
Abstract
Changes in gene expression contribute to reproductive isolation of species, adaptation, and development and may impact the genetic fate of duplicated genes. African clawed frogs (genus Xenopus) offer a useful model for examining regulatory evolution, particularly after gene duplication, because species in this genus are polyploid. Additionally, these species can produce viable hybrids, and expression divergence between coexpressed species-specific alleles in hybrids can be attributed exclusively to cis-acting mechanisms. Here we have explored expression divergence of a duplicated heterodimer composed of the recombination activating genes 1 and 2 (RAG1 and RAG2). Previous work identified a phylogenetically biased pattern of pseudogenization of RAG1 wherein one duplicate—RAG1β—was more likely to become a pseudogene than the other one—RAG1α. In this study we show that ancestral expression divergence between these duplicates could account for this. Using comparative data we demonstrate that regulatory divergence between species and between duplicated genes varies significantly across tissue types. These results have implications for understanding of variables that influence pseudogenization of duplicated genes generated by polyploidization, and for interpretation of the relative contributions of cis versus trans mechanisms to expression divergence at the cellular level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The impact of natural selection on regulatory evolution is best understood through comparative analyses—either between different genetic conditions (such as species, populations, hybrids, strains, or tissues) or between different types of genetic information (such as gene expression level, expression divergence, DNA sequence divergence, breadth of expression, developmental timing of expression, or protein-protein interactions). With the aim of better understanding regulatory evolution across species and tissue types, we have examined expression divergence of a simple system—a heterodimer between the recombination activating gene 1 and 2 proteins (RAG1 and RAG2)—that was duplicated by whole-genome duplication (WGD) in African clawed frogs, genus Xenopus. The RAG1/RAG2 heterodimer is required to carry out V(D)J recombination—the somatic shuffling of “variable,” “diversity,” and “joining” cassettes of DNA that makes possible the adaptive immune response (Bassing et al. 2002; Spicuglia et al. 2006).
WGD occurred in Xenopus about 21–41 million years ago (Chain and Evans 2006), or maybe earlier (Evans et al. 2004), generating a tetraploid ancestor with 36 chromosomes. WGD resulted in two copies (paralogs) of all nuclear genes, although tetraploid genomes that descended from this ancestor are mostly or entirely diploidized (Evans 2008). We refer to these WGD paralogs as α and β. Octoploid and dodecaploid Xenopus species also exist. The octoploid species have 72 chromosomes and were formed on three occasions through allopolyploidization of tetraploid ancestors (Evans 2007). In the absence of gene deletion or pseudogenization, these octoploids are expected to have four paralogs of all nuclear genes (α1, α2, β1, and β2) because two paralogs (α and β) were inherited from each of the tetraploid ancestors.
The Xenopus RAG1/RAG2 heterodimer is an interesting system for further study because of the unexpected way in which pseudogenization sculpted interactions between these proteins after WGD. In different species of Xenopus, multiple paralogs of RAG1 experienced pseudogenization in a way that appears biased by ancestry (Evans 2007; Evans et al. 2005). Analysis of multiple species indicates that between 7 and 16 independent instances of pseudogenization occurred in RAG1β, whereas only 2 instances occurred in RAG1α (Evans 2007). In contrast, RAG2α was deleted from the genome prior to the diversification of extant Xenopus species, so their genomes now encode only RAG2β (Evans 2007). One explanation for nonrandom pseudogenization of RAG1β is that low expression of this paralog made it insufficient to carry out all necessary activities on its own after WGD, whereas expression of RAG1α has been both sufficient and therefore necessary after WGD. We call this the “stoichiometric sufficiency of RAG1α“ (SSRα) hypothesis. To test this, we estimated the expression ratio of RAG1 paralogs (i.e., RAG1α/RAG1β) of Xenopus ancestors from observed expression ratios of eight extant species. This heterodimer is particularly active in bone marrow (Greenhalgh et al. 1993). A prediction consistent with the SSRα hypothesis is that, after accounting for gene deletion, RAG1α paralogs would still be more highly expressed than RAG1β paralogs in extant and in ancestral species of clawed frogs (Prediction 1).
We also examined mechanisms of expression divergence in RAG1 and RAG2, focusing our analysis on two tetraploid species, X. laevis (XL) and X. borealis (XB), and hybrids generated by a cross between these species (HXLXB). XL and XB diverged from a common ancestor roughly 20–40 million years ago (Chain and Evans 2006; Evans et al. 2004). On a genetic level, expression divergence is achieved by mechanisms that operate in cis, which affect transcription in an allele-specific manner, and by mechanisms operating in trans, which affect regulation of both alleles via direct or indirect interaction with cis-regulatory elements. Cis divergence is indicated if alleles are divergently expressed in the same cell. Trans divergence is indicated if alleles are differently expressed when in different cells, but not when in the same cell. These mechanisms drive expression divergence between alleles of a single gene, between alleles of genes generated by duplication (paralogs), and between alleles of genes generated by speciation (orthologs).
Divergent expression of orthologs could be a consequence of either cis- or trans-regulatory divergence, or both (Fig. 1). Divergent expression of paralogs that are coexpressed within a cell necessarily implies cis divergence (Fig. 1). However, if each paralog is expressed in a different tissue type within the same individual, then divergent expression between them could also be achieved by paralog-specific trans factors that interact with unique cis-regulatory sequences on each one. The same is true of paralogs that are expressed in different species: expression divergence between paralog α in species 1 and paralog β in species 2 could be achieved by cis or trans divergence, or both (Fig. 1) (Evans 2008). The relative contributions of these mechanisms can be quantified by comparing expression divergence in parental species and hybrid individuals (Fig. 2) (Wittkopp et al. 2004).

Expression divergence can occur between tissues, the sexes, duplicated genes (paralogs), and between species (orthologs) and is derived from cis- and/or trans-acting factors. In the absence of intraspecific polymorphism, intraspecific expression divergence between the sexes and between tissues must be caused by trans-acting factors because the cis-acting factors are identical within a species. An exception, indicated by an asterisk, is intraspecific expression divergence between the sexes that arises due to differences in allelic copy number at sex-linked loci. Because they share the same cellular environment, divergent expression of paralogs (α, β) that are coexpressed arises from cis-acting mechanisms. If paralogs of a gene are expressed in different species (i.e., not coexpressed), expression divergence between them could occur by cis- or trans-acting mechanisms, or both. Likewise, divergence between orthologs in different species (sp.1, sp.2) can arise by cis- or trans-acting factors, or both. (Modified from Evans 2008)
With respect to the mechanisms of regulatory evolution of the RAG1-RAG2 heterodimer in Xenopus, we predicted that the magnitude of cis divergence between orthologs would be tissue-specific (Prediction 2). We reasoned that cis and trans divergence between orthologs should be detectable only to the extent that trans-acting factors that interact with divergent cis-regulatory elements are expressed in a given tissue. Following the same reasoning, we also expected the magnitude of expression divergence between coexpressed paralogs to vary among tissues (Prediction 3). We expected this because different suites of trans-acting factors may be expressed in different tissues within an individual.
Methods
Prediction 1: Upregulation of RAG1α
We used a pyrosequencing assay (described below; Landry et al. 2005; Wittkopp et al. 2004) to quantify the RAG1α/RAG1β expression ratio in eight species of clawed frog, then used these ratios to estimate the expression ratio of their ancestors in a phylogenetic context. The eight species we examined include four tetraploids (X. borealis, X. muelleri, X. laevis, and X. gilli) and four octoploids (X. itombwensis, X. andrei, X. boumbaensis, and X. amieti). The RAG1β2 paralog of three of these octoploids (X. andrei, X. boumbaensis, and X. amieti) was probably deleted, but RAG1β1 is still expressed in each of these species, even though this paralog is degenerate at the coding level due to nonsense and missense mutations (Evans 2007). In X. itombwensis, RAG1β1 and RAG1β2 are both present in the genome, but both are degenerate at the coding level. All eight of these species, however, still express RNA from at least one RAG1α paralog and at least one RAG1β paralog (Evans 2007).
The program BayesTraits (Organ et al. 2007) was used to reconstruct ancestral expression ratios of RAG1α/RAG1β based on the observed log2-transformed expression ratios in the eight extant species. This reconstruction is based on a Brownian motion model of regulatory evolution in which the variance among the descendant expression ratios increases over time. In other words, a long period of evolution between an ancestor and its descendant species is expected to be associated with more variance in the expression ratios in the descendants than a brief period of evolution. The first step in this analysis is to generate a posterior distribution of the variance parameter given the topology and the observed log2-transformed paralogous expression ratios in bone marrow of the eight species (Table 1).We generated this distribution from 5 million Markov Chain Monte Carlo (MCMC) iterations, sampled every 1000 iterations, after a burn-in of 50,000 iterations. Then another MCMC chain was performed using a chain length of 250 million iterations, sampling every 5000 iterations and using the same burn-in, but this time drawing from the posterior distribution of the variance parameter. We report the mean posterior probability of the null SSRα hypothesis—that there is no ancestral upregulation of RAG1α (log2-transformed ratio ≤0)—based on 10 independent MCMC chains. We did not implement topology scaling parameters such as κ, δ, and λ (Pagel 1997, 1999; Venditti et al. 2006) in this analysis because of concerns that estimates of these parameter values may be inaccurate on a small phylogeny (A. Meade, personal communication).
The reticulating evolutionary history of clawed frogs poses unique challenges to comparative analyses because most approaches for analyzing continuous characters within a phylogenetic context rely on a bifurcating evolutionary relationship among species. However, because sequences are available for essentially all paralogs of all species from RAG1 and RAG2 (Evans 2007; Evans et al. 2005), one can construct a bifurcating phenogram based on the average pairwise genetic distance between each orthologous pair of paralogs and then use this topology for comparative analysis. To accomplish this, an ultrametric phylogeny was estimated with a strict molecular clock enforced based on combined data from RAG1 and RAG2 using the program MrBayes version 3.1.2 (Huelsenbeck and Ronquist 2001). For each pairwise comparison between the eight species in our analysis, we calculated and averaged the patristic distances between each pair of orthologs to generate pairwise genetic distances between each pair of species (Fig. 3c). In these comparisons, averages were performed in a hierarchical way to ensure that each of the individual pairwise distance between orthologs contributed appropriately to the aggregate pairwise distance between the species, including in comparisons involving octoploid species in which the RAG1β2 paralog was deleted. Based on previously published phylogenetic relationships (Evans 2007; Evans et al. 2005), we substituted the genetic distances between evolutionarily equivalent comparisons with the mean genetic distance in each evolutionarily equivalent comparison, for example, between pairs of sister species. The program PHYLIP version 3.6 (Felsenstein 2005) was then used to transform the resulting triangular matrix into a bifurcating phenogram using the UPGMA algorithm. While this phenogram is topologically inaccurate because it does not reticulate, it is nonetheless a reasonable (albeit rough) approximation of genetic distance between species in terms of branch lengths and evolutionary relationships (Fig. 3c; Supplementary Fig. 1).
Because there could be biases in the efficiency of PCR amplification between paralogs, and also because there is an unequal number of RAG1α and RAG1β paralogs in some octoploid species, we standardized the paralogous expression ratios by subtracting the log2-transformed paralogous ratio that was obtained from genomic DNA (gDNA). Biological replicates of these species were not available, but at least two technical replicates were performed for each one. To complement these assays, we also inferred the paralogous expression ratios for XL and XB with three biological replicates from data collected for Predictions 2 and 3 (described next).
Predictions 2 and 3: Magnitude and Mechanisms of Expression Divergence
Expression analysis of interspecies hybrids provides a way to tease apart the contributions of cis- and trans-acting mechanisms to expression divergence between orthologous alleles (Landry et al. 2005; Wittkopp et al. 2004). To quantify the relative contributions of cis- and trans-acting factors in expression divergence between species, we compared the expression ratios of species-specific alleles in XL and XB to the expression ratio of these alleles in HXLXB (Fig. 2). For each locus we tested whether (a) the log2-transformed parental expression ratio was significantly different from the log2-transformed hybrid expression ratio, which would be consistent with trans divergence, and whether (b) the log2-transformed expression ratio in hybrids was significantly different from zero, which would be consistent with cis divergence (Fig. 2) (Landry et al. 2005; Wittkopp et al. 2004).

Characterization of cis and trans contributions to expression divergence between species by comparison of allelic expression ratios in parental and hybrid individuals. Cis-acting elements are depicted as 5′ regulatory regions and trans-acting factors are depicted as transcription factors that bind to these elements. Inferences based on comparisons of expression ratios in parentals and hybrids assume intraspecific variation in expression is small relative to interspecific variation. Species-specific alleles are yellow and green rectangles; only one allele is shown for the parental species but both are shown for hybrids. In this example, red elements upregulate expression and blue ones downregulate it. In hybrid individuals the two-headed arrows indicate that transcription factors from both parental species interact with cis-regulatory elements of the alleles from both parental species. In these comparisons, P refers to the log2-transformed expression ratio of parental alleles in each parental species and H refers to the log2-transformed expression ratio of each parental allele in a hybrid. Cis divergence is indicated when the null hypothesis of P = H is not rejected but the null hypothesis of H = 0 is rejected. Trans divergence is indicated when the null hypothesis of P = H is rejected but the null hypothesis of H = 0 is not rejected (which also implies rejection of the null hypothesis, P = 0). However, if the log2 transformed parental expression ratio is closer to 0 than the hybrid expression ratio, this suggests that cis- and trans-acting factors have acted in opposite directions to stabilize expression levels in each species. When the log2 expression ratio in hybrids is closer to 0 than the parental ratio, it may not be possible to distinguish between scenarios involving synergy and opposition of cis- and trans-acting factors
In hybrids, trans-acting factors from both species can interact with the cis-acting factors of species-specific alleles, so their differential expression in this type of individual can be attributed exclusively to cis divergence (Fig. 2). Under a scenario of exclusively cis divergence, trans-acting factors expressed in a hybrid are the same, even though they are encoded by parental alleles from different species. This is true even though the overall expression level of these transcription factors (in terms of mRNA per cell) may differ between parental and hybrid individuals. Alternatively, divergent expression between species could be due only to trans-acting mechanisms (Fig. 2). Under this scenario, both species-specific alleles would be expressed at the same level in hybrids, even though these alleles are expressed at different levels in each parental species. Other scenarios involving both cis and trans divergence between orthologs are possible. If cis and trans mechanisms both act synergistically to upregulate expression in one species compared to another, then expression of species-specific alleles should be more similar in hybrids than in the parental species (Fig. 2). Alternatively, if cis- and trans- acting factors influence expression divergence in opposite (antagonistic) directions, then expression of species-specific alleles should be more similar in the parental species than in hybrids (Fig. 2) (Landry et al. 2005; Wittkopp et al. 2004; Yan et al. 2002). This is because species-specific cis-trans antagonism is disrupted in hybrids because trans-acting factors of both species interact with cis elements of both parental alleles.
Laboratory procedures for quantification of parental and hybrid expression ratios involve a few ingenious techniques developed by others (Wittkopp et al. 2004). The first challenge is to compare directly the expression intensity of alleles in each parental species while accounting for differences in the total number of cells from each parental species used in each RNA extraction. This is accomplished by homogenizing tissue samples from both parental species to generate a “parental mixture” and then performing both DNA and RNA extractions on this mixture. The parental expression ratio is estimated by measuring expression of alleles from each parental species in the parental mixture, based on species-specific and paralog-specific single-nucleotide polymorphisms (SNPs), and then standardizing this expression ratio by (i) the proportion of tissue from each parental species that was in the parental mixture and (ii) the PCR amplification bias for each species-specific allele. This first proportion (i) is estimated from the ratio of each type of parental DNA in the genomic DNA extraction from the parental mixture. The second proportion (ii) is estimated from the ratio of amplified parental alleles from a genomic DNA extraction from F1 hybrids (which have equal allelic concentrations from each parental species).
Parental mixtures were generated by combining similarly sized tissue samples from each of the parental species, adding a small quantity of RNAse-free water (~100 μl), and homogenizing the mixture by passing it through a 20.5-gauge needle or using a tissue homogenizer (PRO Scientific Inc.). RNA was extracted and converted into cDNA using the RNeasy extraction kit (Qiagen) and the cDNA synthesis Omniscript Reverse Transcriptase kit (Qiagen). Genomic DNA was extracted using the QLAmp kit (Qiagen). The proportion of gDNA or cDNA from each parental species that was in each type of extraction was quantified using a Biotage PSQ96 pyrosequencer (Kruckeberg and Thibodeau 2004; Nilsson and Johansson 2004). After amplification, species-specific alleles of RAG2β were quantified with one pyrosequencing primer, whereas amplified species-specific and paralog-specific alleles of RAG1α and RAG1β of XL and XB were quantified with three different pyrosequencing primers. Primers and SNP information are given in Supplementary Material 1 and are based on previously published sequences of RAG1 and RAG2 (Evans 2007). We performed these assays on RAG1 and RAG2 alleles expressed in adult bone marrow, brain, heart, and liver. For each tissue type, ratios were estimated in F1 hybrids generated from a cross between an XL female and an XB male (two females and two males) or the reciprocal cross (zero females and two males) and compared to the corresponding expression ratios in seven parental mixtures (three parental mixtures between XL and XB females and four parental mixtures between XL and XB males). For some treatments we performed replicate DNA or RNA extractions on an individual and/or replicate PCR amplifications on DNA or RNA.
The manufacturer’s protocol for our RNA extraction kit states that gDNA is efficiently removed from the extraction, but we nonetheless implemented an optional DNAase digestion step in this procedure. We also developed a PCR assay as a further test for gDNA contamination in our RNA extractions. In this assay, the α-globin gene was amplified using the primers alpha.107.for1 5′ TGA CAA GAA ACA CAT CAA GGC AAT TAT GC 3′ and alpha.971.rev 5′ CGA TAT TTG GAW GTC AGA ACA GTA GAT AC 3′. The amplified region includes two small introns, so amplifications from gDNA are larger than those from cDNA. We performed this assay on all of the cDNA extractions we used (including the species examined in Prediction 1 and the tissue crosses between XL and XB; see below). Consistent with the expectation that our RNA extractions did not contain gDNA, the amplifications from the RNA extractions included only the smaller fragment whereas amplifications of the gDNA extractions included only the larger fragment (data not shown).
Expression ratios of species-specific alleles in the parental mixtures and hybrids were standardized using the method of Landry et al. (2005). We fit a regression line between log2-transformed parental genomic ratios from the three pyrosequencing assays (RAG2β, RAG1α, RAG1β) and the median log2-transformed hybrid genomic ratio that was calculated for each primer from at least six biological replicates. The log2-transformed expression ratios from parental mixtures were standardized by subtracting the fitted estimate of parental DNA for each primer pair on the median ratio of parental gDNA that was amplified in hybrids with each primer pair. This accommodates PCR bias, pyrosequencing bias, and differences in concentration of each parental tissue in the parental mixture. The log2-transformed hybrid ratios were standardized by subtracting the median ratio from hybrid gDNA amplified with that primer pair, which accounts for PCR bias and pyrosequencing bias.
Expression Hierarchy
If tissue-specific expression is low, the inferred mechanisms of expression may not be biologically relevant and/or subject to natural selection in that tissue. To explore comparative expression levels across tissue types, we evaluated the hierarchy of expression levels among tissue types with a series of “tissue crosses” comprised of one tissue type from XL with another tissue type from XB. We analyzed a mixture of XL liver with XB heart, XL heart with XB brain, and XL brain with XB bone marrow, and we did these comparisons separately for each sex. The log2-transformed tissue cross-expression ratios were standardized by subtracting the corresponding log2-transformed gDNA ratio of the tissue crosses. The resulting standardized ratios were then used to calculate intraspecific expression ratios between tissue types. For example, the XLliver/XBheart expression ratio was divided by the parental XLliver/XBliver expression ratio to obtain the expression ratio XBliver/XBheart. This allowed us to estimate the expression hierarchy across four tissue types for each gene within each parental species and within each sex.
Statistical Analysis
We used the methods of Landry et al. (2005) to test whether the expression ratios were significantly different from each other and to test whether expression ratios were significantly different from zero. Restricted maximum likelihood (REML) was used to independently estimate variance parameters for each treatment being compared. Analyses were carried out using “proc mixed” in SAS version 9.1.3 (SAS Institute) with modified scripts that were graciously provided by Patricia Wittkopp. Similar to Landry et al. (2005), this approach was justified to avoid bias because the variance in expression ratio among replicates was higher in the parental mixtures than in the hybrids. Student’s t-tests were computed within the mixed procedure and locus-level significance was interpreted after sequential Bonferroni correction for multiple tests (Rice 1989). Calculations included a random effect estimated from the variance between PCR replicates (same extraction but different PCR reaction), which were nested in technical replicates (separate cDNA extractions), and within biological replicates (different individuals). Results from these analyses were very similar to those recovered from regular Student’s t-tests.
Because an inference of cis-only divergence or trans-only divergence is made on the basis of rejection of one hypothesis but inability to reject another (Fig. 2), these inferences suffer from Type II error. Neither inference, therefore, is statistically inconsistent with an inference of cis and trans divergence, which is based on the rejection of two null hypotheses (indicating that P ≠ H and H ≠ 0; Fig. 2). Application of the Bonferroni correction therefore makes the detection of antagonistic or synergistic cis and trans divergence more conservative. We also applied the Bonferroni correction to comparisons between hybrid expression ratios across tissue types. This makes the test for tissue-specific cis divergence more conservative.
Results
Prediction 1: Ancestral Upregulation of RAG1α
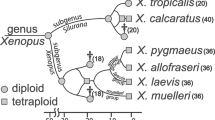
The SSRα hypothesis predicts that RAG1α is expressed more highly than RAG1β in multiple extant species and their ancestors. Consistent with this, we found that RAG1α was expressed more highly than RAG1β in bone marrow of seven of eight species that we examined. This was true even in octoploid species in which the RAG1β2 paralog was deleted (X. andrei, X. boumbaensis, X. andrei); in other words, in these species, even though there are two copies of RAG1α and one copy of RAG1β, expression of RAG1α1 plus RAG1α2 was more than twice as high as expression of RAG1β2 (Table 1). Additionally, in some other tissues this bias was extreme: expression of RAG1α in X. muelleri brain, for instance, was almost 10-fold higher than RAG1β (Table 1). Ancestral reconstruction did not reject the null hypothesis that the log2-transformed paralogous expression ratio of bone marrow was ≤0 in the most recent common ancestor (MRCA) of all Xenopus (node 3; P= 0.0726) (Fig. 3a and b), but it did reject the null hypothesis that this ratio was ≤0 in the MRCA of a subset of these species (node 4; P = 0.0497) (Fig. 3a and b).

Putative allopolyploid evolution of the tetraploids X. laevis (XL), X. gilli (XG), X. borealis (XB), and X. muelleri (XM) and the tetraploid ancestors of Xenopus octoploids. Daggers indicate extinct diploid ancestors or genes. Nodes 1 and 2 correspond to the divergence and union, respectively, of two diploid genomes; Node 3 indicates the MRCA of all Xenopus species; and Node 4 refers to the MRCA of a subset of these species. a A reticulate phylogeny, with ploidy in parentheses. b Nuclear genealogy assuming no recombination and no gene conversion between alleles at different paralogous loci (α and β). The dashed portion of the paralogous lineages in (b) evolved independently in different diploid ancestors. Octoploids, which have 72 chromosomes, inherited the complete genomes of two tetraploid ancestors, each of which had 36 chromosomes, so with no gene deletion, they are expected to have two α and two β paralogs. Some species experienced gene deletion (Evans 2007) so the actual number of paralogs is lower in these species. c Genetic distances between species that evolved through allopolyploidization can be estimated as the average of the branch lengths between each of the MRCAs. For instance, in this example, an allotetraploid evolved from two extinct diploids. This allotetraploid then speciated into two descendant tetraploids (T1 and T2), ancestors of which underwent allopolyploidization to form an octoploid (O). The genetic distance between tetraploid T1 and the octoploid O is equal to the mean of the red and blue branch lengths. Each of these branches can be calculated from patristic distances from a genealogy that includes all paralogs in each species (see text for details)
We inferred species-specific paralogous expression ratios in XL and XB with biological replication from the parental mixtures, and these log2-transformed ratios did not depart significantly from 0 in XL or XB bone marrow, brain, heart, or liver (data not shown). When the ancestral reconstructions were repeated with the expression ratio of these species set to 0, the null hypothesis that the ancestral expression ratio was ≤0 was again rejected for the MRCA of the a subset of these species (node 4; P = 0.0492) (Fig. 3a and b) and it not rejected for the MRCA of all Xenopus (node 3; P = 0.0826) (Fig. 3a and b). Overall, we interpret these results as providing support the SSRα hypothesis—at least for a subset of these species.
Prediction 2: Tissue-Specific Expression Divergence Between Species
Prediction 2 posits that the magnitude and mechanisms of expression divergence between species could vary, depending on which tissue is compared. To test this, we compared expression ratios of species-specific alleles in different tissues of parental and hybrid individuals using the approach of Landry et al. (2005) and Wittkopp et al. (2004). Because tests for parent of origin effects and sex bias were not significant within each tissue type for orthologous ratios of RAG1α, RAG1β, or RAG2β or for paralogous ratios of RAG1 (Supplementary Table 1), we did not perform a separate test for males and females or for each type of hybrid cross (XL or XB mother).
When considering expression divergence of each gene individually, we did not find any unambiguous examples where cis- or trans- divergence was significant but in opposite direction in different tissues, but we did recover support for significantly different magnitudes of tissue-specific expression divergence. Significant or near-significant upregulation of XL RAG2β due to cis divergence was detected in bone marrow and heart, and this was almost significant in brain (H ≠ 0; Table 2). Parsimonious explanations are that cis divergence is (a) driven by the same trans-acting factors in each of these tissues or (b) driven by different tissue-specific trans-acting factors that have overlapping cis targets. In bone marrow and heart, antagonistic trans upregulation of XB RAG2β was also detected (P ≠ H) (Table 2). In RAG1, orthologous expression was generally not significantly different in the parental species (null hypothesis of P = 0 not rejected; Table 2) except in RAG1α in liver, which exhibits antagonism between cis and trans divergence wherein cis divergence upregulates XL and trans divergence upregulates XB. As a result of these antagonistic mechanisms, the parental expression ratio of RAG1α in liver is not significantly different from that in the other tissues (data not shown).
Consistent with one aspect of Prediction 2—that the magnitude of cis divergence could be tissue specific—expression ratios of parental alleles of RAG2β in hybrids were significantly different in multiple pairwise comparisons between tissues, even after correction for multiple tests (Table 3). For example, the XL RAG2β allele was much more highly expressed compared to the XB RAG2β allele in hybrid heart (log2-transformed hybrid ratio = 1.1566) than in hybrid liver (log2-transformed hybrid ratio = 0.0825; Table 2) and this difference is significant (P = 0.0001; Table 3). Likewise, in hybrid liver the expression ratio of XL RAG1α and XB RAG1α is higher than in hybrid bone marrow and brain (Tables 2 and 3), which is also consistent with Prediction 2, but this difference is not significant after correction for multiple tests.
Cis divergence in bone marrow causes upregulation of XL RAG1β compared to XB RAG1β (Table 2). This could also include an antagonistic trans contribution—which could explain why the RAG1β expression ratio is not significantly different in bone marrow from the other tissues—but evidence for trans divergence of RAG1β was not statistically significant in bone marrow (Table 2). As a consequence of tissue-specific cis divergence, upregulation of XL RAG1β compared to XB RAG1β is significantly higher in hybrid bone marrow than in hybrid heart or hybrid liver (Table 3), which is also consistent with Prediction 2.
Prediction 3: Tissue-Specific Paralogous Expression Divergence
We predicted that coexpressed paralogs could have different levels of cis divergence in different tissues (Prediction 3). Consistent with this, tissue specificity of paralogous cis divergence was observed in XL paralogs in hybrid liver compared to hybrid bone marrow (Table 4). In liver, XL RAG1α is significantly upregulated compared to XL RAG1β (log2-transformed hybrid ratio = 0.4625), but in bone marrow XL RAG1β is upregulated (log2-transformed hybrid ratio = −0.8587).
Expression Hierarchy Across Tissue Types
No significant difference was detected among RAG1α, RAG1β, and RAG2β in the hierarchy of expression across tissues, and there was substantial qualitative consistency among these hierarchies (Table 5). Expression was generally highest in bone marrow. The next highest expression was in brain, followed by heart and then liver, except in RAG1β, where expression was higher in liver than heart. These results are consistent with another study (Greenhalgh et al. 1993) that found higher expression of RAG1 and RAG2 in adult bone marrow than in liver. It is not clear from these results whether expression levels in heart and liver are appreciable enough to be biologically significant.
Discussion
In African clawed frogs of the genus Xenopus, genome duplication gave rise to duplicated copies of the RAG1 gene; these paralogs then degraded in a biased fashion wherein one set of closely related paralogs became pseudogenes more frequently than another (Evans 2007; Evans et al. 2005). One explanation for this, which we call the SSRα hypothesis, posits that expression divergence between RAG1 paralogs favors the retention of the one that was upregulated (RAG1α) in an early ancestor. In multiple tissues and species, this hypothesis is supported by higher expression of RAG1α (Table 1) and ancestral reconstructions support upregulation of RAG1α in bone marrow of an ancestor of a subset of the species we examined. We note, however, that there is a “chicken-and-egg” problem in the testing of the SSRα hypothesis because it is not clear whether low expression of RAG1β in octoploids is a cause or a consequence of pseudogenization of RAG1β. Xenopus boumbaensis, X. andrei, X. amieti, and X. itombwensis all have nonsense or frameshift mutations in RAG1β1; X. itombwensis also has a frameshift mutation in RAG1β2, whereas these other octoploids appear to be missing this paralog, probably as a consequence of a single ancestral deletion (Evans 2007). Additionally, one individual of the tetraploid species X. borealis was identified that was heterozygous for a nonsense mutation in RAG1β (Evans 2007). We therefore cannot rule out other explanations for biased RAG1 pseudogenization such as that (a) nonlethal incompatibilities between RAG1β and cofactors in the other subgenome (the “α” subgenome) increase the probability of pseudogenization of this paralog or (b) advantageous interactions between RAG1α and RAG2β (or some other gene) increase the probability of persistence of this paralog compared to RAG1β. These alternative explanations make opposite inferences about the nature of natural selection acting on paralogs of RAG1, i.e., that either negative selection on RAG1β or positive selection on RAG1α contributed to the nonrandom pseudogenization of RAG1β.
We found that the hierarchy of expression of RAG1 paralogs and RAG2β was qualitatively similar (Table 5), which is consistent with these interacting proteins facing similar selective pressures and/or perhaps being coregulated. This result is echoed in fruit flies and yeast, where interacting proteins tend to have similar levels of intraspecific expression polymorphism and their expression levels are positively correlated across strains (Lemos et al. 2004). The overall similarity in the direction of trans divergence across genes, wherein the XB allele is generally upregulated (except in liver; Table 2) is consistent with shared regulation in some tissue types. However, in liver significant divergence between orthologs was detected in parental individuals (RAG2β and in RAG1α) but in opposite directions in each gene: XL RAG2β is upregulated compared to XB RAG2β, possibly by trans divergence, but trans divergence upregulates XB RAG1α compared to XL RAG1α (Table 2). At least some aspects of trans regulation of RAG1 and RAG2, therefore, are unique.
Because gene duplication changes gene dosage, these two phenomena have intertwined implications for development and evolution. An increased understanding of the relative contributions of cis and trans factors in expression divergence, particularly after gene or genome duplication, would offer perspective on fundamental genetic mechanisms that underlie adaptation, reproductive isolation, and phenotypic plasticity. However, conclusions regarding the relative importance of cis and trans mechanisms in expression divergence vary among studies, among genes, and perhaps among species (Cowles et al. 2002; Landry et al. 2005; Wittkopp et al. 2004). Expression analyses in humans, yeast, flies, nematodes, and Eucalyptus suggest an important role for trans-acting factors (Brem et al. 2002; Kirst et al. 2005; Morley et al. 2004; Yvert et al. 2003), whereas other reports on humans, flies, and mice recover an important role for cis-acting factors (Cowles et al. 2002; Wittkopp et al. 2004, 2008; Yan et al. 2002). This issue has important implications for our understanding of the genetic architecture and evolution of gene regulation. For example, if cis divergence is less common than trans divergence, either trans-regulatory factors are a comparatively larger mutational target or they are under comparatively less severe purifying selection (or more extensive positive or diversifying selection) than cis-regulatory elements. Similar to some of these previous studies, our results indicate that cis and trans mechanisms both sculpted expression divergence of the RAG1-RAG2 heterodimer in clawed frogs. Many orthologous expression ratios of RAG1 and RAG2 are not significantly diverged between these parental species, but comparison to hybrid expression uncovers antagonistic interactions between cis and trans divergence, including cis upregulation of XL alleles coupled with trans upregulation of XB alleles in some tissues, such as bone marrow (Table 2).
Purifying selection can lead to a conserved level of expression even when regulatory sequences have changed considerably (Ludwig et al. 2000), and this could contribute to the origin of antagonistic cis and trans divergence. This can happen if compensatory changes occur in different components of the regulatory machinery. In the nematode Caenorhabditis elegans, for example, comparison of mutation accumulation lines with a low effective population size (Ne) had more expression divergence than divergent natural isolates with a larger Ne, suggesting purifying selection on gene regulation in the natural isolates (Denver et al. 2005). Comparison of orthologous divergence in fruit flies, primates, and rodents suggests that the overall level of expression is conserved in many genes, which is also consistent with purifying selection (Jordan et al. 2005; Lemos et al. 2005). Intraspecific polymorphism in expression in Drosophila appears to be largely attributable to trans mechanisms, whereas interspecific expression divergence appears to be primarily a consequence of cis mechanisms, suggesting that purifying selection may minimize trans divergence between species to a greater degree than cis divergence (Wittkopp et al. 2008). Some aspects of regulatory evolution, however, may evolve under relaxed purifying selection or even neutrally (Jordan et al. 2005). Moreover, expression of RAG1 and RAG2 may be under more severe purifying selection in tissues where it is highly expressed, such as bone marrow, than other tissues with lower levels of expression, such as heart. Overall, however, antagonistic cis- and trans-regulatory divergence in multiple tissues, and similarities in the hierarchy of expression of RAG1 and RAG2, suggests the action of purifying selection on regulation of these genes in Xenopus. This could be further tested with comparative studies that quantify the genome-wide rate and probability of cis and trans divergence in expressed duplicates and singletons of Xenopus.
WGD by allopolyploidization can lead to rapid or immediate changes in gene expression that can be nonadditive with respect to parental expression levels (Adams et al. 2003; Albertin et al. 2006; Wang et al. 2006). Additionally, rapid divergence of paralogous expression profiles on a quantitative, spatial, or temporal dimension could contribute to their propensity to continue to encode functional proteins over the long term (Force et al. 1999; Lynch and Force 2000). Regulatory evolution could also be faster if purifying selection is relaxed after gene duplication. In the nematodes Caenorhabdites elegans and C. briggsae, for example, protein and regulatory divergence is weakly correlated in orthologs but not in paralogs (Castillo-Davis et al. 2004). In X. laevis, divergence of paralogous expression profiles across multiple tissues and developmental stages is not correlated with molecular evolution of paralogous sequences, suggesting that selective constraints on protein sequence and expression are not coupled soon after WGD (Chain et al. 2008).
Conclusions
These results are consistent with the prediction that expression bias soon after gene duplication could influence genomic restructuring (pseudogenization and gene deletion) that occurs afterward (Prediction 1). We speculate, therefore, that early changes in gene regulation are particularly significant in sculpting the genetic fates of duplicate genes generated by WGD—even though these fates (pseudogenization, deletion, persistent function) may be realized many millions of years later. Additionally, we found that the magnitude and direction of orthologous and paralogous divergence can be tissue specific (Predictions 2 and 3). This suggests that variation in expression between different cell types within a single tissue—for example, between epithelial and mesenchymal cell types in the liver, as analyzed in this study—could be modulated in unique and nonoverlapping ways. Inferences made about regulatory mechanisms from the entire body (Landry et al. 2005; Wittkopp et al. 2004) or from pooled differentiated cell types within an organ (this study; Cowles et al. 2002; Kirst et al. 2005), then, probably most strongly reflect a combined influence of regulatory mechanisms in (a) the cell type in which a gene is most highly expressed and (b) the cell type that constitutes the highest proportion of the sample. Because of this, the inferred impact of these mechanisms actually might not correspond with those operating in any individual cell. This poses a challenge to the precise dissection of the mechanisms that drive expression divergence at the cellular level.
References
Adams KL, Cronn R, Percifeld R, Wendel JF (2003) Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc Natl Acad Sci USA 100:4649
Albertin W, Balliau T, Brabant P, Chévre A-M, Eber F, Malosse C, Thiellement H (2006) Numerous and rapid nonstochastic modifications of gene products in newly synthesized Brassica napus allotetraploids. Genetics 173:1101–1113
Bassing CH, Swat W, Alt FW (2002) The mechanism and regulation of chromosomal V(D)J recombination. Cell 109:S45–S55
Brem RB, Yvert G, Clinton R, Kruglyak L (2002) Genetic dissection of transcriptional regulation in budding yeast. Science 296:752–755
Castillo-Davis CI, Hartl DL, Achaz G (2004) cis-regulatory and protein evolution in orthologous and duplicate genes. Genome Res 14:1530–1536
Chain FJJ, Evans BJ (2006) Multiple mechanisms promote the retained expression of gene duplicates in the tetraploid frog Xenopus laevis. PLoS Genetics 2:e56
Chain FJJ, Ilieva D, Evans BJ (2008) Duplicate gene evolution and expression in the wake of vertebrate allopolyploidization. BMC Evol Biol 8:43
Cowles CR, Hirschhorn JN, Altshuler D, Lander ES (2002) Detection of regulatory variation in mouse genes. Nat Genet 32:432–437
Denver DR, Morris K, Streelman JT, Kim SK, Lynch M, Thoman WK (2005) The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans. Nature Genet 37:544–548
Evans BJ (2007) Ancestry influences the fate of duplicated genes millions of years after duplication in allopolyploid clawed frogs (Xenopus). Genetics 176:1119–1130
Evans BJ (2008) Genome evolution and speciation genetics of allopolyploid clawed frogs (Xenopus and Silurana). Frontiers Biosci 4687–4706
Evans BJ, Kelley DB, Tinsley RC, Melnick DJ, Cannatella DC (2004) A mitochondrial DNA phylogeny of African clawed frogs: phylogeography and implications for polyploid evolution. Mol Phylogen Evol 33:197–213
Evans BJ, Kelley DB, Melnick DJ, Cannatella DC (2005) Evolution of RAG-1 in polyploid clawed frogs. Mol Biol Evol 22:1193–1207
Felsenstein J (2005) PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle
Force A, Lynch M, Pickett B, Amores A, Yan YL, Postlethwait JH (1999) Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151:1531–1545
Greenhalgh P, Olesen CEM, Steiner LA (1993) Characterization and expression of recombination activating genes (RAG-1 and RAG-2) in Xenopus laevis. J Immunol 151:3100–3110
Huelsenbeck JP, Ronquist F (2001) MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Jordan IK, Marino-Ramirez L, Koonin EV (2005) Evolutionary significance of gene expression divergence. Gene 345:119–126
Kirst M, Basten CJ, Myburg AA, Zeng ZB, Seberoff RR (2005) Genetic architecture of transcript-level variation in differentiating xylem of a eucalyptus hybrid. Genetics 169:2295–2303
Kruckeberg KE, Thibodeau SN (2004) Pyrosequencing technology as a method for the diagnosis of multiple endocrine neoplasia type 2. Clin Chem 50:522–529
Landry CR, Wittkopp PJ, Taubes CH, Ranz JM, Clark AG, Hartl DL (2005) Compensatory cis-trans evolution and the dysregulation of gene expression in interspecific hybrids of Drosophila. Genetics 171:1813–1822
Lemos B, Meiklejohn CD, Hartl DL (2004) Regulatory evolution across the protein interaction network. Nature Genet 36:1059–1060
Lemos B, Meiklejohn CD, Cáceres M, Hartl DL (2005) Rates of divergence in gene expression profiles of primates, mice, and flies: stabilizing selection and variability among functional categories. Evolution 59:126–137
Ludwig MZ, Bergman C, Patel NH, Kreitman M (2000) Evidence for stabilizing selection in a eukaryotic enhancer element. Nature 403:564–567
Lynch M, Force A (2000) The probability of duplicate gene preservation by subfunctionalization. Genetics 154:459–473
Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, Cheung VG (2004) Genetic analysis of genome-wide variation in human gene expression. Nature 430:743–747
Nilsson TK, Johansson CA (2004) A novel method for diagnosis of adult hypolactasia by genotyping of the −13910 C/T polymorphism with Pyrosequencing technology. Scand J Gastroenterol 39:287–290
Organ CL, Shedlock AM, Meade A, Pagel M, Edwards SV (2007) Origin of avian genome size and structure in non-avian dinosaurs. Nature 446:180–184
Pagel M (1997) Inferring evolutionary processes from phylogenies. Zoologica Scripta 26:331–348
Pagel M (1999) Inferring the historical patterns of biological evolution. Nature 401:877–884
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Spicuglia S, Franchini DM, Ferrier P (2006) Regulation of V(D)J recombination. Curr Opin Immunol 18:158–163
Venditti C, Meade A, Pagel M (2006) Detecting the node-density artifact in phylogeny reconstruction. Syst Biol 55:637–643
Wang J, Tian L, Lee H-S, Wei NE, Jiang H, Watson B, Madlung A, Osborn TC, Doerge RW, Comai L, Chen ZJ (2006) Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172:507–517
Wittkopp PJ, Haerum BK, Clark AG (2004) Evolutionary changes in cis and trans gene regulation. Nature 430:85–88
Wittkopp PJ, Haerum BK, Clark AG (2008) Regulatory changes underlying expression differences within and between Drosophila species. Nature Genet 40:346–350
Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW (2002) Allelic variation in human gene expression. Science 297:1143
Yvert G, Brem RB, Whittle J, Akey JM, Foss E, Smith EN, Mackelprang R, Kruglyak L (2003) Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nature Genet 35:57–64
Acknowledgments
We thank Patricia Wittkopp for statistical advice and SAS scripts, Andrew Meade and Mark Pagel for advice on using BayesTraits, Adam Smith for assistance with pyrosequencing, Darcy Kelley and Elizabeth Leininger for providing some of the F1 hybrid individuals, Mohammad Iqbal Setiadi and Frédéric Chain for assistance with animal propagation, and Frédéric Chain and two anonymous reviewers for helpful comments on the manuscript. This research was supported by the Canadian Foundation for Innovation, the National Science and Engineering Research Council, the Ontario Research and Development Challenge Fund, and McMaster University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Anderson, D.W., Evans, B.J. Regulatory Evolution of a Duplicated Heterodimer Across Species and Tissues of Allopolyploid Clawed Frogs (Xenopus). J Mol Evol 68, 236–247 (2009). https://doi.org/10.1007/s00239-009-9197-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-009-9197-6