
Abstract
The paralytic shellfish poisoning (PSP) toxins, saxitoxin, and its derivatives, are produced by a complex and unique biosynthetic pathway. It involves reactions that are rare in other metabolic pathways, however, distantly related organisms, such as dinoflagellates and cyanobacteria, produce these toxins by an identical pathway. Speculative explanations for the unusual phylogenetic distribution of this metabolic pathway have been proposed, including a polyphyletic origin, the involvement of symbiotic bacteria, and horizontal gene transfer. This study describes for the first time the identity of one gene, sxt1, that is involved in the biosynthesis of saxitoxin in cyanobacteria. It encoded an O-carbamoyltransferase (OCTASE) that was proposed to carbamoylate the hydroxymethyl side chain of saxitoxin precursor. Orthologues of sxt1 were exclusively present in PSP-toxic strains of cyanobacteria and had a high sequence similarity to each other. L. wollei had a naturally mutated sxt1 gene that encoded an inactive enzyme, and was incapable of producing carbamoylated PSP-toxin analogues, supporting the proposed function of Sxt1. Phylogenetic analysis revealed that OCATSE genes were present exclusively in prokaryotic organisms and were characterized by a high rate of horizontal gene transfer. OCTASE has most likely evolved from an ancestral O-sialoglycoprotein endopeptidase from proteobacteria, whereas the most likely phylogenetic origin of sxt1 was an ancestral α-proteobacterium. The phylogeny of sxt1 suggested that the entire set of genes required for saxitoxin biosynthesis may spread by horizontal gene transfer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Paralytic shellfish poisoning (PSP) is a life-threatening affliction that results from the consumption of water or seafood that is contaminated by saxitoxin and its analogues (Kao and Levinson 1986). These toxins are among the most potent algal toxins and considered a serious toxicological health risk that may affect humans, animals, and ecosystems worldwide (Kaas and Henriksen 2000; Pereira et al. 2000). They act by blocking voltage-gated sodium channels (Kao and Levinson 1986), which prevents the transduction of neuronal signals, and thus cause muscular paralysis. In addition, they modulate voltage-gated calcium and potassium channels of heart muscle cells, resulting in a depression of the cardiac output (Su et al. 2004; Wang et al. 2003).
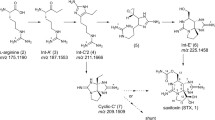
Saxitoxin is the parent compound of more than 30 naturally occurring analogues. It provides a tricyclic perhydropurine core with an O-carbamoylated methyl side chain. The biosynthesis of PSP toxins is complex and unique (Fig. 1), and involves biochemical reactions that are rare in other metabolic pathways. They include a Claisen condensation of an amino acid to a carboxylic acid, amidino transfer, unconventional heterocyclization, and O-carbamoylation (Shimizu 1993). Despite their unique biosynthesis, these toxins are produced in organisms that span two kingdoms of life. By far the most notorious producers of PSP toxins are certain species of marine dinoflagellates, the causative organisms of ‘red tides’ (Hallegraeff 1995). It has also been firmly established that certain species of freshwater cyanobacteria produce these toxins (Sivonen 1996). In addition, there are cases where PSP toxins have been detected in organisms, where dinoflagellates and cyanobacteria were unlikely sources for these toxins. These cases included freshwater and marine species of puffer fish (Nakashima et al. 2004; Sato et al. 1997; Zaman et al. 1997), the amphibian, Atelopus zetekii (Yotsu-Yamashita et al. 2004), the red algae, Jania sp. (Kotaki et al. 1983; Oshima et al. 1984), and bacteria isolated from dinoflagellate cells (Gallacher et al. 1997; Kodoma et al. 1988).

Biosynthesis pathway of saxitoxin (Shimizu 1993). SAM, S-adenosylmethionine
The unique biosynthesis of these metabolites, combined with their unusual phylogenetic distribution, makes PSP toxins intriguing metabolites. The phylogenetic origin of their biosynthetic genes, whether symbiotic bacteria may play a role in the production of these toxins in dinoflagellates (Gallacher et al. 1997; Kodoma et al. 1990), and whether saxitoxin biosynthesis genes may spread via horizontal gene transfer (Daly 2004; Plumley 2001) have been much debated. So far, the identity of genes involved in the biosynthesis of PSP toxins has been elusive, despite intensive research efforts (Hackett et al. 2005; Pomati et al. 2004; Pomati et al. 2006; Pomati and Neilan 2004). In this study, we describe the identity and phylogeny of a saxitoxin biosynthesis gene, sxtI, that was identified in PSP toxin-producing cyanobacteria. This gene encoded an O-carbamoyltransferase (OCTASE), which was related to enzymes that are involved in the production of nodulation factors (Jabbouri et al. 1998) and antibiotics (Coque et al. 1995). The assigned function of SxtI is to transfer a carbamoyl group from carbamoyl phosphate to the hydroxymethyl side chain of saxitoxin precursor (Kellmann and Neilan 2007). It is thus one of the key enzymes in saxitoxin biosynthesis, and was used to examine the possible evolution of this metabolic pathway.
Materials and Methods
Organisms, Culture Conditions, and DNA Extraction
Cyanobacterial strains (Table 1) were grown in Jaworski medium (Thompson et al. 1988) at 26°C under continuous illumination (10 μmol m−2 s−1). Total genomic DNA was extracted from cyanobacterial cells by lysozyme/SDS/proteinase K lysis following phenol-chloroform extraction as described previously (Neilan 1995). DNA in the supernatant was precipitated with 0.6 vol isopropanol, washed with 70% ethanol, dissolved in TE buffer (10:1), and stored at −20°C.
PCR Amplification and Sequencing
PCR primers that were used in this study are listed in Table 2. The degenerate PCR primers NOD-F and NOD-R were designed using cyanobacterial OCTASE gene sequences from the NCBI nucleotide sequence database (Fig. 2). They targeted the most conserved regions in OCTASE and were used to detect members of this gene family in PSP-toxic cyanobacteria.

Alignment of two conserved regions in the nucleotide sequence of cyanobacterial O-carbamoyltransferase genes. The translated consensus and primer sequences are shown
A standard PCR was performed in 20-μl reaction volumes containing 1 × Taq polymerase buffer, 2.5 mM MgCl2, 0.2 mM deoxynucleotide triphosphates, 10 pmol forward and reverse primers, between 10 and 100 ng genomic DNA, and 0.2 U Taq polymerase (Fischer Biotech, Perth, Australia). Thermal cycling was performed in a GeneAmp PCR System 2400 Thermocycler (Perkin Elmer Corp., Norwalk, CT, USA). Cycling began with a denaturing step at 94°C for 3 min, followed by 30 cycles of denaturing at 94°C for 10 s, primer annealing at between 45° and 65°C for 20 s, and DNA strand extension at 72°C for 1 min. Amplification was completed by a final extension step at 72°C for 7 min. DNA was separated by agarose gel electrophoresis in TAE buffer (40 mM Tris-acetate, 1 mM EDTA, pH 7.8) and visualized by UV translumination after staining in ethidium bromide (0.5 μg/ml). PCR products were ligated into the pGEM-Teasy vector (Promega Catalog No. A1360), and a clone library was prepared in Escherichia coli DH5α according to the manufacturer’s instructions. Colony PCR, using the vector-directed MpF and MpR primers, was then used for the amplification and sequencing of inserts. Batches of four clones were sequenced until no new sequences were obtained. A colony PCR consisted of a standard PCR reaction as described above, with the exception that a tiny amount of colony, containing the plasmid with an insert, was used as the template.
Automated DNA sequencing was performed using the PRISM Big Dye cycle sequencing system and a model 373 sequencer (Applied Biosystems Inc., Foster City, CA, USA). Sequence data were analyzed using ABI Prism-Autoassembler software, and percentage similarity and identity to other translated sequences determined using BLAST in conjunction with the National Center for Biotechnology Information (NIH, Bethesda, MD, USA). Upon sequencing of the entire sxt1 gene by adapter-mediated PCR, as described below, a nondegenerate PCR primer pair, Sxt1-F/Sxt1-R (Table 2), was designed to specifically amplify sxt1 orthologues. Sequences obtained in this study were submitted to the NCBI database. Accession numbers were EU439556 to EU439565 for sxt1 sequences and EU439566 and EU439568 for 16S rDNA sequences.
Adapter-Mediated PCR (Panhandle PCR)
The sequence of unknown regions that were flanking candidate genes was determined by an adapter-mediated PCR method (Siebert et al. 1995) that was modified as described by Moffitt and Neilan (2004). Short adapter DNA was ligated to digested genomic DNA, and a specific genomic outward-facing primer was then used with an adapter primer to amplify a region of the genome. Twenty picomoles of T7 adapter was added to each reaction mixture, containing 1 μg of genomic DNA, 10 U of blunt-ended restriction enzyme, and 5 U of T4 ligase (Promega) in 1 × One Phor All buffer (Amersham/Pharmacia). The one-step digestion and ligation reaction mixture was incubated at room temperature overnight.
The single-stranded end of the adapter was blocked in a solution containing 1 × PCR buffer (Fischer Biotech), 4 mM MgCl2, and 12.5 μM ddNTP with 1 U Taq DNA polymerase (Fischer Biotech). Thermal cycling was performed in a PCR Sprint temperature cycling system machine (Hybaid Ltd.) with an initial step at 70°C for 15 min, followed by 10 cycles of DNA denaturation at 95°C for 10 s, DNA reannealing at 40°C for 1 min, and extension of the strand with ddNTP at 70°C for 1 min. Following PCR cycles, the reaction mixture was incubated with 1 U of shrimp alkaline phosphatase (Boehringer Mannheim, Göttingen, Germany) at 37°C for 20 min, and the enzyme was heat-inactivated at 85°C for 5 min.
The flanking region PCR mixture contained 1 to 2 μl of adapter-ligated DNA, 10 pmol of adapter primer, and 10 pmol of a genome-specific oligonucleotide primer. PCR cycling was performed as described above, with DNA strand extension at 72°C for 5 min. The primer annealing temperature was decreased by 1°C at each cycle, from 65o to 55°C, followed by primer annealing at 55°C for a further 25 cycles.
Phylogenetic Analysis
Multiple sequence alignments of 16S rDNA and OCTASE protein sequences were prepared using Clustalw and the IUB DNA and Gonnet 250 protein weight matrix, respectively (Thompson et al. 1997). Multiple sequence alignment of OCTASE genes was based on the corresponding protein sequence alignment, using the EMBOSS tools, transeq and tranalign (Rice et al. 2000). Sequence alignments were manually confirmed. All alignments were bootstrapped with 1000 resampling events. Phylogenetic trees were reconstructed from a pairwise distance matrix (Jukes and Cantor 1969) using the neighbor-joining method (Saitou and Nei 1987). Phylogenetic trees and sequence alignments were reproduced using the software NJPlot (Perrière and Gouy 1996) and TeXshade (Beitz 2000),respectively. In addition to the neighbor-joining method, the phylogenies of OCATSE and 16S rRNA genes were determined by the maximum likelihood method, using the Phylip software package (Felsenstein 1989). Alignments were bootstrapped with 100 resampling events using SEQBOOT, and phylogenies were inferred by maximum likelihood using DNAML. A consensus tree was constructed using CONSENSE. Because the topologies of maximum likelihood and neighbor-joining trees were not significantly different, only neighbor-joining trees are shown.
Results
Identification of an O-Carbamoyltransferase Gene in PSP-Toxic Cyanobacteria
The biosynthetic pathway of saxitoxin (Shimizu 1993) and biochemical studies (Kellmann and Neilan 2007) have suggested that an OCTASE may be involved in saxitoxin biosynthesis, where it transfers a carbamoyl group to the hydroxymethyl side chain of saxitoxin precursor (Table 3). A pair of degenerate PCR primers, NOD-F/NOD-R, was designed that targeted two sequence blocks that are highly conserved in OCTASE genes (Fig. 2). A PCR product of the expected size (~1 kb) was obtained using the NOD-F/NOD-R primer pair and genomic DNA from the PSP-toxic strains Anabaena circinalis AWQC131C, Aphanizomenon flos-aquae NH-5, Cylindrospermopsis raciborskii T3, and Lyngbya wollei (Farlow) as a template. Sequencing of cloned PCR products, in conjunction with BLAST searching, demonstrated that each PCR product consisted of a single sequence with homology to OCTASE. A PSI-BLAST search with Sxt1 from C. raciborskii T3 yielded 269 homologous hits from the database. Of these, six sequences were from archaea, and three sequences from bacteriophages, however, the overwhelming majority of sequences were found in eubacteria. The majority of eubacterial sequences (170) were from proteobacteria, in particular, from α-proteobacteria (60 sequences). The most similar BLAST hit (70.6% to 71.2% identities) was a hypothetical OCTASE from Trichodesmium erythraeum (accession no. ZP_00673599). The amplified gene was designated sxt1, and its complete sequence was obtained from all four PSP-toxic species by adapter-mediated PCR. The sxt1 genes from A. circinalis AWQC131C, Aph. flos-aquae NH-5, and C. raciborskii T3 were 1839 bp in length, and had between 90.4% and 97.4% nucleotide sequence identity to each other. The sxt1 gene from L. wollei was only 1071 bp long. Its first 912 bp had 92.9% identity to sxt1 from C. raciborskii T3, bp 913 to 1059 and bp 1177 to 1189 (C. raciborksii T3 numbering) were deleted, and the 3′-end was truncated after bp 1227.
A sxt1-specific primer pair (Sxt1-F/Sxt1-R) was designed to PCR-screen 10 PSP-toxic and 18 non-PSP-toxic strains of A. circinalis, Aph. flos-aquae, C. raciborskii, and L. wollei. A PCR product of the expected size (1669 bp) was obtained, using genomic DNA from PSP-toxic strains of A.circinalis, Aph. flos-aquae, C. raciborskii, and L. wollei as a template. In contrast, a PCR product was not obtained, when genomic DNA from non-PSP-toxic strains was used as a template Table 1. The sxt1 gene was thus present in all PSP-toxic strains, whereas it was absent from all tested non-PSP-toxic strains. The nucleotide sequence identities between partial sxt1 genes (956 bp) from A. circinalis strains were at least 99.8%.
The phylogeny of sxt1 and other cyanobacterial OCTASE genes was compared to the corresponding 16S rRNA gene phylogeny (Fig. 3a and b). Sixty-four strains of cyanobacteria have a completely sequenced genome, however, OCTASE genes were represented in only 11 strains: 2 Oscillatoriales, 4 Chroococcales, and 5 Prochlorales members. In addition, the Prochlorococcus phage P-SSM2 had two OCTASE genes in its genome. The function of any of these genes was not known.


Phylogenetic affiliations of partial O-carbamoyltransferase (a) and 16S rRNA genes (b) from cyanobacteria. The phenograms were reconstructed from a pairwise distance matrix (Jukes and Cantor 1969) using the neighbor-joining method (Saitou and Nei 1987). PSP-toxic strains are indicated by boldface. The scale represents the number of substitutions per 100 nucleotides. Bootstrap values (1000 resampling cycles) >500 are shown and represent the statistical significance at each node
sxt1 sequences grouped into a single cluster that was separated from other sequences. It was divided into two branches, one containing sequences from A. circinalis and Aph. flos-aquae, and the other containing sequences from C. raciborskii and L. wollei (Fig. 3). OCTASE genes from other cyanobacteria and cyanophage P-SSM2 were more divergent from each other than sxt1 sequences.
The 16S rDNA phylogeny divided cyanobacteria into four main groups according to their taxonomy, with the exception of Synechococcus sp. RCC307 (Fig. 3b). Group 1 consisted of Nostocales members, such as Anabaena, Aphanizomenon, Umezakia, and Cylindrospermopsis. Group 2 contained all members related to Oscillatoriales, such as Lyngbya and Trichodesmium, whereas group 3 consisted of the Chroococcales members Cyanothece, Chrocosphaera, and Synechocystis. Group 4 consisted of Prochlorales strains of Prochlorococcus marinus, as well as Synechococcus sp. RCC307. The latter strains has been classified as a Chroococcales member, based on morphology, however, the genus Synechococcus is known to be heterogeneous (Honda et al. 1999) and includes strains that have to be reclassified.
The phylogenies of cyanobacterial OCTASE and 16S rRNA genes were not congruent. The most apparent incongruence was the grouping of sxt1 from L. wollei and C. raciborskii T3 (Fig. 3a), whereas their 16S rRNA genes were located on separate branches (Fig. 3b). The 16S rRNA gene phylogeny divided A. circinalis into two distinct branches (Fig. 3b), as described previously (Beltran and Neilan 2000), however, sxt1 genes from all PSP-toxic A. circinalis were placed in a single group (Fig. 3a). Prochlorococcus strains and Synechococcus sp. RCC307 were closely related according to their 16S rDNA phylogeny, however, the OCTASE genes from Prochlorococcus marinus MED4 and MIT9303, as well as from Synechococcus sp. RCC307, were highly divergent from each other and all other sequences (Fig. 3a). Two OCTASE genes from the P. marinus phage P-SSM2 were only distantly related to cyanobacterial OCTASEs, and formed an outgroup.
Structure and Function of Sxt1
Unfortunately, methods for the genetic manipulation of PSP-toxin producing organisms are lacking, and sxt1 could not be functionally characterized by site-directed mutagenesis. Bioinformatic tools were therefore applied to infer the function of sxt1, which was strongly supported by the natural sxt1 null mutant of L. wollei. sxt1 from A. circinalis AWQC131C, Aph. flos-aquae NH-5, and C. raciborskii T3 encoded a product that was 612 amino acid residues long, however, sxt1 from L. wollei, which had deletions and was truncated at its 3′-end, encoded a product of only 356 amino acid residues. Sxt1 from A. circinalis AWQC131C, Aph. flos-aquae NH-5, and C. raciborskii T3 had at least 90.5% identical and 95.4% similar amino acid residues, respectively, whereas Sxt1 from L. wollei had at least 89.5% identical and 95.1% similar amino acid residues, respectively, over the first 304 residues.
Iterated PSI-Blast searching revealed that OCTASEs, such as Sxt1, were related to O-sialoglycoprotein endopeptidase (OSGP; EC 3.4.24.57). Structural homology searching (FUGUE v2.s.07) detected significant structural relatedness of C. raciborskii T3 Sxt1 to OSGP from Pyrococcus abyssi (2IVNA; Z-score, 13.99). A structural alignment of Sxt1 and other OCTASEs to 2IVNA revealed that the zinc- and ATP-binding sites of OSGP (Aravind and Koonin 1999) were conserved in OCTASE (Fig. 4). The C-terminus of OCTASEs extended beyond that of OSGPs (Fig. 4) and provided two highly conserved sequence blocks, E-x-G-P-R-[AS]-L-[CG]-x-R-S-[ILV]-[FILV]-x(2)-[APS] and H-x-D-x-[ST]-[ACGSTV]-R-[AILPV]-Q-x-[ILV]. These motifs were signature sequences of the OCTASE family (IPB003696B, IPB003696D), according to a BLOCKS search (Henikoff et al. 2000), and may be part of the catalytic site. Sxt1 from L. wollei lacked the two OCTASE signature sequences, due to the C-terminal truncation, and it had a deletion in its ATP-binding site (Fig. 4). It was therefore presumed that L. wollei Sxt1 was nonfunctional. L. wollei was unable to produce carbamoylated analogues of saxitoxin (Onodera et al. 1997). The observed phenotype combined with the presumed null mutation of sxt1 is therefore consistent with the predicted function of Sxt1 in saxitoxin biosynthesis.

Structural sequence alignment of O-sialoglycoprotein endopeptidase and O-carbamoyltransferase to the crystal structure of Pyrococcus abyssii O-sialoglycoprotein endopeptidase (2IVNA)
Discussion
Saxitoxin has an O-linked carbamoyl group at its side chain, which is rare in nature, however, O-carbamoyl groups are known from certain bacterial secondary metabolites, such as antibiotics (Coque et al. 1996) and nodulation factors (Jabbouri et al. 1995, 1998). In all cases that have been investigated, O-carbamoyl groups are formed via a carbamoyl transfer from carbamoyl phosphate to a hydroxylated carbon in the recipient molecule (Coque et al. 1996). These reactions are invariably catalyzed by a member of the OCTASE family, which includes NolO, NodU, and CmcH. A previous study (Kellmann and Neilan 2007), where supplementation with carbamoylphosphate stimulated saxitoxin production in vitro, suggested that an OCTASE was involved in the biosynthesis of saxitoxin.
In this study, an OCTASE gene, sxt1, was identified by PCR in PSP-toxic strains of cyanobacteria. Ten PSP-toxic and 18 non-PSP-toxic strains of cyanobacteria were PCR-screened for the presence of sxt1. The sxt1 gene was present in all PSP-toxic strains and absent from all non-PSP-toxic strains. Among the tested strains were closely related strains of A. circinalis (7 PSP-toxic, 4 non-PSP-toxic) and C. raciborskii (1 PSP-toxic, 12 non-PSP-toxic), as well as more distantly related PSP-toxic (L. wollei and Aph. flos-aquae) and non-PSP-toxic cyanobacteria (Aph. ovalisporum and U. natans). sxt1 homologues had high (≥90%) nucleotide sequence identities, and grouped on a single phylogenetic branch that was separated from other cyanobacterial OCTASEs, indicating that sxt1 genes were orthologous. The majority of PSP-toxic cyanobacteria were members of the order Nostocales, however, OCTASE genes were not previously known from any Nostocales member that had a sequenced genome. The fact that an involvement of OCTASE in saxitoxin biosynthesis has been predicted, combined with the correlation between PSP toxicity of closely and distantly related cyanobacteria strains and the presence of sxt1 in their genomes, is strong support for an involvement of sxt1 in PSP-toxin biosynthesis.
Structure and Function of Sxt1
Little is known regarding the structure and function of OCTASE. Iterated PSI-BLAST searching and a structural homology search (FUGUE) revealed relatedness of Sxt1 to O-sialoglycoprotein endopeptidase (OSGP). The latter enzyme family is represented in all organisms whose genomes have been sequenced so far and, where tested, were essential for cell survival (Aravind and Koonin 1999). A structural alignment of Sxt1 and selected OCTASEs to the known crystal structure of P. abyssii OSGP (2IVNA) revealed that the zinc- and ATP-binding sites were conserved in Sxt1. Biochemical studies demonstrated that CmcH required ATP for enzyme activity (Brewer et al. 1980), whereas the zinc-binding site in NolO was essential for the activity of this enzyme (Madinabeitia et al. 2002). Sxt1 and other OCTASEs had a C-terminus that extended beyond that of OSGP, which provided two highly conserved sequence regions. A BLOCKS search (Henikoff et al. 2000) identified these two regions as signature sequences for OCTASE (IPB003696B, IPB003696D). They are likely part of the catalytic site in OCTASEs. Sxt1 from L. wollei was approximately half the size of other Sxt1 orthologues, due to a deletion in its ATP-binding site, and a truncated C-terminus that provided the two OCTASE signature sequences (Fig. 4). This enzyme is therefore, in all likelihood, nonfunctional, and represented a natural knockout mutant of sxt1. The lack of capability of L. wollei to produce any carbamoylated analogues of saxitoxin (Onodera et al. 1997) therefore confirmed strong support for the predicted function of Sxt1 in saxitoxin biosynthesis.
Phylogeny and Evolution of Sxt1
OCTASE genes were represented in the genomes of eubacteria, archaea, and two bacteriophages (P-SSM2 and X15), however, they were absent from eukaryotic genomes. The largest number of OCTASE genes was found in proteobacteria (170 of a total of 269), whereas only six and three sequences were detected in the genomes of archaea and bacteriophages, respectively. OCTASE is thus a true prokaryotic gene family. Deduced gene products had structural and sequence homology to OSGP, which is represented in the genomes of every sequenced organism and essential for cell survival. OSGP is therefore most likely ancestral to OCTASE, which may have evolved from an ancestral prokaryotic OSGP. The fact that the greatest diversity of OCTASE genes was found in proteobacteria may indicate that this gene family has its phylogenetic origin in this division of bacteria.
It has been generally accepted that horizontal gene transfer is a major driving force in the evolution of bacterial genomes (Gogarten and Townsend 2005). The variable phylogenetic presence of OCTASE genes, combined with a phylogeny that is incongruent with the organismic phylogeny, is a strong indicator that this gene family has an evolutionary history with frequent horizontal gene transfers (Lerat et al. 2005). This is supported by the fact that OCTASE genes are often encoded on mobile genetic elements, such as plasmids, and in viral genomes.
Only a small proportion of cyanobacterial strains (11 strains of 64) that have a completely sequenced genome harbored OCTASE genes. They were limited to strains from the orders Chroococcales, Prochlorales, and Oscillatoriales, whereas sxt1 was the first OCTASE gene that has been detected among Nostocales members. The phylogeny of Sxt1 and other cyanobacterial OCTASE genes was incongruent with the organismic phylogeny, as determined by 16S rDNA (Fig. 3a and b), and suggested that there may have been several horizontal OCTASE gene transfers among cyanobacterial species. OCTASE genes from two Oscillatoriales, T. erythraeum and Lyngbya sp. PCC 8106, grouped with sequences from two Chroococcales, C. watsonii and Cyanothece sp. CCY0110. Thus there may have been genetic exchange between strains of the Chroococcales and Oscillatoriales. On the other hand, the separation of the OCTASE gene from Synechococcus sp. RCC307 (Prochlorales according to 16S rDNA phylogeny) from other sequences may indicate that this strain has acquired the gene in an independent event (Fig. 3a). The phylogeny of sxt1 may suggest that this gene has spread to cyanobacterial phyla, which previously did not include any PSP-toxic member. PSP-toxic cyanobacteria identified so far belong to the Nostocales, apart from one member of the Oscillatoriales, L. wollei (Fig. 3b). sxt1 from L. wollei grouped, however, with an orthologue from C. raciborksii T3 (Fig. 3a). It is therefore likely that L. wollei may have acquired sxt1 from a Nostocales member. A. circinalis has evolved into two phylotypes, one consisting predominantly of PSP-toxic strains, and the other of non-PSP-toxic strains, apart from one strain, A. circinalis AWQC134C (Beltran and Neilan 2000). sxt1 did not resolve according to the phylotype, suggesting that it has been acquired in A. circinalis AWQC134C after it speciated into its phylotype. In the case of both L. wollei and A. circinalis AWQC134C, sxt1 may have moved by horizontal gene transfer to strains from phylogenetic groups that otherwise did not include any PSP-toxic members. The biosynthesis of PSP toxins may require 12 or more different genes (Kellmann and Neilan 2007), and this study suggests that all genes required for their synthesis may spread by horizontal gene transfer. Considering the prokaryotic origin sxt1, which represents a key saxitoxin biosynthesis gene, it is reasonable to assume that PSP-toxin biosynthesis has evolved in a prokaryotic organism and subsequently spread via horizontal gene transfer to other organisms. It was not clear from the present study whether PSP-toxin biosynthesis may have evolved in ancestral cyanobacteria after sxt1 was acquired, or whether cyanobacteria have acquired the entire metabolic pathway from a noncyanobacterial source. If PSP-toxin biosynthesis is carried out by orthologous enzymes in dinoflagellates, then they must have acquired the required genes from a bacterial source. Sequencing of the entire gene clusters from PSP-toxic cyanobacteria and screening of EST libraries from PSP-toxic dinoflagellates with sxt1-specific probes are under way to determine the evolution of this intriguing metabolic pathway.
Conclusion
A gene that was designated sxt1 has been identified, which is involved in the biosynthesis of PSP toxins. It encoded an enzyme that is related to OCTASE. Several lines of evidence were presented that supported the proposed function of the sxt1 gene product. The involvement of OCTASE in saxitoxin biosynthesis has been predicted previously, based on precursor incorporation studies and biochemical assays. sxt1 was exclusively detected in PSP-toxic strains of cyanobacteria, whereas it was not present in closely related, non-PSP-toxic strains. sxt1 genes had high sequence similarity to each other, and clustered on a phylogenetic branch that was divergent from OCTASE genes of other organisms, indicating that they represented orthologous genes. A natural knockout mutant of sxt1 was identified in L. wollei, which had a deleted ATP-binding site and truncated C-terminus. This species produced only decarbamoylated analogues of saxitoxin, which strongly supported the proposed function of Sxt1. The OCTASE gene family may have evolved in ancestral proteobacteria by gene duplication of a member of OSGP and subsequently spread through multiple horizontal gene transfer events to a diverse range of prokaryotes, including cyanobacteria. Cyanobacterial OCTASE genes were derived from multiple lineages and may have been acquired during independent horizontal gene transfer events. The most likely phylogenetic origin of sxt1 orthologues was proteobacteria. This study thus suggests that a key PSP-toxin biosynthesis gene may have evolved in a prokaryotic organism and subsequently spread via horizontal gene transfer to cyanobacteria.
References
Aravind L, Koonin EV (1999) Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J Mol Biol 287:1023–1040
Beitz E (2000) TeXshade: shading and labeling of multiple sequence alignments using LaTeX2e. Bioinformatics 16:135–139
Beltran EC, Neilan BA (2000) Geographical segregation of the neurotoxin-producing cyanobacterium Anabaena circinalis. Appl Environ Microbiol 66:4468–4474
Brewer S, Taylor P, Turner M (1980) An adenosine triphosphate-dependent carbamoylphosphate-3-hydroxymethylcephem O-carbamoyltransferase from Streptomyces clavuligerus. Biochem J 185:555–564
Coque JJ, Perez-Llarena FJ, Enguita FJ, Fuente JL, Martin JF, Liras P (1995) Characterization of the cmcH genes of Nocardia lactamdurans and Streptomyces clavuligerus encoding a functional 3′-hydroxymethylcephem O-carbamoyltransferase for cephamycin biosynthesis. Gene 162:21–27
Coque JJ, Enguita FJ, Cardoza RE, Martin JF, Liras P (1996) Characterization of the cefF gene of Nocardia lactamdurans encoding a 3′-methylcephem hydroxylase different from the 7-cephem hydroxylase. Appl Microbiol Biotechnol 44:605–609
Daly JW (2004) Marine toxins and nonmarine toxins: convergence or symbiotic organisms? J Nat Prod 67:1211–1215
Du L, Sanchez C, Chen M, Edwards DJ, Shen B (2000) The biosynthetic gene cluster for the antitumor drug bleomycin from Streptomyces verticillus ATCC15003 supporting functional interactions between nonribosomal peptide synthetases and a polyketide synthase. Chem Biol 7:623–642
Felsenstein J (1989) PHYLIP. Phylogeny inference package. Cladistics 5:164–166
Gallacher S, Flynn KJ, Franco JM, Brueggemann EE, Hines HB (1997) Evidence for production of paralytic shellfish toxins by bacteria associated with Alexandrium spp. (Dinophyta) in culture. Appl Environ Microbiol 63:239–245
Gogarten JP, Townsend JP (2005) Horizontal gene transfer, genome innovation and evolution. Nat Rev Microbiol 3:679–687
Hackett JD, Scheetz TE, Yoon HS, Soares MB, Bonaldo MF, Casavant TL, Bhattacharya D (2005) Insights into a dinoflagellate genome through expressed sequence tag analysis. BMC Genom 6:80
Hallegraeff GM (1995) Harmful algal blooms: A global overview. In: Hallegraeff GM, Anderson DM, Cembella AD (eds) Manual on harmful marine microalgae. UNESCO, Paris, pp 1–22
Haydock SF, Appleyard AN, Mironenko T, Lester J, Scott N, Leadlay PF (2005) Organization of the biosynthetic gene cluster for the macrolide concanamycin A in Streptomyces neyagawaensis ATCC 27449. Microbiology 151:3161–3169
Henikoff JG, Greene EA, Pietrokovski S, Henikoff S (2000) Increased coverage of protein families with the blocks database servers. Nucleic Acids Res 28:228–230
Honda D, Yokota A, Sugiyama J (1999) Detection of seven major evolutionary lineages in cyanobacteria based on the 16S rRNA gene sequence analysis with new sequences of five marine Synechococcus strains. J Mol Evol 48:723–739
Jabbouri S, Fellay R, Talmont F, Kamalaprija P, Burger U, Relic B, Prome JC, Broughton WJ (1995) Involvement of nodS in N-methylation and nodU in 6-O-carbamoylation of Rhizobium sp. NGR234 nod factors. J Biol Chem 270:22968–22973
Jabbouri S, Relic B, Hanin M, Kamalaprija P, Burger U, Prome D, Prome JC, Broughton WJ (1998) nolO and noeI (HsnIII) of Rhizobium sp. NGR234 are involved in 3-O-carbamoylation and 2-O-methylation of Nod factors. J Biol Chem 273:12047–12055
Jukes TH, Cantor CR (1969) Evolution of protein molecules. In: Munro HN (ed) Mammalian protein metabolism. Academic Press, New York, pp 21–132
Kaas H, Henriksen P (2000) Saxitoxins (PSP toxins) in Danish lakes. Water Res 34:2089–2097
Kao CY, Levinson SR (1986) Tetrodotoxin, Saxitoxin, and the molecular biology of the sodium channel. In: Boland B, Cullinan J, Cohn T (eds) Annals of the New York Academy of Science. New York Academy of Science, New York, pp 1–445
Kellmann R, Neilan BA (2007) Biochemical characterisation of paralytic shellfish toxin biosynthesis in vitro. J Phycol 43:497–508
Kellmann R, Michali TK, Jeon YJ, Pickford R, Pomati F, Neilan BA (2008) Biosynthetic intermediate analysis and functional homolgy reveal a saxitoxin gene cluster in cyanobacteria. Appl Environ Microbiol 74:4044–4053
Kharel MK, Basnet DB, Lee HC, Liou K, Woo JS, Kim BG, Sohng JK (2004) Isolation and characterization of the tobramycin biosynthetic gene cluster from Streptomyces tenebrarius. FEMS Microbiol Lett 230:185–190
Kodoma M, Ogata T, Sato S (1988) Bacterial production of saxitoxin. Agr Biol Chem 52:1075–1077
Kodoma M, Ogata T, Sakamoto S, Sato S, Honda T, Miwatani T (1990) Production of paralytic shellfish toxins by a bacterium Moraxella sp. isolated from Protogonyaulax tamarensis. Toxicon 28:707–714
Kotaki Y, Tajiri M, Oshima Y, Yasumoto T (1983) Identification of a calcareous red alga as the primary source of paralytic shellfish toxins in coral reef crabs and gastropods. Bull Jpn Soc Sci Fish [Nippon Suisan Gakkaishi] 49:283–286
Lerat E, Daubin V, Ochman H, Moran NA (2005) Evolutionary origins of genomic repertoires in bacteria. PLoS Biol 3:e130
Madinabeitia N, Bellogin RA, Buendia-Claveria AM, Camacho M, Cubo T, Espuny MR, Gil-Serrano AM, Lyra MC, Moussaid A, Ollero FJ, Soria-Diaz ME, Vinardell JM, Zeng J, Ruiz-Sainz JE (2002) Sinorhizobium fredii HH103 has a truncated nolO gene due to a -1 frameshift mutation that is conserved among other geographically distant S. fredii strains. Mol Plant Microbe Interact 15:150–159
Mao Y, Varoglu M, Sherman DH (1999) Molecular characterization and analysis of the biosynthetic gene cluster for the antitumor antibiotic mitomycin C from Streptomyces lavendulae NRRL 2564. Chem Biol 6:251–263
Moffitt MC, Neilan BA (2004) Characterization of the nodularin synthetase gene cluster and proposed theory of the evolution of cyanobacterial hepatotoxins. Appl Environ Microbiol 70:6353–6362
Nakashima K, Arakawa O, Taniyama S, Nonaka M, Takatani T, Yamamori K, Fuchi Y, Noguchi T (2004) Occurrence of saxitoxins as a major toxin in the ovary of a marine puffer Arothron firmamentum. Toxicon 43:207–212
Neilan BA (1995) Identification and phylogenetic analysis of toxigenic cyanobacteria by multiplex randomly amplified polymorphic DNA PCR. Appl Environ Microbiol 61:2286–2291
Neilan BA, Jacobs D, DelDot T, Blackall LL, Hawkins PR, Cox PT, Goodman AE (1997) rRNA sequences and evolutionary relationships among toxic and nontoxic cyanobacteria of the genus Microcystis. Int J Syst Bacteriol 47:693–697
Onodera H, Satake M, Oshima Y, Yasumoto T, Carmichael Wayne W (1997) New saxitoxin analogues from the freshwater filamentous cyanobacterium Lyngbya wollei. Nat Toxins 5:146–151
Oshima Y, Kotaki Y, Harada T, Yasumoto T (1984) Paralytic shellfish toxins in tropical waters. In: Ragelis E (ed) Seafood toxins. American Chemical Society, Washington, DC, pp 160–170
Pereira P, Onodera H, Andrinolo D, Franca S, Araujo F, Lagos N, Oshima Y (2000) Paralytic shellfish toxins in the freshwater cyanobacterium Aphanizomenon flos-aquae, isolated from Montargil reservoir, Portugal. Toxicon 38:1689–1702
Perrière G, Gouy M (1996) WWW-Query: an on-line retrieval system for biological sequence banks. Biochemie 78:364–369
Plumley FG (2001) Purification of an enzyme involved in saxitoxin synthesis. J Phycol 37:926–928
Pomati F, Neilan BA (2004) PCR-based positive hybridization to detect genomic diversity associated with bacterial secondary metabolism. Nucleic Acids Res 32:e7
Pomati F, Burns BP, Neilan BA (2004) Identification of an Na(+)-dependent transporter associated with saxitoxin-producing strains of the cyanobacterium Anabaena circinalis. Appl Environ Microbiol 70:4711–4719
Pomati F, Kellmann R, Burns BP, Cavaliere R, Neilan BA (2006) Comparative gene expression studies of PSP-toxins producing and non-toxic Anabaena circinalis strains and effects of lidocaine hydrochloride. Environ Int 32:734–748
Rascher A, Hu Z, Buchanan GO, Reid R, Hutchinson CR (2005) Insights into the biosynthesis of the benzoquinone ansamycins geldanamycin and herbimycin, obtained by gene sequencing and disruption. Appl Environ Microbiol 71:4862–4871
Rice P, Longden I, Bleasby A (2000) EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet 16:276–277
Saitou N, Nei M (1987) The neighbour-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sato S, Kodoma M, Ogata T, Saitanu K, Furuya M, Hirayama K, Kakinuma K (1997) Saxitoxin as a toxic principle of a freshwater puffer Tetraodon fugi in Thailand. Toxicon 35:137–140
Shimizu Y (1993) Microalgal metabolites. Chem Rev 93:1685–1698
Siebert PD, Chenchik A, Kellogg DE, Lukyanov KA, Lukyanov SA (1995) An improved PCR method for walking in uncloned genomic DNA. Nucleic Acids Res 23:1087–1088
Sivonen K (1996) Cyanobacterial toxins and toxin production. Phycologia 35:12–24
Steffensky M, Muhlenweg A, Wang ZX, Li SM, Heide L (2000) Identification of the novobiocin biosynthetic gene cluster of Streptomyces spheroides NCIB 11891. Antimicrob Agents Chemother 44:1214–1222
Su Z, Sheets M, Ishida H, Li F, Barry WH (2004) Saxitoxin blocks L-type ICa. J Pharmacol Exp Ther 308(1):324–329
Thompson AS, Rhodes JC, Pettman I (1988) Catalogue of strains. Natural Environment Research Council Culture Collection of Algae and Protozoa, p 22
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Wang J, Salata JJ, Bennett PB (2003) Saxitoxin is a gating modifier of HERG K+ channels. J Gen Physiol 121:583–598
Yotsu-Yamashita M, Kim YH, Dudley SC Jr, Choudhary G, Pfahnl A, Oshima Y, Daly JW (2004) The structure of zetekitoxin AB, a saxitoxin analog from the Panamanian golden frog Atelopus zeteki. Proc Natl Acad Sci USA 101:4346–4351
Yu TW, Bai L, Clade D, Hoffmann D, Toelzer S, Trinh KQ, Xu J, Moss SJ, Leistner E, Floss HG (2002) The biosynthetic gene cluster of the maytansinoid antitumor agent ansamitocin from Actinosynnema pretiosum. Proc Natl Acad Sci USA 99:7968–7973
Zaman L, Arakawa O, Shimosu A, Onoue Y (1997) Occurrence of paralytic shellfish poison in Bangladeshi freshwater puffers. Toxicon 35:423–431
Acknowledgments
The Australian Research Council is thanked for its financial support. Wayne Carmichael, Peter Baker, and Martin Saker are thanked for providing cyanobacterial samples.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00239-009-9210-0
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Kellmann, R., Michali, T.K. & Neilan, B.A. Identification of a Saxitoxin Biosynthesis Gene with a History of Frequent Horizontal Gene Transfers. J Mol Evol 67, 526–538 (2008). https://doi.org/10.1007/s00239-008-9169-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-008-9169-2