
Abstract
Prokaryotic organisms preferentially utilize less energetically costly amino acids in highly expressed genes. Studies have shown that the proteome of Saccharomyces cerevisiae also exhibits this behavior, but only in broad terms. This study examines the question of metabolic efficiency as a proteome-shaping force at a finer scale, examining whether trends consistent with cost minimization as an evolutionary force are present independent of protein function and amino acid physicochemical property, and consistently with respect to amino acid biosynthetic costs. Inverse correlations between the average amino acid biosynthetic cost of the protein product and the levels of gene expression in S. cerevisiae are consistent with natural selection to minimize costs. There are, however, patterns of amino acid usage that raise questions about the strength (and possibly the universality) of this selective force in shaping S. cerevisiae’s proteome.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Functional selection is typically considered to be the dominant force shaping proteome evolution. Mutations that give rise to changes in protein structure can lead to alterations of function that affect fitness (Nei 1975). An evolutionary force that is less gene-centric and more global in nature, metabolic efficiency, recently has been shown to influence prokaryotic proteome evolution (Akashi and Gojobori 2002; Heizer Jr. et al. 2006; Swire 2007). Relationships between the expressivity and the average amino acid biosynthetic cost of an organism’s proteins have been put forth as evidence of metabolic efficiency as a proteome-wide evolutionary force. Highly expressed proteins would be expected to avoid the use of biosynthetically expensive amino acids and preferentially utilize biosynthetically inexpensive amino acids.
Others have searched for evidence of this selective force in eukaryotes (Urrutia and Hurst 2003; Seligmann 2003; Kahali et al. 2007; Swire 2007) but have limited their analysis to single proxies for expressivity (Swire actually avoided the use of expression data altogether), have employed the use of protein biosynthesis cost measures that are not specific to the species under study, and have not determined whether the effects are limited to specific amino acids (hydrophobic vs. hydrophylic) or to proteins from specific functional categories (though Kahali et al. did show that it was independent of protein secondary structure, and Swire did show consistency across functional categories for one organism, S. cerevisiae). The work described here examines in detail the question of whether metabolic efficiency affects proteome evolution and employs multiple surrogates for expressivity, determines if the effects are independent of amino acid physicochemical property and protein function, and explicitly considers the differences in amino acid biosynthetic costs associated with both aerobic and anaerobic metabolism in yeast. The results reveal the existence of significant and persistent trends in amino acid usage. Some of these trends are consistent with an evolutionary pressure to reduce the metabolic cost for proteins. Others raise new questions about the universality of such a selective force in the yeast proteome.
Methods
While a useful concept in the abstract, “expressivity” is difficult to quantify—especially on a proteome-wide scale. For this reason six separate predictors of expressivity were employed: protein abundance, transcript abundance, and adherence to codon usage bias, under both aerobic and anaerobic growth conditions. All comparisons between biosynthetic cost and expressivity were performed using like data (i.e., aerobic cost vs. aerobic expression data and anaerobic cost vs. anaerobic expression data).
Protein Abundance Data
Aerobic protein abundance data were drawn from the supplemental material of analysis by Ghaemmaghami et al. (2003). This study grew 1.7-ml cultures in a 96-well format to log phase. Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE)/western blot analysis was utilized to examine total cell extracts.
Anaerobic transcript abundance data were obtained from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), accession number GSM177360. The study performed a quantitative proteomic analysis of anaerobic and aerobic yeast cultures. The results were in the form of a protein expression ratio (anaerobic/aerobic). For this work, expression values for each protein were determined by multiplying the aerobic expression data described above (as generated by Ghaemmaghami et al. 2003] by the protein expression ratio. Proteins shown to be expressed under aerobic-only conditions were set to 0. Likewise, proteins shown to be expressed under anaerobic-only conditions were set to 0, as a ratio multiplication of aerobic data would be meaningless. The total number of proteins with anaerobic expression data was 626.
Transcript Abundance Data
Aerobic transcript abundance data were obtained from the supplementary materials of Holstege et al. (1998). Anaerobic transcript abundance data were obtained from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), accession number GSE5926. The study measured anaerobic transcriptional response to weak organic acids in chemostat cultures of S. cerevisiae. Reference (untreated) data were utilized in this study (average expression values of three reference data sets).
Adherence to Codon Usage Bias
Each gene’s adherence to synonymous codon usage bias was established by calculating the degree to which the gene has adapted its codon usage to that exhibited by highly expressed genes. A codon adaptation index (CAI) value was determined for each gene in the S. cerevisiae genome using the techniques developed by Sharp and LI (1987). The set of highly expressed genes utilized to determine the relative adaptiveness (or weight) for each codon was the top 1% of genes drawn from the respective transcript abundance data (aerobic transcript abundance for aerobic CAI and anaerobic transcript abundance for anaerobic CAI). Transcript abundance was chosen over protein abundance due to the relatively small number of proteins with anaerobic expression values (626; see Protein Abundance Data, above).
Sequence Data
Sequence data were obtained from the National Center for Biotechnology Information (NCBI; http://www.ncbi.nih.gov/Genomes/) for all 16 chromosomes of S. cerevisiae (Goffeau et al. 1996). Genes with fewer than 100 codons (not including the start and stop codon) were removed from consideration to minimize sampling effects and potential length biases (Eyre-Walker 1996).
Genes that were recent additions to the genome (horizontally transferred) were removed from consideration since they may not reflect yeast’s codon usage bias (dos Reis et al. 2003). The techniques employed by Garcia-Vallvé et al. (2003) were utilized to identify candidates for horizontal gene transfer (HGT). While it has been shown that eukaryotes experience HGT (Jain et al. 1999), it has been noted that the GC content in yeast is unusually malleable (genes acquired by HGT are expected to yield rapidly to the factors that govern GC-content trends in yeast’s genome), such that all genes that have been horizontally transferred may not have been identified by this approach (Hall et al. 2005). In summary, genes that are extraneous in terms of GC content and codon usage (excluding highly expressed genes and those that code for proteins that deviate from organismal amino acid content) were considered candidates for HGT. Clusters of high- or low-GC-content genes (identified by a sliding 11-gene window) were also considered likely to have been acquired. In this implementation the GC-content values (GCT, GC1, GC2, and GC3) were determined by considering all chromosomes combined. This was due to the relatively uniform GC content found across the chromosomes (μ = 38.4%, σ = 0.36%).
To prevent oversampling effects, only one paralogue was retained from each set of paralogous genes. Paralogues were identified using unfiltered BLAST (http://www.ncbi.nlm.nih.gov/BLAST/) searches (Altschul et al. 1990) against the S. cerevisiae proteome. Proteins with >60% amino acid identity in these proteomic searches were considered to be paralogous. Only the single paralogue with the highest expression level (as predicted by CAI) was retained. Analysis was performed both with and without genes containing introns, with similar results. Genes (304) containing introns were included in this study. A total of 4459 genes were included in this study after culling was completed (of 5858 coding sequences initially considered, 320 were removed due to being <100 codons, 671 due to likelihood of relatively recent HGT, 403 due to membership in paralogue clusters, and 5 because of differences between the predicted amino acid sequence of the gene and the amino acid sequence provided in the accompanying GenBank file).
Protein Production Cost
The approach taken to calculate biosynthetic costs was first employed by Craig and Weber (1998), and by Akashi and Gojobori (2002), and it exploited the near-universality of biosynthetic pathways to determine the number of high-energy phosphate bonds (~PO4) required to synthesize amino acids. To ensure that the results reflect actual biosynthetic costs to the organism, the energy lost (that could have been produced if the precursors had not been removed from energy metabolism) is added to the total number of high-energy phosphate bonds expended. The majority of amino acid synthesis cost can be attributed to potential energy lost by diverting metabolic intermediates to amino acid production. Wagner (2005) performed this analysis for both fermentative and respiratory conditions for the S. cerevisiae proteome, and the values he derived are utilized in this work. It should be noted that the conversion of reducing power to the common currency of high-energy phosphate bonds is not straightforward under anaerobic respiration. Wagner’s calculations assume 0H per ATP (under ethanol fermentation). There are alternative pathways for amino acid production (http://pathway.yeastgenome.org/biocyc/), however, their costs tend to be similar to those described here (Tables 1 and 2). The protein production costs were determined by calculating the average (per amino acid) number of high-energy phosphate bonds (~PO4) required for the synthesis of the protein’s constituent amino acids. The biosynthesis costs associated with start codons were not considered, as these costs are constant across all proteins. Similarly, stop codons do not code for an amino acid and were not included in the analysis.
Statistical Analyses
To compare the average biosynthetic cost of S. cerevisiae’s proteins and their three expressivity measures (CAI, transcript abundance, and protein abundance), a Spearman (1904) rank correlation was performed (significance set at α = 0.05). To determine whether the effects of cost selection are experienced by proteins independent of their function, correlations between average amino acid biosynthetic cost and expression level were determined on genes in each of the 15 functional categories (as listed in the Comprehensive Yeast Genome Database [Güldener et al. 2005]). Those genes that were labeled “classification not yet clear-cut” or “unclassified proteins” or that were in functional categories containing fewer than 50 genes were excluded.
Individual amino acid usage was examined to determine whether cost selection is experienced consistently across all amino acids (biosynthetically inexpensive amino acids should exhibit preferential use in highly expressed genes, while biosynthetically expensive amino acids should exhibit avoidance). To determine whether amino acid usage is consistent with biosynthetic cost, a Spearman rank correlation was calculated between the gene’s usage of each amino acid and the gene’s expression level. A Mantel-Haenszel (1959) test was used to determine whether functional category was a confounding factor in the amino acid usage analysis. The Mantel-Haenszel test stratifies the amino acid usage data by functional category into 2 × 2 contingency tables where the counts of the amino acid (for a given functional category) are reported as highly or weakly expressed, and as a count of the target amino acid and a count of all other amino acids. Genes falling below the median expression value were designated “weakly expressed,” while those falling above the median were designated “highly expressed.” The threshold for significance for both tests was set at α = 0.05 with a sequential Bonferroni correction.
Results
Correlation Between Gene Expressivity and Amino Acid Production Cost
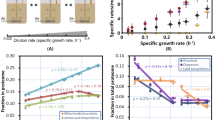
Statistically significant negative Spearman rank correlations (p < 0.05; Table 3, overall values; Fig. 1) were found between all expression measures (degree to which translationally preferred codons are used, degree of transcript abundance, and degree of protein abundance) and the average biosynthetic cost per encoded amino acid for both aerobic and anaerobic pathways in S. cerevisiae.

Comparison of average aerobic and anaerobic cost in high-energy phosphate bonds (~PO4) and CAI, protein abundance, and transcript abundance in Saccharomyces cerevisiae. Each point represents binned protein data containing approximately one-twentieth of the proteome’s amino acids. The exact number varies slightly among bins, to avoid dividing the amino acids for a single protein between two bins. Error bars represent standard errors of the means of the bins. X-axis values calculated separately for aerobic and anaerobic data (e.g., aerobic expression data used in conjunction with aerobic cost data)
Correlation Between Expressivity and Cost in Functional Categories
To demonstrate whether cost selection is experienced by proteins independent of their function, the protein data have been separated by functional category (as determined using the Comprehensive Yeast Genome Database [CYGD] [Güldener et al. 2005]). Spearman rank correlations were calculated for expression data vs. amino acid metabolic cost, both aerobic and anaerobic, for the proteins in each of the functional categories. The database classifies proteins into 1 of 15 different functional categories. Significance is determined in the traditional manner by calculating a t-statistic based on the Spearman rank correlation coefficient and the number of proteins in the analysis. The t-statistic is then used to determine a p-value. No single category appears to be responsible for the correlation between the expression data and the average amino acid production cost (Table 4). All 15 categories exhibited either negative or statistically insignificant correlations between the average amino acid cost and the expression measures.
Correlation Between Gene Expressivity and Cost in Hydrophilic, Hydrophobic, and Ambivalent Amino Acids
To determine whether selection for cost is independent of amino acid physicochemical property, the 20 common amino acids were separated into three physicochemical categories: hydrophilic, hydrophobic, and ambivalent. Membership in each class was taken from Zubay (1998). Hydrophobic amino acids are found primarily in the protein core, while hydrophilic amino acids tend to be polar and charged amino acids. Ambivalent amino acids are amphipathic or borderline residues. A Spearman rank correlation test was employed to determine whether cost selection trends exist for amino acids in each of these three classes (Table 3 and Fig. 2; Fig. 2 utilizes transcript abundance only).

Comparison of transcript abundance and average aerobic and anaerobic cost in high-energy phosphate bonds (~PO4) among hydrophilic, hydrophobic, and ambivalent amino acids in S. cerevisiae. Each point represents binned protein data containing approximately one-twentieth of the proteome’s amino acids. The exact number varies slightly among bins, to avoid dividing the amino acids for a single protein between two bins. Error bars represent standard errors of the means of the bins. X-axis values calculated separately for aerobic and anaerobic data (e.g., aerobic expression data used in conjunction with aerobic cost data)
For the hydrophobic (Phe, Leu, Ile, Met, and Val) and ambivalent (Trp, Tyr, Cys, Ala, Ser, Gly, Pro, and Thr) amino acids, statistically significant negative correlations between expressivity and amino acid cost were seen for all but one of the expression measures and cost structures (p < 0.05; Table 3) (the exception was protein abundance vs. anaerobic cost—hydrophobic amino acids). Hydrophilic amino acids (His, Arg, Lys, Gln, Glu, Asn, and Asp), however, display only two expression correlations that were significant (anaerobic, CAI r S = −0.167, p < 0.0005; and anaerobic, transcript abundance r S = −0.031, p < 0.05).
Amino Acid Utilization and Gene Expression Levels
The proportional usage of amino acids in proteins was compared to expressivity to determine whether amino acid usage was consistent with respect to biosynthetic cost (Tables 1 and 2). These results are similar to those obtained previously by one of the authors of this work (Akashi 2003). The findings are somewhat different in that the r S values found herein are based on unbinned data and are, therefore, smaller in magnitude, and the Mantel-Haenszel test is stratified by a different set of functional categories (Güldener et al. 2005). Cases where the Spearman rank correlation is not significant but the Mantel-Haenszel Z statistic is significant (such as for Lys) can be explained by the nature of the Mantel-Haenszel test. The test is used here to determine whether trends that may be present are driven by genes from a subset of the functional categories, not whether (and to what extent) a correlation exists. Also, the data used in the Mantel-Haenszel test are binary (2 × 2 contingency tables populated with target and nontarget amino acid counts in highly expressed and weakly expressed genes), whereas the Spearman rank correlations are performed using a ranking on expression and average cost data.
The results demonstrate that three low-cost amino acids tend to increase in usage with expressivity, both overall and within each of the 15 functional categories examined. Ala, Gly, and Val exhibited statistically significant positive trends (p < 0.0005 for all r S and Mantel-Haenszel Z-scores except for anaerobic/Val, using protein abundance, for which p < 0.005). Val is the most biosynthetically expensive of these three, and there are four amino acids whose costs (either aerobic or anaerobic) were less than that of Val that did not follow expected trends (Tables 1 and 2). Cys, Arg, Asn, and Ser are biosynthetically inexpensive (aerobically less biosynthetically expensive than Val) and have usage values that exhibited a significantly negative trend with respect to expressivity (p < 0.0005 for both r S and Mantel-Haenszel Z-scores for all three expression measures). Ser is biosynthetically inexpensive (anaerobically less biosynthetically expensive than Val) and has a usage value that exhibited a significantly negative trend with respect to expression measures under anaerobic conditions (p < 0.0005 for both r S and Mantel-Haenszel Z-scores for all three expression measures). These unexpected negative trends were generally consistent across all functional categories (as evidenced by the Mantel-Haenszel tests) but were not enough to nullify the overall trend in the S. cerevisiae proteome of using less expensive amino acids in highly expressed proteins (Table 3), suggesting that the trend to biosynthetic conservation is driven primarily by small, aliphatic amino acids. With these three amino acids removed the overall Spearman rank correlations between all six expression measures (transcript abundance, protein abundance, and CAI, measured under both aerobic and anaerobic conditions) and their respective biosynthetic costs (aerobic and anaerobic) become significantly positive (+0.136, +0.135, and +0.091 [aerobic] and +0.160, +0.146, and +0.123 [anaerobic] for CAI, protein abundance, and transcript abundance, respectively; p < 0.0005 for all). Conservation of amino acid biosynthetic costs is considered equally in both aerobic and anaerobic metabolic modes. This is reasonable in light of the fact that these costs are highly correlated (r = 0.514, p < 0.05) (costs taken from Tables 1 and 2).
Consistency in Amino Acid Utilization
The inconsistencies in amino acid usage described above differ markedly from the results of Swire (2007). Using overall amino acid biosynthetic costs of proteins rather than estimates of expression levels as a predictor, Swire showed that usage of expensive amino acids tends to increase with overall protein cost (positive gradient), while usage of inexpensive amino acids tends to decrease (negative gradient). Biases in expression estimates or amino acid usage trends driven by structural constraints may underlie the incongruence between Swire’s results and our findings. In particular, aerobic amino acid costs are correlated with amino acid hydrophobicity scores (r = 0.52, p = 0.02) (hydrophobicity values taken from Black and Mould [1991]). If yeast proteins with high average hydrophobicity consistently use amino acids that exhibit high hydrophobicity (and proteins with low average hydrophobicity consistently use amino acids with low hydrophobicity), gradient consistency analysis may reveal amino acid usage consistent with cost minimization. It should also be noted that gradient consistency tests for the S. cerevisiae proteome using anaerobic metabolic costs show inconsistent amino acid usage (r 2 = 0.08, p = 0.2) (to make accurate comparisons with Swire results, the p-value was calculated using Kendall’s tau nonparametric approach).
Discussion
Evidence of selection for cost minimization may consist of weak correlations (Table 3; see also Tables 1 and 2) and subtle trends, globally, across the entire proteome (Fig. 1), and such patterns can be obscured by competing evolutionary forces. The results described here, and in the work of others (Kahali et al. 2007), show overall negative correlations between protein synthesis cost and expression level in yeast (Table 3). Protein size varies inversely with expressivity (Akashi 2003; Urrutia and Hurst 2003; Seligmann 2003), yielding a metabolic cost savings to the organism. It is these trends that have been cited as evidence that metabolic efficiency is a selective force at work shaping proteome evolution in S. cerevisiae. However, there is more to the story, in that some trends appear to be inconsistent with a cost minimizing selective force. To the extent possible (for continued protein functionality), one would expect the cost minimization trends to be exhibited across all amino acids, regardless of physicochemical property and independent of protein functional category. Utilization of metabolically inexpensive amino acids should increase in highly expressed genes, while usage of expensive amino acids should decrease. Indeed, this has been shown to be true for prokaryotic proteomes (Akashi and Gojobori 2002; Heizer Jr. et al. 2006).
The yeast proteome, however, exhibits some characteristics inconsistent with cost minimization. For example, when hydrophilic amino acids are considered, only two of the correlations are significant (transcript abundance/aerobic, r S = −0.03, p < 0.05, and CAI/anaerobic, r S = −0.167, p < 0.0005, between biosynthetic cost and expressivity; Table 3). The rest of the trends are not significant (p > 0.05). A possible explanation for this behavior is the predominance of ribosomal protein coding genes in the set of highly expressed genes. Ribosomal proteins interact with negatively charged RNA species and this necessitates the utilization of an unusually large number of positively charged amino acids such as Asp, Glu, and Lys. The average mole percentages (22.1 mol%; σ = 4.8%) of these three amino acids is higher in proteins involved in protein synthesis compared to all other yeast proteins (19.8 mol%, σ = 5.1%). Escherichia coli displays a similar trend for protein synthesis proteins (e.g., 17.5 mol% of Asp, Glu, and Lys; σ = 3.7%) vs. for all other E. coli proteins (15.0 mol%; σ = 5.0%).
Along with the unusual biosynthetic cost vs. expressivity trends in hydrophilic amino acids, there are some unexplained amino acid frequency relationships. The most biosynthetically expensive amino acid (aerobic, Trp) exhibits no significant trend in usage with respect to expressivity (Table 1; to be considered significant, both r S and Mantel-Haenszel Z-score must be <0.05; not significant for all three expression measures). The most biosynthetically expensive amino acid utilizing anaerobic costs is Met, which also exhibits no significant trend (Table 2). The other aromatic amino acids (Tyr and Phe) which also are aerobically biosynthetically expensive generally show no significant trend in usage vs. expression data acquired under aerobic conditions (Tyr has a significant negative trend with respect to aerobic transcript abundance; however, the Mantel-Haenszel statistic is not significant, indicating that the trend is not consistent across functional categories) (Tables 1 and 2). Additionally, there are several amino acids with a low biosynthetic cost that exhibit negative correlations with all three expressivity measures (Ser for anaerobic costs and Arg, Asn, Cys, His, and Ser, e.g., for aerobic) (Tables 1 and 2).
There are a number of possible explanations. One is that the trend is driven by only a few highly variable amino acids (i.e., Val, Gly, and Ala). The choice of replacement amino acids may be more constrained in the complex environments in which yeast and other eukaryotes find themselves. There are more protein-to-protein interfaces and more overall specificity requirements in eukaryotic proteomes (Brocchieri and Karlin 2005; Warringer and Blomberg 2006). With these three amino acids removed the overall Spearman rank correlations between all six expression measures (transcript abundance, protein abundance, and CAI, measured under both aerobic and anaerobic conditions) and their respective biosynthetic costs (aerobic and anaerobic) become significantly positive. With Val, Gly, and Ala included in the analysis the overall aerobic trends are −0.048, −0.049, and −0.079, and the anaerobic trends are −0.179, −0.178, and −0.132, for CAI, protein abundance, and transcript abundance, respectively (p ≤ 0.05 for all). With Val, Gly, and Ala removed the trends become +0.136, +0.135, and +0.091 (aerobic) and +0.160, +0.146, and +0.123 (anaerobic) for CAI, protein abundance, and transcript abundance, respectively (p < 0.0005 for all). The codons that code for Val, Gly, and Ala are GTN, GGN, and GCN, respectively. Akashi (2003) noted the strong positive trend in yeast for these amino acids and showed that a similar trend for GNN amino acids exists in Caenorhabditis elegans, Drosophila melanogaster, E. coli, and Bacillus subtilis (Gutierrez et al. 1996). The fact that these amino acids do not contain nitrogen or sulfur in their side chains may also be an advantage. Furthermore, it is possible that unrecognized synthetic, reclamation, or uptake mechanisms for an amino acid produce a perceived cost to the organism that differs substantially from its assumed biosynthetic cost. And factors other than properties of amino acids might underlie some of the amino acid vs. expression trends in S. cerevisiae (e.g., the strong negative correlation for serine usage with expression is mostly caused by reduced usage of the twofold family in highly expressed genes; the fourfold family shows a weak negative trend [Akashi 2003]). The presence of alternative pathways for aerobic and anaerobic lifestyles may also contribute to amino acid abundance behavior. The cost variation among amino acids is lower under anaerobic metabolism. Aerobic costs vary from 9.5 to 75.5 high-energy phosphate bonds, while anaerobic costs are in the range of 1 to 24 (Tables 1 and 2). Lower cost variability under anaerobic conditions should translate directly to less selective advantage for utilization of lower-cost amino acids (Gln, Phe, Trp, and Tyr aerobically and Gln, Met, Phe, Thr, Trp, and Tyr anaerobically all have nonsignificant changes in usage relative to all three measures of expressivity as determined by Spearman rank correlation coefficients and Mantel-Haenszel Z-scores [both must have p < 0.05 to be considered significant]; Tables 1 and 2).
While the trend to biosynthetic conservation is somewhat limited by physicochemical property (hydrophilic amino acids do not consistently follow the selection-for-cost trend; Table 3) and inconsistent with respect to amino acid usage (trends are driven primarily by small, aliphatic amino acids; Tables 1 and 2), the effects do appear to be relatively consistent across functional category. Correlations in all functional categories for all expression measures between biosynthetic cost and expressivity were either significantly negative or not significant (for functional categories with more than 50 genes, see Table 4).
Trends are also consistent between aerobic and anaerobic growth conditions. Overall Spearman rank correlations are significant and negative for aerobic biosynthetic costs vs. all aerobic expression measures, and for anaerobic costs vs. all anaerobic expression data (aerobic cost vs. aerobic expression data, rS = −0.048, −0.049 and −0.079 for CAI, protein abundance, and transcript abundance, respectively, and p < 0.005, <0.05, and <0.0005; anaerobic cost vs. anaerobic expressivity, r S = −0.179, −0.178, and −0.132 for CAI, protein abundance, and transcript abundance, respectively, with all p-values <0.0005; Table 3).
Of note are the generally stronger negative trends in the anaerobic data (all but two of the Spearman rank correlations are higher for anaerobic biosynthetic cost vs. anaerobic expression data; Table 3). This could be related to S. cerevisiea’s strong preference for fermentation over oxidative phosphorylation (Dickinson and Schweizer 1999). It is difficult to draw clear conclusions regarding comparisons between the aerobic and the anaerobic data, as aerobic and anaerobic amino acid biosynthetic costs are themselves correlated (r = 0.514, p < 0.05). E. coli and B. subtilis are facultative as well, and this relationship could have a bearing on them as it does in yeast. Many of the observed statistically significant correlations explain less than 5% of the variance in the available data, implying that selection for preferred codons is relatively weak and requires a genome-wide analysis to be detected. This is not surprising given the rough estimates of expression and other factors (especially functional constraint) that affect amino acid usage. We attempted to account for effects of anaerobic costs on aerobic comparisons (and vice versa); partial correlations were performed to control for cost and expression data from alternative lifestyles, but all trends became nonsignificant (data not shown).
Given these mixed results, cost minimization would appear, at best, to be a weak selective force in yeast (all r S values ≤ 0.192 in magnitude; Table 3) that is easily obscured by other influences. Examples of such influences could include those related to constraints on amino acid usage due to the complexity of eukaryotic systems, avoidance of nitrogen and sulfur containing amino acids, and the predominant usage in highly expressed genes of one family of codons (for instance, the twofold family over the fourfold). It is also possible that the cost minimization trend is simply a vestige of a time when S. cerevisiae was more actively maintaining cost minimization. Other factors such as translational selection, folding efficiency, and perhaps even the differences between perceived and calculated biosynthetic cost also could play important roles. Expression data available today (transcript abundance, protein abundance, and codon usage bias) are approximations of the underlying protein production rates. It may be difficult to determine the contribution of amino acid cost minimization to S. cerevisiae without more accurate estimates of both costs and expression rates.
References
Akashi H (2003) Translational selection and yeast proteome evolution. Genetics 164(4):1291–1303
Akashi H, Gojobori T (2002) Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc Natl Acad Sci USA 99(6):3695–3700
Altschul S, Gish W, Miller W, Myers E, Lipman D (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Black SD, Mould DR (1991) Development of hydrophobicity parameters to analyze proteins which bear post- or cotranslational modifications. Anal Biochem 193(1):72–82
Brocchieri L, Karlin S (2005) Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Res 33(10):3390–3400
Craig CL, Weber RS (1998) Selection costs of amino acid substitutions in cole1 and colia gene clusters harbored by Escherichia coli. Int J Biol Macromol 24:109–118
Dickinson J, Schweizer M (1999) The metabolism and molecular physiology of Saccharomyces cerevisiae. Taylor & Francis, London
dos Reis M, Wernisch L, Savva R (2003) Unexpected correlations between gene expression and codon usage bias from microarray data for the whole Escherichia coli K-12 genome. Nucleic Acids Res 31(23):6976–6985
Eyre-Walker A (1996) Synonymous codon bias is related to gene length in Escherichia coli: selection for translational accuracy? Mol Biol Evol 13:864–872
Garcia-Vallvé S, Guzman E, Montero MA, Romeu A (2003) HGT-DB: a database of putative horizontally transferred genes in prokaryotic complete genomes. Nucleic Acids Res 31(1):187–189
Ghaemmaghami S, Huh W-K, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS (2003) Global analysis of protein expression in yeast. Nature 425(6959):737–741
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG (1996) Life with 6000 genes. Science 274(5287):546–567
Güldener U, Münsterkötter M, Kastenmüller G, Strack N, van Helden J, Lemer C, Richelles J, Wodak SJ, García-Martínez J, Pérez-Ortín JE, Michael H, Kaps A, Talla E, Dujon B, André B, Souciet JL, De Montigny J, Bon E, Gaillardin C, Mewes HW (2005) CYGD: the Comprehensive Yeast Genome Database. Nucleic Acids Res 33(Database Issue):D364–D368
Gutierrez G, Marquez L, Maryn A (1996) Preference for guanosine at first codon position in highly expressed Escherichia coli genes. A relationship with translational efficiency. Nucleic Acids Res 24:2525–2527
Hall C, Brachat S, Dietrich FS (2005) Contribution of horizontal gene transfer to the evolution of Saccharomyces cerevisiae. Eukaryot Cell 4(6):1102–1115
Heizer EM Jr, Raiford DWIII, Raymer ML, Doom TE, Miller RV, Krane DE (2006) Amino acid cost and codon-usage biases in 6 prokaryotic genomes: a whole-genome analysis. Mol Biol Evol 23(9):1670–1680
Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA (1998) Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95(5):717–728
Jain R, Rivera MC, Lake JA (1999) Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci USA 96(7):3801–3806
Kahali B, Basak S, Ghosh TC (2007) Reinvestigating the codon and amino acid usage of S. cerevisiae genome: a new insight from protein secondary structure analysis. Biochem Biophys Res Commun 354(3):693–699
Mantel N, Haenszel W (1959) Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 22:719–748
Nei M (1975) Molecular population genetics and evolution. Front Biol 40:I–288
Seligmann H (2003) Cost-minimization of amino acid usage. J Mol Evol 56(2):151–161
Sharp PM, Li WH (1987) The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15:1281–1295
Spearman C (1904) General intelligence objectively determined and measured. Am J Psychol 15:201–293
Swire J (2007) Selection on synthesis cost affects interprotein amino acid usage in all three domains of life. J Mol Evol 64(5):558–571
Urrutia AO, Hurst LD (2003) The signature of selection mediated by expression on human genes. Genome Res 13(10):2260–2264
Wagner A (2005) Energy constraints on the evolution of gene expression. Mol Biol Evol 22(6):1365–1374
Warringer J, Blomberg A (2006) Evolutionary constraints on yeast protein size. BMC Evol Biol 6:61
Zubay G (1998) Biochemistry. William C, Brown, New York
Acknowledgments
This study was supported in part by Grant MBC-0132097 to R.V.M. and by NSF Grant DEB-0521964 to H.A. We also thank an anonymous reviewer for numerous helpful comments and suggestions regarding an early version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Raiford, D.W., Heizer, E.M., Miller, R.V. et al. Do Amino Acid Biosynthetic Costs Constrain Protein Evolution in Saccharomyces cerevisiae?. J Mol Evol 67, 621–630 (2008). https://doi.org/10.1007/s00239-008-9162-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-008-9162-9