. cinnamonensis and B. linens), the Enterobacteriales (P. agglomerans and P. atrosepticum), and the β-proteobacteria (B. cepacia). S. cinnamonenensis was found to contain two PhzA/B proteins. Homologues were not located in any of the remaining 154 γ-proteobacteria species (Supplementary Table 1) or any other bacterial species represented in the database used in this analysis. P. aeruginosa PA01 is known to contain two phz operons (Mavrodi et al. 2001). Multiple copies of phzA/B were also found in P. aeruginosa UCBPP-PA14, suggesting that this may also contain two phz clusters; all were included for comparative purposes. Maximum likelihood (ML), LogDet, and Bayesian phylogenies were reconstructed from an alignment of all PhzA and PhzB proteins (Fig. 2).

PhzA/B maximum likelihood/LogDet/Bayesian phylogeny. Maximum likelihood and LogDet bootstrap scores are displayed above branches. Bayesian posterior probabilities are shown below selected branches. Pseudomonad PhzA and PhzB homologues are found in separate clades. Nonpseudomonad homologues are branched beside PhzA proteins. According to the AU test this inference is not robust, however
The resultant phylogenies place the P. agglomerans, S. cinnamonenensis, B. linens, B. cepacia, and P. atrosepticum PhzA/B representatives beside the pseudomonad phzB homologues with varying degrees of support (Fig. 2; 100% bootstrap support [BP] and 0.60 Bayesian posterior probability [BPP], respectively.
A constrained tree that grouped pseudomonad phzA and phzB homologues as sister-group taxa, to the exclusion of the nonpseudomonad phzA/B homologues, was reconstructed (not shown). A second constrained tree that grouped the nonpseudomonad taxa with pseudomonad phzA homologues was also reconstructed. The approximately unbiased test of phylogenetic tree selection (AU test) (Shimodaira 2002) showed that the topologies of the unconstrained (Fig. 2) and the two constrained trees (not shown) are not significantly different. Therefore, one cannot confidently infer that the nonpseudomonad organisms represented in this analysis have gained a pseudomonad phzB homologue rather than a phzA copy as inferred by the phylogeny (Fig. 2). However, the patchy phylogenetic distribution coupled with the high sequence similarity suggests that the nonpseudomonad organisms represented in Fig. 2 may have gained a pseudomonad copy of phzA/B through HGT.
Both phylogenies infer that there is strong phylogenetic support showing the differentiation between the two phzB copies in P. aeruginosa PA01 and P. aeruginosa UCBPP-PA14 (Fig. 2; 98% BP and 0.77 BPP). Neither, ML, LogDet, nor Bayesian phylogenies could differentiate between the two phzA genes found in these organisms, however.
PhzC Phylogeny
Using the PhzC amino acid sequence from P. aeruginosa PAO1 as a query sequence, 192 bacterial PhzC homologues from 172 organisms were located. Homologues were confined to the phyla of Actinobacteria and Proteobacteria, except for one Bacteroidetes representative (Salinibacter ruber DSM 13855). B. linens and B. cepacia, S. cinnamonensis, and M. abscessus all contain a homologue. However, homologues were not located in the closely related plant pathogen P. atrosepticum or P. agglomerans.
PhzC encodes a 3-deoxy-d-arabino-heptulosonate-7-phosphate (DAHP) synthase and redirects intermediate products from primary metabolism into phenazine biosynthesis (Mavrodi et al. 1998). DAHP synthase is also the first step in the shikimate pathway and catalyzes the condensation of phosphoenolpyruvate and erythrose-4-phosphate (Mavrodi et al. 1998).
A PhzC ML phylogeny for all 192 homologues was reconstructed (Supplemental Fig. 3). For display purposes a second reduced phylogeny was also reconstructed for representative homologues (Fig. 3).

PhzC maximum likelihood phylogeny. Bootstrap scores are displayed above selected branches. The pseudomonad core PhzC homologues are found in clade A. B. cepacia located in clade C with a number of xanthomonad species. B. linens is clustered with other Actinobacteria in clade D. All nonpseudomonad core Phz proteins are highlighted with arrows
Both M. abscessus and S. cinnamonenensis PhzC proteins are found beside the pseudomonad core PhzC proteins in a strongly supported clade (Fig. 3 and Supplemental Fig. 3; clade A, 100% BP). This inference is consistent with the Blast analysis (Table 1). Pseudomonad species contain multiple DAHP synthase homologues. For example, as well as containing the two phzC genes found in core phz operons, P. aeruginosa PAO1 and UCBPP-PA14 have a third DAHP family member, which groups with other pseudomonad homologues (Supplemental Fig. 3; clade B, 100% BP). This Pseudomonas-specific clade is found within a large proteobacterial clade (Supplemental Fig. 3). Interestingly, the clade (clade A) that contains the pseudomonad core PhzC proteins shares a sister-group relationship with an Actinomycetales-specific clade (Fig. 3 and Supplemental Fig. 3; 90% and 61% BP) to the exclusion of the large proteobacterial-specific clade (Supplemental Fig. 3). The most parsimonious explanation for this inference is that the last common ancestor of the Pseudomonas species represented in this analysis gained a phzC gene from an Actinomycetales source that has been subsequently retained after multiple Pseudomonas speciation events. Interestingly, M. abscessus has a second phzC gene but it is grouped with homologues from other mycobacterial species (Supplemental Fig. 3); this is also consistent with the Blast analysis (Table 1). A constrained phylogeny that places the M. abscessus homologue found in clade A (Supplemental Fig. 3), with the other Mycobacterial species was reconstructed (not shown). The AU test of phylogenetic tree selection, showed that the original unconstrained tree (groups M. abscessus with pseudomonad core PhzC) receives the optimal likelihood tree score, and the difference in likelihood score compared to the constrained tree is significant (p < 0.00001).
Overall there are three likely scenarios to explain this inference.
-
1.
M. abscessus may have obtained the core PhzC protein via HGT from S. cinnamonensis or an unsampled Actinomycetales bacterium.
-
2.
The M. abscessus core PhzC may be the result of a gene duplication followed by a phase of accelerated evolution. The M. abscessus core PhzC gene may have then been transferred to S. cinnamonensis and a pseudomonad ancestral species.
-
3.
Finally, a pseudomonad ancestral species may have originally gained an Actinomycetales copy of phzC from clade D (Fig. 3 and Supplementary Fig. 3) through HGT. This Actinomycetales/Pseudomonas homologue may have consequently been transferred to M. abscessus and S. cinnamonensis via interphyla gene transfer.
The B. cepacia phzC homologue is grouped beside xanthomonad species (Fig. 3 and Supplemental Fig. 3; clade C, 100% BP). This finding is consistent with the Blast analysis that showed Xanthomonas axonopodis to be B. cepacia’s top database hit (Table 1). This Burkholderia/xanthomonad clade does not share a sister-group relationship with either the large proteobacterial- or Actinomycetales-specific clades (Supplemental Fig. 3). Of the 19 complete Burkholderia genomes included in this analysis, only B. cepacia has a phzC homologue. Scenarios to explain this phylogenetic inference include the fact that B. cepacia may have gained phzC from a xanthomonad source. Similarly phzC may have originated through gene duplication in B. cepacia and spread via HGT to an ancestral xanthomonad species, or vice versa. However, a second xanthomonad species Stenotrophomonas maltophila is found in clade C (Supplemental Fig. 3), therefore a single transfer from a xanthomonad donor into B. cepacia is most likely. Interestingly phzC in B. cepacia is separated from the remaining core Burkholderia phenazine genes by two genes (Fig. 1).
The B. linens phzC homologue is grouped beside other actinobacterial species (Fig. 3 and Supplemental Fig. 3; clade D). This inference is again consistent with the Blast analysis that showed actinobacterial species to be B. linens’s top DAHP synthase database hit (Table 1). Based on this phylogeny B. linens phzC is similar to that found in other Actinomycetale species and does not appear to have undergone interphyla gene transfer.
PhzD Phylogeny
Homologues for 107 PhzD proteins from 92 organisms were located through the database search. Homologues were located in the Firmicutes, Actinobacteria, and Proteobacteria. S. cinnamonensis, P. agglomerans, B. cepacia, P. atrosepticum, B. linens, and M. abscessus, all contain a copy of phzD. B. linens actually has two homologues. Therefore the phzD gene is universal to all putative phenazine operons of interest (Fig. 1).
The phzD gene encodes a protein belonging to the isochorismatase enzyme family (Mavrodi et al. 1998). In phenazine biosynthesis PhzD transforms 2-amino-4-deoxychorismic acid to 2,3-dihydro-3-hydroxyanthranilic acid (DHHA). The structure of PhzD is similar to a subfamily of α/β-hydrolase enzymes (Parsons et al. 2003). However, PhzD lacks a nucleophilic cysteine found in its relatives and catalyzes unrelated chemistry (Parsons et al. 2003). All PhzD homologues used in this analysis lack the nucleophilic cysteine. They have the characteristic isochorismatase glycine at the analogous position instead (not shown).
Complete and representative PhzD ML phylogenies were reconstructed (Supplemental Fig. 4 and Fig. 4). The pseudomonad core PhzD proteins are grouped together in a strongly supported clade (Fig. 4 and Supplemental Fig. 4; clade A). A large strongly supported proteobacterial clade is evident and does not include the pseudomonad species (Supplemental Fig. 4; clade B, 88% BP). The B. cepacia, M. abscessus, and S. cinnamonensis isochorismatase enzymes are grouped among the pseudomonad species with moderate support (Fig. 4; 61% BP). The grouping of these three species with the pseudomonad species is unsurprising, as they are >60% identical at the amino acid level (Table 1). Two additional actinobacterial (Thermobifida fusca and Frankia EAN1pec) species are found at the base of this clade. The most parsimonious explanation for the grouping of these proteins (Fig. 4; clade A) involves gene transfers from an actinobacterial donor into the last common ancestor of all pseudomonads and a second independent transfer into B. cepacia. An alternative explanation is that an ancestral pseudomonad species gained an actinobacterial phzD homologue, and this has subsequently been transferred back into a number of actinobacterial species; this would have required at least four independent transfers into actinobacterial species, however.

PhzD maximum likelihood phylogeny. Bootstrap scores are displayed above selected branches. The pseudomonad core PhzD homologues are found in clade A. Nonpseudomonad core Phz proteins are highlighted with arrows. The majority of proteobacterial species represented in this analysis are located in clade B
The B. linens, P. agglomerans, and P. atrosepticum PhzD homologues are grouped beside the pseudomonad representatives, but this inference is poorly supported (Fig. 4 and Supplemental Fig. 4). The weak phylogenetic support for these and other inferences is likely the result of a short PhzD protein (<200 amino acids) with low levels of phylogenetic information. A ML phylogeny was also constructed from a nucleotide equivalent alignment (third positions removed). Similar inferences and support values were also observed (not shown). Therefore based on this phylogeny one cannot confidently elucidate the evolutionary history of the core phzD genes in B. linens, P. agglomerans, and P. atrosepticum.
PhzE Phylogeny
Database searches found homologues for 69 PhzE enzymes in 61 species. The majority of homologues were located in the Proteobacteria (α = 30, γ = 21, β = 1, δ = 2) and Actinobacteria, although members of the Cyanobacteria (Anabaena PCC 7120 and Nostoc punctiforme) and Deinococcus-Thermus (Deinococcus geothermalis and Thermus thermophilus) phyla are also present. Multiple species have paralogous copies of PhzE (Supplemental Table 2). S. cinnamonensis, P. agglomerans, B. cepacia, P. atrosepticum, B. linens, and M. abscessus all contain a copy of phzE. Therefore, as with phzD, the phzE gene is universal to all putative phenazine operons studies in this analysis (Fig. 1). The PhzE protein is approximately three times larger than PhzD (> 600 amino acids); therefore it should be a more reliable phylogenetic marker.
PhzE is involved in the first step of phenazine biosynthesis. At the sequence level it is similar to anthranilate synthase and converts chorismate, the end product of the shikimate pathway, to 2-amino4-deoxychorismic acid anthranilate (McDonald et al. 2001).
ML phylogenies based on all 69 PhzE homologues (Supplemental Fig. 5) and selected representatives were reconstructed (Fig. 5). As with other Phz proteins, the pseudomonad core PhzE enzymes are found in a highly supported clade (Fig. 5 and Supplemental Fig. 5; clade A, 100% BP). The B. cepacia, M. abscessus, and S. cinnamonensis homologues form a moderately supported sister-group relationship with the pseudomonad proteins (Fig. 5; 55% BP). These phylogenetic inferences are consistent with the Blast analysis that showed pseudomonad proteins to be B. cepacia, M. abscessus, and S. cinnamonensis top database hits (Table 1). The phylogeny reconstructed using representative homologues places P. agglomerans, B. linens, and P. atrosepticum in a large clade that contains the pseudomonad PhzE homologues (Fig. 5; 99% BP). This inference contradicts the phylogeny based on all 69 PhzE homologues, which places these three species at the base of clade B (Supplemental Fig. 5). The PhzE proteins from P. agglomerans, B. linens, and P. atrosepticum have a high evolutionary rate illustrated by their long-branch lengths (Fig. 5). Therefore the PhzE phylogeny may be suffering from long-branch attraction artifacts (LBA). To help rule out LBA, Bayesian and LogDet phylogenies based on the reduced dataset were reconstructed (Fig. 5). The Bayesian phylogeny utilized a heterogeneous site model that performs well compared to other methods against phylogenetic artifacts, such as long-branch attraction (Lartillot et al. 2007). The resultant phylogenies infer that the PhzE homologues from P. agglomerans, B. linens, and P. atrosepticum do indeed form a sister-group relationship with the core pseudomonad, B. cepacia, S. cinnamonensis, and M. abscessus PhzE proteins (Fig. 5; clade A, 1.0 BPP and 61% BP).

PhzE maximum likelihood phylogeny. Maximum likelihood and LogDet bootstrap scores are displayed below branches. Bayesian posterior probabilities are shown above selected branches. The pseudomonad core PhzE homologues are found in clade A. Nonpseudomonad core Phz proteins are highlighted with arrows and are also located in clade A. The remaining organisms in clade B are actinobacterial
Interestingly, P. agglomerans, P. atrosepticum, B. cepacia, and Pseudomonas are all proteobacteria. The remaining species grouped in clade B are all Actinobacteria (Fig. 5 and Supplemental Fig. 5). All remaining proteobacterial species are grouped together in a separate clade (Supplemental Fig. 5; clade C, 100% BP). A constrained phylogeny with Proteobacteria- and Actinobacteria-specific clades was reconstructed (not shown). The AU test of phylogenetic tree selection showed that the original unconstrained tree (Supplemental Fig. 5) has a significantly better (p < 0.00001) likelihood score than the constrained phylogeny. There are a number of possible explanations for this finding. For example, an ancestral Actinobacteria may have gained a proteobacterial PhzE from a close relative of one of the proteobacterial species represented in clade B (Fig. 5). More likely is the suggestion that the core phzE gene present in the proteobacterial species (Pseudomonas, B. cepacia, P. agglomerans, and P. atrosepticum) in clade B (Fig. 5 and Supplemental Fig. 5) originated from an Actinomycetale source. However, as with phzC and phzD an interphyla transfer of phzE from an actinobacterial donor into one of the non-Actinobacterial species followed by multiple transfers into the remaining species clustered in clade A (Fig. 5 and Supplemental Fig. 5) is also plausible.
As already mentioned, both phzD and phzE are universal to all putative phenazine operons of interest (Fig. 1). Furthermore, in relative terms they are congruent with one another with respect to the branching order of all species containing a putative phz operon (Figs. 5, 6). However, certain PhzD inferences, in particular, the phylogenetic placement of P. agglomerans, P. atrosepticum, and B. linens beside the other core PhzD proteins is not strongly supported, probably because of the low phylogenetic signal in this protein. Therefore, it would be interesting to determine if there is coevolution between PhzD and PhzE. This hypothesis cannot be tested in an AU framework, however, as both PhzD and PhzE phylogenies have different constituent taxa.

PhzF maximum likelihood phylogeny. Bootstrap scores are displayed above selected branches. All nonpseudomonad core Phz proteins are highlighted with arrows. The pseudomonad core PhzF homologues are found in clade A. B. cepacia, P. atrosepticum, and P. agglomerans form a sister-group relationship with the pseudomonad PhzF homologues (clade A). A number of Burkholderia homologues are also present and are located in clade B. B. linens is grouped with other actinobacterial species in clade C
PhzF Phylogeny
A database search located 114 PhzF homologues in 99 prokaryotic organisms. The majority of PhzF homologues are found in Firmicutes and proteobacterial species. However, phzF genes were also located in Cyanobacteria, Actinobacteria, and a small number of Archaeal organisms (Natronomonas pharaonis and halobacterial species). Homologues were not located in the genomes of M. abscessus or S. cinnamonensis.
PhzF is a key enzyme in the biosynthesis of phenazines. Biochemical analysis has shown that PhzF is the first step in the dimerization of DHAA to to the broad-spectrum antibiotic PCA (Parsons et al. 2004b). It is similar in structure to the lysine biosynthetic enzyme diaminopimelate epimerase but lacks the same catalytic residues (Blankenfeldt et al. 2004). The PhzF active site has been previously characterized (Blankenfeldt et al. 2004; Parsons et al. 2004b), and all PhzF proteins used in this analysis contain the active center residues.
ML phylogenies for full and representative PhzF homologues were reconstructed. A strongly supported core pseudomonad clade is evident (Fig. 6 and Supplementary Fig. 6; clade A, 100% BP). B. cepacia, P. agglomerans, and P. atrosepticum all form sister-group relationships with the pseudomonad phzF homologues (Fig. 6 and Supplemental Fig. 6; clade A, 100% BP). Interestingly homologues from multiple Burkholderia species are also present. These are grouped together in a strongly supported clade that does not include the B. cepacia PhzF protein (Fig. 6 and Supplemental Fig. 6; clade B). A constrained phylogeny that groups all Burkholderia homologues together was reconstructed (not shown). The AU test of phylogenetic tree selection inferred that the original unconstrained tree (Fig. 6) has a significantly better likelihood score than the constrained tree (p < 0.00001). This is consistent with the hypothesis that B. cepacia has gained a PhzF homologue either via HGT or through gene duplication. If it has gained a gene through duplication, this has subsequently been transferred to all other species found in clade A (Fig. 6 and Supplemental Fig. 6). Overall it is not possible to definitely determine the origin of the core phzF copies found in clade A, as it does not form a robust sister-group relationship with other clades (Supplemental Fig. 6). However, based on the strongly supported phylogenetic inference, all PhzF proteins found in clade A have a common evolutionary history (Fig. 6 and Supplemental Fig. 6). Multiple scenarios are possible. For example, the core phzF gene may have originated de novo in pseudomonad species and subsequently transferred via HGT to B. cepacia, P. agglomerans, and P. atroserticum. Conversely the phzF gene may have originated in B. cepacia, P. agglomerans, or P. atroserticum and consequently transferred to all species and the last common ancestor of the pseudomonad species in clade A (Fig. 6 and Supplemental Fig. 6). Another possibility is that all organisms in clade A gained the phzF gene from a bacterial genus that is not represented in the genome database yet. Regardless of the direction of the transfer event, it is clear that all core phzF genes in clade A (Fig. 6) share a common evolutionary history.
The evolutionary history of the B. linens phzF homologue is intriguing. It is found in an Actinomycetales-specific clade (Fig. 6 and Supplemental Fig. 6; clade C), which is itself grouped within a large, predominantly proteobacterial clade (Fig. 6 and Supplemental Fig. 6). Furthermore the Actinomycetales-specific clade is grouped beside a strongly supported α/β-proteobacterial clade (Fig. 6 and Supplemental Fig. 6; clade D). Therefore both PhzE phylogenies (Fig. 6 and Supplemental Fig. 6) led to the conclusion that an ancestral Actinomycetales species most likely gained a PhzE homologue from a proteobacterial source. For completeness a constrained tree that places the B. linens PhzF protein beside the core PhzF copies found in clade A (Fig. 6) was reconstructed (not shown). According to the AU test this constrained phylogeny is significantly worse than the original unconstrained tree (Fig. 6). Therefore phylogenetic analysis infers that B. linens has not acquired its phzF gene from the same donor source as the species found in clade A. Furthermore the B. linens phzF is located outside the putative phenazine operon (Fig. 1). This leads to the hypothesis that the phzF homologue found in B. linens is most likely not a core phzF homologue and has a different evolutionary history relative to B. cepacia, P. atrosepticum, P. agglomerans, and pseudomonad species phzF found in clade A (Fig. 6 and Supplemental Fig. 6).
PhzG Phylogeny
Following database searches, 202 PhzG homologues were located in 192 bacterial species. The majority of PhzG homologues used in this analysis are found in the proteobacterial phylum, although representatives from Actinobacteria, Cyanobacteria, and Deinococcus are also present. PhzG homologues were located in S. cinnamonensis, P. agglomerans, B. cepacia, P. atrosepticum, and M. abscessus. A homologue was not located for B. linens.
PhzG is a flavin-dependent oxidase (Parsons et al. 2004a). Even though the three-dimensional crystal structure of PhzG has been solved, the specific reaction catalyzed by PhzG in phenazine biosynthesis is unknown (Parsons et al. 2004a). Structural data have shown that PhzG is very similar to the gene product (pyriodoxine-5′-phosphate oxidase) of Escherichia coli pdxH. It has been proposed that PhzG arose through duplication of pdxH (Parsons et al. 2004a).
PhzG phylogenies were reconstructed based on full and representative datasets (Supplementary Fig. 7 and Fig. 7). Pseudomonad core PhzG proteins are clustered together and form a strongly supported sister-group relationship with M. abscessus, S. cinnamonensis, P. agglomerans, P. atrosepticum, and B. cepacia (Fig. 7 and Supplementary Fig. 7; clade A). This is in agreement with the initial Blast analysis (Table 1).

PhzG maximum likelihood phylogeny. Bootstrap scores are displayed above selected branches. All nonpseudomonad core Phz proteins are highlighted with arrows. The pseudomonad core PhzG homologues are found in clade A. B. cepacia, P. atrosepticum, P. agglomerans, S. cinnamonensis, and M. abscessus all form a sister-group relationship with the pseudomonad PhzG homologues (clade A). Additional pseudomonad homologues are found in clade B. The majority of Burkholderia species represented in this analysis are found in clade C
Further examination of PhzG phylogenies shows that a second pseudomonad clade is evident (Fig. 7 and Supplemental Fig. 7; clade B). Homologues from multiple Burkholderia species are also present, including a second B. cepacia PhzG homologue (Fig. 7 and Supplementary Fig. 7; clade C). Similarly there is also an actinobacterial-specific clade (Fig. 7 and Supplemental Fig. 7; clade D). Further investigation revealed that all homologues found in clades B, C, and D are actually PdxH proteins. A constrained phylogeny that grouped all Burkholderia species together (including the B. cepacia homologue found in clade A) was constructed (not shown). Similarly, constrained phylogenies that grouped all pseudomonad proteins and all actinobacterial species together were also constructed (not shown). According to the AU test, the original unconstrained tree receives a significantly better likelihood score than all three constrained trees (p < 0.00001). These results show that the core B. cepacia, S. cinnamonens, and M. abscessus and PhzG proteins in clade A do not match their species phylogeny. As with PhzF, multiple scenarios are possible for these inferences, including a de novo origin of PhzF through gene duplication in any one of the species found in clade A, followed by gene transfer to the remaining species in that clade (Fig. 7 and Supplemental Fig. 7).
Discussion
Phenazines are secondary metabolites with broad-spectrum antibiotic activity. In pseudomonad species, a conserved seven-gene operon is involved in the production of these antibiotics from chorismic acid. Initially it was thought that this seven-gene operon was unique to pseudomonad species, however, further analyses have located phz-like operons in a diverse range of phenazine producing bacterial species. Subsequent analyses of phz genes in nonpseudomonad species uncovered a high degree of sequence similarity to pseudomonad homologues. This has led to the hypothesis that the Pseudomonas phz operon may have crossed the species/phylum barrier into other bacterial species via HGT.
In this analysis I wished to eluciadate the evolutionary history of the phz operon and determine if there is evidence of interphylum gene transfer. Comparative analyses were performed on core phenazine genes and their host genomes to determine if common sequence features of HGT could be detected. In general, GC content (Table 2), codon usage (Supplemental Fig. 1), and 3:1 dinucleotide frequencies (Supplemental Table 4) were consistent between the host genome and the core phz genes. A small number of exceptions did exist, however. For example, an analysis of the P. agglomerans phenazine operon revealed an atypical GC content relative to its genome (Table 2). Furthermore, 3:1 dinucleotide frequencies and GC contents of phzA/B and phzD genes of P. atrosepticum are divergent from the rest of its genome (Supplemental Table 4). These two anomalies in sequence composition have been shown to be associated with HGT. P. atrosepticum and P. agglomerans are closely related plant pathogens. If HGT of core phz genes has occurred into these species, it was most likely into their last common ancestor. However, the P. atrosepticum phenazine operon is embedded within its genome, while the P. agglomerans operon is associated with an extra chromosomal plasmid. Operon transfer via plasmids has been observed previously (Rosas-Magallanes et al. 2006). Therefore two independent transfer events cannot be ruled out.
Phylogenetic reconstructions undertaken in this analysis show that the phz operon found in pseudomonad and nonpseudomonad species (Fig. 1) shares a common evolutionary history and has most likely moved between species through HGT. For example Figs. 2–7 show that the majority of phz genes located in the phz operon (Fig. 1) share sister-group relationships; these are summarized in Table 3. For example, with respect to the core pseudomonad phz operon (Fig. 1), five (phzADEFG) of B. cepacia’s six phz genes share a sister-group relationship with pseudomonad core phz genes. Similarly five S. cinnamonensis (phzACDEG), P. agglomerans (phzADEFG), and P. atrosepticum (phzADEFG) core phz genes are grouped among pseudomonad homologues (Table 3). These inferences agree with Blast-based database searches which show that these nonpseudomonad core phz genes have top hits to pseudomonad species and sequence similarity is also relatively high (Table 1). The most parsimonious solution for these observations is that a phenazine operon has independently transferred into these distantly related species. The supposition that phz genes were gained by some species through HGT is credible, as a ISXo8 transposase is found within 2 kb of the M. abscessus phenazine cluster (not shown). Furthermore, a DNA insertion element (transpoase Bcep18194_B1576) is located beside the B. cepacia phz operon and several transposes lie in the vicinity of the B. linens core phz genes (not shown). The presence of transposases in these species is significant, because if their phz operon came from another bacterial donor, the vectors may have facilitated this transfer event.
Determining the phz operon donor species is not a facile task, however. Examination of phz phylogenetic inferences shows that the phz operon has had a checkered history. As has already been mentioned, previous researchers assumed that nonpseudomonad species have gained the phz operon from a pseudomonad source (Giddens et al. 2002). This assertion assumes that the phz operon originated de novo in Pseudomonas, followed by gene transfer to divergent species. If this scenario is the correct one, we would expect to see all phz core genes clustered together in a phylogeny (this is true for the majority of phz genes), and assuming artifact-free trees (e.g., long-branch attraction), the core phz genes should form a sister-group relationship with pseudomonad or closely related proteobacterial paralogues/homologues. This is definitely not the case for phzC, phzD, and phzE, where the corresponding phylogenies infer that the core phz genes have originated from an actinobacterial donor (Figs. 3–5). However, it is striking that most phz core genes display similar orientations (Fig. 1) and phylogenetic histories. Therefore, if we assume that the phz operon has been transferred en masse from a pseudomonad to divergent bacterial species, scenarios such as subsequent transfers back into actinobacterial species can be invoked to explain this hypothesis. However, it is important to note that these explanations are applicable to all species containing a phz operon (Fig. 1). Therefore one cannot confidently determine the donor species.
Interestingly there is one B. cepacia gene that does not share a sister-group relationship with the remaining PhzC proteins. Phylogenetic and Blast-based analyses suggest that B. cepacia gained phzC from a xanthomonad source (Fig. 3, Table 1). The operon structure of B. cepacia is very similar to that of Pseudomonas aeruginosa (Fig. 1). The one exception is the chromosomal position of phzC. The phzC in B. cepacia has been acquired from a xanthomonad source. It would be interesting to know the original integration site of the xanthomonad phzC in the B. cepacia genome. If it were different from its current location (Fig. 1), this would suggest that natural selection is shuffling the gene arrangement of B. cepacia through recombination to bring the xanthomonad phzC into the phenazine operon.
Overall this study has shown that the phz operon found in a number of diverse bacterial genomes has a common ancestry and has most likely moved between species through interspecies/phylum gene transfer. These findings, while novel, are unsurprising, as it has been proposed that genes encoding nonessential catabolic processes are more likely to be transferred than essential housekeeping genes (Jain et al. 1999). The acquisition of an operon associated with antibiotic properties could obviously increase the fitness of the recipient organism.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402 (Online)
Bell KS, Sebaihia M, Pritchard L, Holden MT, Hyman LJ, Holeva MC, Thomson NR, Bentley SD, Churcher LJ, Mungall K, Atkin R, Bason N, Brooks K, Chillingworth T, Clark K, Doggett J, Fraser A, Hance Z, Hauser H, Jagels K, Moule S, Norbertczak H, Ormond D, Price C, Quail MA, Sanders M, Walker D, Whitehead S, Salmond GP, Birch PR, Parkhill J, Toth IK (2004) Genome sequence of the enterobacterial phytopathogen Erwinia carotovora subsp. atroseptica and characterization of virulence factors. Proc Natl Acad Sci USA 101:11105–11110
Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O’Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147
Blankenfeldt W, Kuzin AP, Skarina T, Korniyenko Y, Tong L, Bayer P, Janning P, Thomashow LS, Mavrodi DV (2004) Structure and function of the phenazine biosynthetic protein PhzF from Pseudomonas fluorescens. Proc Natl Acad Sci USA 101:16431–16436
Doolittle WF (1999) Phylogenetic classification and the universal tree. Science 284:2124–2129
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Eisen JA (2000) Assessing evolutionary relationships among microbes from whole-genome analysis. Curr Opin Microbiol 3:475–480
Giddens SR, Feng Y, Mahanty HK (2002) Characterization of a novel phenazine antibiotic gene cluster in Erwinia herbicola Eh1087. Mol Microbiol 45:769–783
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Haagen Y, Gluck K, Fay K, Kammerer B, Gust B, Heide L (2006) A gene cluster for prenylated naphthoquinone and prenylated phenazine biosynthesis in Streptomyces cinnamonensis DSM 1042. Chembiochem 7:2016–2027
Hassett DJ, Woodruff WA, Wozniak DJ, Vasil ML, Cohen MS, Ohman DE (1993) Cloning and characterization of the Pseudomonas aeruginosa sodA and sodB genes encoding manganese- and iron-cofactored superoxide dismutase: demonstration of increased manganese superoxide dismutase activity in alginate-producing bacteria. J Bacteriol 175:7658–7665
Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S (2003) Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol 21:526–531
Imamura N, Nishijima M, Takadera T, Adachi K, Sakai M, Sano H (1997) New anticancer antibiotics pelagiomicins, produced by a new marine bacterium Pelagiobacter variabilis. J Antibiot (Tokyo) 50:8–12
Jain R, Rivera MC, Lake JA (1999) Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci USA 96:3801–3806
Jain R, Rivera MC, Moore JE, Lake JA (2003) Horizontal gene transfer accelerates genome innovation and evolution. Mol Biol Evol 20:1598–1602
Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McLnerney JO (2006) Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol 6:29
Kurland CG, Canback B, Berg OG (2003) Horizontal gene transfer: a critical view. Proc Natl Acad Sci USA 100:9658–9662
Lartillot N, Philippe H (2004) A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol 21:1095–1099
Lartillot N, Brinkmann H, Philippe H (2007) Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol Biol 7(Suppl 1):S4
Lawrence JG, Ochman H (1997) Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol 44:383–897
Lockhart P, Steel M, Hendy M, Penny D (1994) Recovering evolutionary trees under a more realistic model of sequence. Mol Biol Evol 11:605–612
Martin K, Morlin G, Smith A, Nordyke A, Eisenstark A, Golomb M (1998) The tryptophanase gene cluster of Haemophilus influenzae type b: evidence for horizontal gene transfer. J Bacteriol 180:107–118
Mavrodi DV, Ksenzenko VN, Bonsall RF, Cook RJ, Boronin AM, Thomashow LS (1998) A seven-gene locus for synthesis of phenazine–1-carboxylic acid by Pseudomonas fluorescens 2–79. J Bacteriol 180:2541–2548
Mavrodi DV, Bonsall RF, Delaney SM, Soule MJ, Phillips G, Thomashow LS (2001) Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PAO1. J Bacteriol 183:6454–6465
Mavrodi DV, Blankenfeldt W, Thomashow LS (2006) Phenazine compounds in fluorescent Pseudomonas spp. biosynthesis and regulation. Annu Rev Phytopathol 44:417–445
McDonald M, Mavrodi DV, Thomashow LS, Floss HG (2001) Phenazine biosynthesis in Pseudomonas fluorescens: branchpoint from the primary shikimate biosynthetic pathway and role of phenazine-1,6-dicarboxylic acid. J Am Chem Soc 123:9459–9460
McInerney JO (1998) GCUA: general codon usage analysis. Bioinformatics 14:372–373
Medigue C, Rouxel T, Vigier P, Henaut A, Danchin A (1991) Evidence for horizontal gene transfer in Escherichia coli speciation. J Mol Biol 222:851–856
Ochman H, Lawrence JG, Groisman EA (2000) Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304
Ohnishi Y, Ishikawa J, Hara H, Suzuki H, Ikenoya M, Ikeda H, Yamashita A, Hattori M, Horinouchi S (2008) Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J Bacteriol 190:4050–4060
Parsons JF, Calabrese K, Eisenstein E, Ladner JE (2003) Structure and mechanism of Pseudomonas aeruginosa PhzD, an isochorismatase from the phenazine biosynthetic pathway. Biochemistry 42:5684–5693
Parsons JF, Calabrese K, Eisenstein E, Ladner JE (2004a) Structure of the phenazine biosynthesis enzyme PhzG. Acta Crystallogr D Biol Crystallogr 60:2110–2113
Parsons JF, Song F, Parsons L, Calabrese K, Eisenstein E, Ladner JE (2004b) Structure and function of the phenazine biosynthesis protein PhzF from Pseudomonas fluorescens 2–79. Biochemistry 43:12427–12435
Rosas-Magallanes V, Deschavanne P, Quintana-Murci L, Brosch R, Gicquel B, Neyrolles O (2006) Horizontal transfer of a virulence operon to the ancestor of Mycobacterium tuberculosis. Mol Biol Evol 23:1129–1135
Sato A, Takahashi S, Ogita T, Sugano M, Kodama K (1995) Marine natural products. Annu Rep Sankyo Res Lab 47:1–58
Shimodaira H (2002) An approximately unbiased test of phylogenetic tree selection. Syst Biol 51:492–508
Sidow A, Nguyen T, Speed TP (1992) Estimating the fraction of invariable codons with a capture-recapture method. J Mol Evol 35:253–260
Snedecor GW, Cochran WG (1995) Statistical methods, 8th edn. Iowa State University Press, Ames
Thollesson M (2004) LDDist: a Perl module for calculating LogDet pair-wise distances for protein and nucleotide sequences. Bioinformatics (Oxford, England) 20:416–418
Turner JM, Messenger AJ (1986) Occurrence, biochemistry and physiology of phenazine pigment production. Adv Microb Physiol 27:211–275
Woo PC, To AP, Lau SK, Yuen KY (2003) Facilitation of horizontal transfer of antimicrobial resistance by transformation of antibiotic-induced cell-wall-deficient bacteria. Med Hypotheses 61:503–508
Acknowledgments
I wish to thank two anonymous referees whose comments significantly improved the manuscript. I wish to acknowledge the financial support of the Irish Health Research Board (HRB). I also wish to acknowledge the SFI/HEA Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Fitzpatrick, D.A. Lines of Evidence for Horizontal Gene Transfer of a Phenazine Producing Operon into Multiple Bacterial Species. J Mol Evol 68, 171–185 (2009). https://doi.org/10.1007/s00239-009-9198-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-009-9198-5