
Abstract
Freshwater sponges include six extant families which belong to the suborder Spongillina (Porifera). The taxonomy of freshwater sponges is problematic and their phylogeny and evolution are not well understood. Sequences of the ribosomal internal transcribed spacers (ITS1 and ITS2) of 11 species from the family Lubomirskiidae, 13 species from the family Spongillidae, and 1 species from the family Potamolepidae were obtained to study the phylogenetic relationships between endemic and cosmopolitan freshwater sponges and the evolution of sponges in Lake Baikal. The present study is the first one where ITS1 sequences were successfully aligned using verified secondary structure models and, in combination with ITS2, used to infer relationships between the freshwater sponges. Phylogenetic trees inferred using maximum likelihood, neighbor-joining, and parsimony methods and Bayesian inference revealed that the endemic family Lubomirskiidae was monophyletic. Our results do not support the monophyly of Spongillidae because Lubomirskiidae formed a robust clade with E. muelleri, and Trochospongilla latouchiana formed a robust clade with the outgroup Echinospongilla brichardi (Potamolepidae). Within the cosmopolitan family Spongillidae the genera Radiospongilla and Eunapius were found to be monophyletic, while Ephydatia muelleri was basal to the family Lubomirskiidae. The genetic distances between Lubomirskiidae species being much lower than those between Spongillidae species are indicative of their relatively recent radiation from a common ancestor. These results indicated that rDNA spacers sequences can be useful in the study of phylogenetic relationships of and the identification of species of freshwater sponges.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sponges (Porifera) are ancient metazoans which mainly inhabit marine ecosystems but which also have colonized freshwater environments (Willmer 1990; Nielsen 2001). Their phylogeny and taxonomy are complicated due to the limited number and the low information content of morphological features employed for taxonomy (Hooper and van Soest 2002). The freshwater sponges consist of six extant and one fossil family belonging to the suborder Spongillina, which are classified together with marine families having diactinal megascleres to comprise the order Haplosclerida Topsent (Manconi and Pronzato 2002). According to several lines of molecular evidence, freshwater sponges do not belong to haplosclerids and their affinity has not been determined (Borchiellini et al. 2004; Nichols 2005; Addis and Peterson 2005; Redmond et al. 2007; Itskovich et al. 2007). However according to Sperling et al. (2007) freshwater sponges group with the marine Haplosclerids.
The family Spongillidae is cosmopolitan and includes 21 genera with more than 150 species (Penney and Racek 1968; Manconi and Pronzato 2002). The family Lubomirskiidae is restricted to Lake Baikal (Siberia), the deepest and oldest lake in the world, which houses a great number of endemic species and is a place of intense evolutional processes (Timoshkin 1995). Recently the number of species described in Lubomirskiidae has increased significantly, with 13 species and 1 subspecies known (Efremova 2001, 2004).
The taxonomy of sponges is based on features of the skeleton including its structural elements, megascleres and microscleres. Some species produce asexual resting bodies (gemmules), which contain specialized microscleres (gemmoscleres). For classification of freshwater sponges, the presence and shape of their microscleres and the structure of their gemmules and gemmoscleres are the most important diagnostic features. However, in three of the six extant freshwater sponge families (Lubomirskiidae, Malawispongiidae, Metschnikowiidae), such important morphological diagnostic features as microscleres, gemmules, and, accordingly, gemmoscleres are not present (Manconi and Pronzato 2002). Therefore, the shape of megascleres and features of skeletal organization only can be used for their classification, although these characters are highly variable. Since gemmules are facilities for survival under adverse habitat conditions, sponges produce gemmules during a restricted time of the year (Manconi and Pronzato 2002). As a result, even in Spongillidae species known to produce gemmules, these are often absent in collected samples, making their species identification more difficult. These factors determine the importance of using molecular biological methods to investigate the phylogeny of freshwater sponges and the development of convenient molecular markers for their species identification.
Many authors have considered freshwater sponges to be polyphyletic and believed that the sponges of ancient lakes and cosmopolitan freshwater sponges are derived from different ancestors (Marshall 1885; Brien 1970; Volkmer-Ribeiro and De Rosa-Barbosa 1978; Volkmer-Ribeiro and Watanabe 1983; Volkmeir-Ribeiro 1990). Earlier molecular studies revealed a possible monophyly of freshwater sponges, although species not from all existing families were examined (Itskovich et al. 1999; Efremova et al. 2002; Schröder et al. 2003; Addis and Peterson 2005; Itskovich et al. 2006; Meixner et al. 2007; Itskovich et al. 2007). Recent studies also allowed some proposals to be made about the interrelationships of the group. Addis and Peterson (2005) revealed that Corvomeyenia sp. (Metaniidae) and Trochospongilla pennsylvanica (Spongillidae) are basal taxons among Spongillina. Addis and Peterson (2005) revealed also paraphyly of Ephydatia with respect to the Lubomirskiidae and propose to subsume Lubomirskiidae to Spongillidae. Data of Meixner et al. (2007) support results of Addis and Peterson (2005) and indicate that Malawispongiidae is polyphyletic because two analyzed species of this family clustered in a different clades. Since these first phylogenetic analyses were based on the conservative 18S rRNA gene and the slowly evolving COXI gene, they were informative at the family level and above. Thus, phylogenetic relationships within freshwater sponge families remain unclear.
The ribosomal internal transcribed spacer (ITS) region is convenient for phylogenetic studies because of its high variability and the possibility of verifying and aligning sequences according to the conservative parts of ribosomal RNAs. Verification of the obtained sequences is extremely important for sponges, because they are inhabited by a great number of symbionts, including intracellular symbionts (Wilkinson 1978). The ITS region is one of the most variable parts of the genome and is suitable for analyses of closely related species (Coleman and Vacquier 2002; Coleman 2003). ITS spacers have been successfully used for phylogenetic reconstructions in plants, insects, and animals including another diploblast, Cnidaria (Yoon et al. 2001; Bargues et al. 2001, 2003; Young and Coleman 2004).
Recent studies on sponges phylogeny based on ITS region comparisons focused on marine sponge populations (Wörheide et al. 2002; Duran et al. 2004) and marine family Aplysinidae (Schmitt et al. 2005). In a study by Addis and Peterson (2005), analyses of the ITS2 sequences of two species of Lubomirskiidae and three species of Spongillidae revealed possible paraphyly of Spongillidae in relation to Lubomirskiidae. However, due to the limited taxon sampling, this conclusion was preliminary.
In this work we analyzed ITS1 and ITS2 regions of 11 species of Lubomirskiidae, 13 species of Spongillidae, and 1 species of Potamolepidae to investigate the phylogenetic relationships between and within these families and to develop molecular markers for species identification in sponges.
Materials and Methods
Sample Collection and DNA Extraction
Species of freshwater sponges belonging to the family Spongillidae were collected in lakes, rivers, and canals of Honhsu island (Japan). Samples were collected manually from shallow depths. Samples of the family Lubomirskiidae were collected during expeditions to Lake Baikal in 1993–2000. The collection sites are situated in the litoral zones of southern, central, and northern Baikal. Sampling was carried out by scuba diving and by dredge surveys from depths of 1 to 200 m. More than 1300 samples of the family Lubomirskiidae were collected. Specimens of Echinospongilla brichardi (Potamolepidae) were collected from Lake Tanganyika (Zambia). For molecular analyses, samples of 11 species of Lubomirskiidae, 13 species of Spongillidae, and 1 species of Potamolepidae were used. The taxonomy and collection sites are listed in Table 1. All specimens were photographed alive. Data on ecology, habitat, and texture were recorded. One part of each sample was fixed in 70% ethanol for taxonomic identification, and another part was frozen in liquid nitrogen for molecular analysis or was used for DNA extraction immediately. Species identification was performed based on microscopic study of the skeleton. Spicule and skeleton preparation were performed as previously described (Masuda et al. 1999; Efremova 2001). Total genomic DNA extraction was performed from the choanosome, reducing as much as possible foreign material by the standard phenol method (Sambrook et al. 1989), by the CTAB method (Gustincich et al. 1991), and by use of the Genomic-tip 100 G Kit (Qiagen). DNA was analyzed by electrophoresis in 0.6% agarose, and its concentration was measured by a spectrophotometer. Purification of DNA was carried out using Nucleosil sorbent (Macherey–Nagel).
Polymerase Chain Reaction, Cloning, and Sequencing
Based on the conservative regions 18S rRNA and 28S rRNA of marine sponge species from GenBank, the following primers were designed using the program GeneTools (Resenchuk 1991).
-
Forward primer (fw13): 5′-TACACACCGCCCGTCGCTACTA-3′
-
Reverse primer (1278): 5′-CTYYGACGTGCCTTTCCAGGT-3′
Fragments about 1060 bp long, including a 3′ end of 18S rRNA, ITS1, 5.8S rRNA, and a 5′ end of 28S rRNA, were amplified. Twenty-five microliters of PCR reaction mix contained the following: 10× PCR buffer (Promega), 2.5 μl; MgCl2 (25 mM), 3 μl; each primer (10 pmol/μl), 0.5 μl; dNTP mix (100 mM each), 1 μl; DNA (~0.1 μg), 1 μl; Taq DNA polymerase (5 U/μl) (Promega), 0.2 μl; and ddH2O, to 25 μl. Cycle parameters were: initial denaturation at 94°C for 120 s, followed by 40 cycles of denaturation at 94°C for 60 s, anneling at 55°C for 60 s, and extension at 72°C for 60 s, followed by a final extension of 8 min at 72°C. Amplifications were carried out in a Perkin-Elmer thermocycler. Each PCR reaction was purified through a QIAquick Spin column (Qiagen) and cloned into pGEM-T (Promega). Sequencing of the clones was carried out by an ABI 373A automatic DNA Sequencer (Applied Biosystems) with the BigDye Termination Cycle Sequencing Kit (Applied Biosystems) according to protocols. Chromatograms were analyzed by BioEdit (Hall 1999). From one to three clones were sequenced from each sample.
Sequence Alignments and Tree Reconstructions
Sequences were manually aligned using the SeaView program (Galtier et al. 1996). ITS secondary structures were generated as described by Mai and Coleman (1997). After structural elements were identified in the transcripts, the alignment was refined and screened for compensating base changes (CBCs). To elucidate the folding pattern of secondary structure elements the Web mfold server (version 3.2 [Zuker 2003]) was used. Alignment is available upon request.
Phylogenetic trees were inferred with maximum-likelihood (ML), distance (neighbor joining; NJ), and maximum parsimony (MP) optimality criteria using PAUP* 4.0b10 (Swofford 2002) and Bayesian inference (BI) using MrBayes v3.0b3 (Huelsenbeck and Ronquist 2001). Echinospongilla brichardi (Potamolepidae) was used as outgroup. The root was inferred from analyses derived using 18S rDNA (Itskovich et al. 2007). The optimal substitution model (for ML and NJ analyses) was selected via hierarchical likelihood ratio tests using Modeltest 3.04 (Posada and Crandall 1998). Results revealed that the TVM model incorporating estimates of the proportion of invariable sites and gamma (TVM + I + G model) is the best model for our data. Distances for NJ analyses were calculated by ML. ML and MP analyses used heuristic searches with a branch-swapping algorithm (tree bisection-reconnection). In BI, two parallel MCMC runs were carried out for 1 million generations sampled every 100 generations, for a total of 10,000 samples. The first 500 samples were discarded as “burn-in.” The robustness of the trees was estimated by bootstrap percentages (BP; Felsenstein 1985) using 1000 (NJ), 500 (MP), and 100 (ML) replications and by posterior probabilities (PPs) in BI. BP < 50% and PP < 0.90 were not taken into account. In MP, the stepwise addition option (10 heuristic searches with random taxon input order) was used for each bootstrap replicate. The ML-bootstrap used a single heuristic search (starting tree via stepwise addition) per replicate.
Results
ITS Alignment and Secondary Structure
Sequences of the ITS region of 11 species of Lubomirskiidae, 13 species of Spongillidae, and 1 species of Potamolepidae were obtained. For some species of Spongillidae, several samples from different localities were analyzed, and the common amount of analyzed samples was 39.
While the internal transcribed spacer 1 (ITS1) ranged from 207 nt in Echinospongilla brichardi to 281 nt in Baikalospongia recta and Baikalospongia fungiformis, with a mean length of 259 nt (SD, ±22.8), the lengths of ITS1 sequences in the majority of taxa studied fell within the range of 245–257 nt, with a G + C content of 57.3% (SD, ±0.02). It should be noted that in all species studied, the base frequencies were unevenly distributed over the length of the ITS1, with the 3′ part being notably A + T rich.
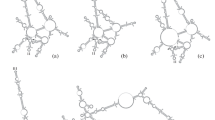
The sequences were rather variable, with only 145 nt conserved among 90% of 16 accessions of Spongillidae, Echinospongilla and two Lubomirskiidae, Baikalospongia bacillifera (BK248) and Lubomirskia baicalensis (BK460). Identification of ITS1 positional homology was difficult without considering the secondary structure which we obtained for all sequences. The thermodynamically predicted model of the sponge ITS1 transcript identified three stem-loop domains (termed I-1, I-2, and I-3; Fig. 1). Approximately two-thirds of the nucleotides were involved in the formation of the stem-loops. The only long single-stranded AU-rich region (55–70 nt) extended between stem 2 and stem 3. It had only a few relatively short conserved motifs and its complete alignment was not possible due to length variations between sequences. Stem-loop 2 was the longest and most conserved of all stem-loops. In most taxa its terminal part bifurcated into two nearly equal short (8 ± 1-bp) parts, but in Eunapius sp. 2, E. sinensis, and Echinospongilla brichardi, this bifurcation was lacking. Interestingly, in all Lubomirskiidae sequences stem 2 was 12 bp longer then in other taxa due to insertion in the central part (Fig. 1). The remaining two stems were much shorter and had rather well-conserved primary and secondary structures. In some species mfold predicted several short (3- to 4-bp) stems between stem 2 and stem 3, but their structure was inconsistent, so the stems were disregarded.

Proposed secondary structure model of the ITS1 (a) and ITS2 (b) transcripts of Baikalospongia bacillifera (Porifera; Spongillina; Lubomirskiidae). Some compensating base pair changess (CBC) and hemi-CBCs preserving base pairing in helices are shown. Square brackets denote insertion in helix 2 of ITS1 in Baikalospongia bacillifera (see Results). Nucleotide positions conserved in 90% of 16 accessions of Spongillidae, Echinospongilla, and two Lubomirskiidae, Baikalospongia bacillifera (BK248) and Lubomirskia baicalensis (BK460), are in boldface
The amount of sequence diversity and the stability of the predicted secondary structures across several sponge genera suggested that compensatory base changes (CBCs) in the stem regions must be frequent to maintain functionality in these structures. Examination of the secondary structure-based alignment revealed compensating nucleotide changes in stems 1 and 2. Expansion of stem 2 in Lubomirskiidae (Fig. 1) was found in the pairing portion of the stem and insertion of 12 nt at two distant complementary parts of the primary sequence did not disturb the pairing. The structure of stem 3 could not be verified with the CBCs, therefore we consider it tentative.
The shortest ITS2 sequences were found in Eunapius spp. (244–246 nt), while Lubomirskiidae spp. again were characterized by the longest sequences (319–332 nt). An average ITS2 length was 298 ± 30.6 nt, with a G + C content of 58.8% (SD, ±0.04%). For the Lubomirskiidae sequences a somewhat lower percentage of GC (53–56%) was typical and for the remaining taxa this value was respectively higher (>60%). Primary sequence conservation in the ITS2 was lower than found in ITS1, with 120 nt conserved in 90% of 16 accessions of Spongillidae, Echinospongilla, and two Lubomirskiidae (see above), and this conservation was confined to their central parts. Numerous indels furthermore hampered straightforward sequence alignment.
Folding of sequences produced the ITS2 secondary structure presented in Fig. 1. This structure in the sponges investigated, as in many eukaryotes, displayed four stem-loop regions (II-1, -2, -3, and -4). Helix 3 was the longest and helix 4 the most variable in length of the four stem-loops. In Lubomirskiidae sequences the latter had a string of 5 or 6 A-U pairs at the base lacking in all other species. Helix 2 and its flanking sequences exhibited the highest degree of primary sequence conservation. It revealed a pyrimidine mismatch pairing that was previously reported in many distantly related groups of organisms (Mai and Coleman 1997; Joseph et al. 1999; Coleman and Vacquier 2002; Goertzen et al. 2003). In helix 3, also relatively conserved in primary sequence, the conservation increased toward its distal part, where the longest string of invariant nucleotides is located (Fig. 1). Variation in the secondary structure of this helix referred to the position and size of bulges. The region of the highest sequence variability in the ITS2 related to the spacer between stem 3 and stem 4 and the latter stem (Fig. 1). The length of this stem varied greatly and no motifs common to all taxa were found.
An alignment of ITS1 based on the predicted secondary structure resulted in 228 characters available for phylogenetic analyses. Due to greater sequence variation, the ITS2 alignment contributed 222 positions. Position homology of highly variable portions of the spacer between stem 2 and stem 3 in ITS1 as well as stem 1, the spacer between stem 3 and stem 4, and the basal portion of stem 4 in ITS2 remained questionable, and these areas were excluded from analyses.
Genetic distances in pairwise comparisons between all analyzed sequences were calculated according to Kimura’s two-parameter model. The average sequence divergence between the sequences obtained from 13 Spongillidae species was 31%. Between 11 Lubomirskiidae sequences the mean level of sequence divergence was extremely low (3.6%) in comparison with Spongillidae. Since for some Spongillidae species ITS sequences were obtained from several samples from different localities, we could estimate the level of intraspecies variability of this DNA region. The genetic distance between different specimens of was 0.1% for H. multidentata, 1.5% for E. fluviatilis, 0.2% for R. cerebellata, and 0.6% for S. lacustris. For tree reconstructions the consensus sequences for each analyzed species of Spongillidae were used.
Dendrograms obtained with ML, distance (NJ), and MP optimality criteria and Bayesian inference (BI) had similar topologies, and that drawn using ML is shown in Fig. 2. There were small differences in the clustering of closely related species within the family Lubomirskiidae, where the bootstrap support was also very low, and therefore these relationships were unresolved.

Phylogenetic tree showing relationships between representatives of several sponge genera (Porifera; Spongillina) based on comparisons of 27 ITS sequences (602 aligned positions). The topology was inferred by maximum likelihood (TVM + I + G model). Nodes are characterized by bootstrap percentages (BP > 50%) and Bayesian posterior probabilities (PP > 0.9): ML/NJ(TVM + I + G)/MP/BI. Highly significant (≥99% BP in all methods and 1.00 PP) branches are thicker. Echinospongilla brichardi (Potamolepidae) was used as outgroup
Our results do not support the monophyly of Spongillidae since Trochospongilla latouchiana formed a robust clade with the outgroup Echinospongilla brichardi (Potamolepidae). There are several well-supported monophyletic groups among the species of the family Spongillidae. Eunapius, Spongilla, and Radiospongilla formed unresolved basal polytomy, while Heterorotula and Ephydatia formed a robust clade that comprised all Lubomirskiidae. The family Lubomirskiidae was strongly monophyletic in all phylogenetic trees (ML, 100%; NJ, 100%; MP, 100%; BI, 1.0).
Species of Eunapius fell in a highly supported clade in the obtained trees (100–99% bootstrap; BI, 1.0) consisting of Eunapius coniferus, E. ryuensis, E. sinensis, Eunapius sp1, and и Eunapius sp2. Three species of the genus Radiospongilla (R. sendai, R. cerebellata, R. crateriformis) formed a moderately supported clade (ML, 75%; NJ, 59%; MP, 77%; BI, 1.0).
Ephydatia muelleri was basal to all Lubomirskiidae species (ML, 74%; NJ, 84%; MP, 83%; BI, 0.98). Two additional species of Ephydatia—E. a fluviatilis and E.a muelleri—Heterorotula multidentata, and all Lubomirskiidae species formed a monophyletic group (ML, 69%; NJ, 88%; MP, 83%; BI, 0.99).
The positions of Spongilla lacustris and, Radiospongilla were unresolved, branching from the base of the group that included E. mulleri, Heterorotula, and all Lubomirskiidae, but with no bootstrap support (51% only in MP).
Phylogenetic relationships within the family Lubomirskiidae remained unresolved because of the low sequence divergence except for one monophyletic group including Rezinkovia echinata, Baikalospongia fungiformis, Lubomirskia fusifera, Baikalospongia sp., and Lubomirskiidae sp., which had high support (ML, 87%; NJ, 95%; MP, 82%; BI, 1.0). An undescribed species (Lubomirskiidae sp., voucher BK267) with spicules which were morphologically similar to those of Spongillidae clustered within the Lubomirskiidae clade (ML, 100%; NJ, 100%; MP, 99%; BI, 1.0).
Discussion
ITS1 and ITS2 Secondary Structure Models
Our putative model of the ITS1 secondary structure revealed three helical domains and a relatively long single-stranded AU-rich region separating the last two helices (Fig. 1). A relatively high proportion of paired nucleotides contrasted with previous predictions made for ITS1 secondary structure in green algae (Coleman et al. 1998), embryophytes (Goertzen et al. 2003), and yeasts (van Nues et al. 1994), in which a major portion of the transcript was said to be single-stranded, with only a limited number of nucleotides involved in the formation of usually short stems. At the same time the compensatory base changes (CBCs) and hemi-CBCs in the longest stem 2 and extensive insertion in stem 1 found in the Lubomirskiidae sequences (Fig. 1) retain their secondary structure and confirm our prediction. Although thermodynamic-based models may overestimate some secondary structure elements and sequence covariation analysis may be superior in producing more realistic models (Goertzen et al. 2003), the latter approach requires a considerable number of sequences that are presently not available in the sponges. Keeping this in mind, we consider the proposed ITS1 secondary structure model as preliminary but adequate to guide alignments.
The secondary structure of the ITS2 transcript in sponges (see Fig. 1) is more representative of ITS2 secondary structures in a wide range of organisms (Coleman 2003, 2007; Wolf et al. 2005). Typical of these structures are a pyrimidine mismatch pairing in stem 2 and a string of conserved nucleotides at the 5′ side of helix 3 (Fig. 1). Comparison of our model with those presented by Schmitt et al. (2005) for the marine Aplysinidae reveals great similarity in overall structure and individual stem architecture, despite the fact that our ITS2 sequences are ca. 100 nt longer and generally somewhat less GC rich.
Our results suggest that the utility of rDNA spacer sequences for phylogeny reconstruction in sponges, particularly ITS1, could be expanded. To our knowledge, the present study is the first one in which ITS1 sequences were successfully aligned using verified secondary structure models and, in combination with ITS2, used to infer relationships between the freshwater sponges. It appears that the generally accepted hypervariability of ITS1 was overestimated. We demonstrate that the spacer sequences were easy to align between distantly related genera and some regions are almost invariant.
Phylogeny of Spongillidae
Spongillidae are the largest freshwater sponge family, including more than half the existing species of the suborder Spongillina (Manconi and Pronzato 2002). The taxonomy of Spongillidae is based on the shape of microscleres and gemmoscleres, whereas megascleres are mainly similar in shape and size in different species (Penney and Racek 1968; Manconi and Pronzato 2002). It was suggested based on paleontological data that the family Spongillidae is monophyletic and the genus Radiospongilla is ancestral to the rest of Spongillidae (Racek and Harrison 1975). However, this conclusion is not supported by our results based on ITS sequences because Radiospongilla did not form a basal branch among six studied genera of the Spongillidae. Trochospongilla latouchiana grouped with Echinospongilla brichardi from the family Potamolepidae and the family Lubomirskiidae was found in a clade containing all remaining spongillidae sequences. Our data suggest that the taxonomy of the freshwater sponges at the family level should be reevaluated after additional molecular data from more comprehensive taxon sampling have become available.
At the genus level branching of the phylogenetic trees is, in general, in accordance with the morphology-based taxonomy of this family, with the monophyly of Eunapius receiving high bootstrap support and the genus Radiospongilla receiving moderate support (Fig. 2). For Spongilla, only one species was analyzed, so we could not test monophyly of this genus.
However, the monophyly of the genus Ephydatia was not supported—E. fluviatilis and E. muelleri did not cluster together, with E. fluviatilis found in a clade with H. multidentata. This branching supports the paraphyly of Ephydatia in relation to Heterorotula and all analyzed species of the family Lubomirskiidae. Our data support findings of Addis and Peterson (2005) and Meixner et al. (2007) which also revealed paraphyly of Ephydatia with respect to the Lubomirskiidae. The derived characters for the clade including Ephydatia, Heterorotula, and Lubomirskiidae are absence of microscleres, radially arranged birotulate gemmoscleres, and amphioxea megascleres.
Previous studies of other markers (18S rRNA and COXI gene) revealed close relationships between Spongilla and Eunapius (Addis and Peterson 2005; Meixner et al. 2007). At the morphological level, these species have a tangential arrangement of gemmoscleres (oxeas or strongyles) in the gemmular theca that represents a synapomorphy for this clade. However, according to our results the position of Spongilla lacustris was unresolved.
Systematic study of the genus Eunapius remains difficult (Masuda and Satoh 1992). E. coniferus and E. ryuensis had identical ITS sequences. However, according to morphological data these species are clearly separated from each other. Here we analyzed five specimens of Eunapius that could not be identified based on morphology and determined that four of them (vouchers BW0114, BW0101, JP9, and BW0104), labeled as Eunapius sp. 2 (Fig. 2), had almost-identical sequences of ITS1 and ITS2 (1% divergence), whereas the sequence of the fifth sample (voucher BW0118), marked as Eunapius sp. 1 (Fig. 2), differed significantly. These samples were distinct from the other analyzed species of the genus Eunapius. Differences in the shape of spicules (Masuda, unpublished) and molecular data suggest that these sponges should be described as new species belonging to genus Eunapius.
The general agreement of morphology-based taxonomy and phylogeny presented here supports the usefulness of the ITS region for phylogenetic studies of sponges. There is a serious problem of species identification for freshwater sponges, because the absence of gemmules often makes it impossible to determine their exact taxonomic position. Sequences of ITS1 and ITS2 showed the existence of substitution between the sequences of all different species in this genomic region. At the same time intraspecific differences were never more than 1.5% and never exceeded interspecific variability. This indicates the possibility of effectively using this region for species identification of freshwater sponges.
For several Spongillidae species, most of the ITS1 and ITS2 sequences of specimens from different localities had nucleotide substitutions. ITS sequences have been used successfully for studies of the population history of marine sponges (Duran et al. 2004). However, in marine sponges intragenomic variability of ITS was revealed by Wörheide et al. (2004), which can interfere with population -level studies. It is possible that the ITS region could also be used for population studies of freshwater sponges, but as in the case of marine species, screening for intragenomic polymorphism should be carried out first.
Phylogeny of Lubomirskiidae
Sponges of the family Lubomirskiidae are endemic to Lake Baikal with the somewhat questionable exception of the species Baikalospongia dzhegatajensis from Lake Chagytai (Rezvoi 1936), whose affinity wih this family has yet to be confirmed by molecular data. The family Lubomirskiidae was separated by Rezvoi (1936) on the basis of their anatomy, skeletal peculiarities, and absence of the gemmules. He considered Lubomirskiidae to be monophyletic and to probably be descendants of marine families. Therefore, he concluded that the origin of Lubomirskiidae bears no relation to Spongillidae. In the first work on the molecular phylogeny of freshwater sponges based on partial sequences of the 18S rRNA gene, it was shown that Lubomirskiidae and Spongillidae are closely related and have a common origin (Itskovich et al. 1999). In another study the possible paraphyly of Ephydatia in relation to Lubomirskiidae was suggested based on analyses of three different genes from a several species of Spongillidae and Lubomirskiidae (Addis and Peterson 2005). Phylogenetic analyses based on 18S and COXI genes including E. fluviatilis, E. muelleri, L. baicalensis, L. abietina, B. intermedia, B. bacillifera, B. fungiformis, and S. papyracea revealed that these species form a common clade, and phylogenetic relationships within this clade are unresolved (Itskovich et al. 2007; Meixner et al. 2007). Addis and Peterson (2005) suggested that Lubomirskiidae should be abandoned and all lubomirskiid species should be included in the genus Ephydatia. The evidence from our tree indicates that Ephydatia muelleri and all lubomirskiid species are sister groups with moderate significance and Ephydatia is also paraphyletic in relation to Heterorotula. Other nongemmulating sponge species from ancient Lake Ohrid, Ochridaspongia sp. (Malawispongiidae), were also related in origin to genus Ephydatia (Meixner et al. 2007). Therefore in at least two ancient lakes, Baikal and Ohrid, endemic sponge fauna were more closely related to Ephydatia. Molecular data suggest that the composition of families within the suborder Spongillina needs urgent revision, but we suggest not changing taxonomy Spongillina before more molecular data are available.
Until the present study, the monophyly of Lubomirskiidae was uncertain because only six species were analyzed and slow-evolving genes were used. In this work, for the first time we analyzed 11 of 13 existing species representing all four genera of Lubomirskiidae. According to the results of our analyses of the ITS region, the family Lubomirskiidae is a robust monophyletic entity.
Some assumptions about the phylogenetic relationships within Lubomirskiidae were made by Rezvoi (1936) based on different skeletal structures. In his opinion, the genus Baikalospongia is the most primitive, because it has an irregular skeleton, without main and secondary bunches. He considered that the genus Swartschewskia, which has a regular net of bunches in the superficial layer, and the genus Lubomirskia, with a firm highly differentiated skeleton, are more evolutionarily advanced (Fig. 3). In our phylogenetic trees all Lubomirskiidae had considerably lower interspecies genetic distances than the distances between Spongillidae species. Due to the lower genetic distances, the phylogeny within Lubomirskiidae was unresolved. Within Lubomirskiidae there was one moderately supported monophyletic group including Rezinkovia echinata, Baikalospongia fungiformis, Lubomirskia fusifera, Baikalospongia sp., and Lubomirskiidae sp. This group included species of three genera and is not supported morphologically. Distribution of the four existing genera according to the present classification is not supported by the molecular data. Recently it was suggested by analyses of 18S rRNA and COXI genes that, among Lubomirskiidae, Swartschewskia papyracea may have diverged from a common ancestor earlier than other species (Efremova et al. 2002; Schröder et al. 2003; Itskovich et al. 1999, 2006, 2007). Therefore these data do not support the opinion of Rezvoi (1936) regarding the primitive status of the genus Baikalospongia. According to our ITS region analyses, S. papyracea may be placed in one group with other species, and it is not distinguished by a larger genetic distance. This result may be related to the inconstancy of the evolution rate of the ITSs in comparison with the coding parts of genome used in the previous studies. However, since there are nucleotide differences between the analyzed species, ITS sequences can be used for molecular identification for both Spongillidae and Lubomirskiidae species. The ITS1 and ITS2 sequences are more suitable for species identification within Lubomirskiidae than the sequences of the 18S rRNA and COXI genes because on these parts of the genome most species of Lubomirskiidae were found to have identical sequences (Efremova et al. 2002; Schröder et al. 2003; Itskovich et al. 2007).
Differences in the form of spicules and skeletal patterns are the reasons for giving specific status to the Lubomirskiidae species (Efremova 2001, 2004) (Fig. 3). Calculations based on the ITS1 and ITS2 sequences show that the average genetic distance between Baikalian sponge species is greater than the interpopulation differences within other species. In marine species the level of intraspecific variability of ITS1 and ITS2 is 0.46–1.7% within Crambe crambe (Duran 2004) and 0.1–1.6% within Leucetta chagonensis (Wörheide 2004). For species of Spongillidae, according to our data the intraspecific variability of ITS1 and ITS2 is equal to 0.1–1.5%. The mean level of genetic divergence between Lubomirskiidae species is about 3.6%. The maximum distance is 8.6% (between Baikalospongia sp. and Lubomirskia incrustans) and the minimum distance is 0.2% (between Swartschewskia papyracea and Lubomirskia abietina). Although among the analyzed species there were no species with identical ITS sequences, the differences between them sometimes were restricted to a few substitutions and deletions. However, analyses of other gene regions (18S and COXI) revealed a clear separation of at least one species (S. papyracea) from others (Itskovich et al. 1999, 2006, 2007; Efremova et al. 2002; Schröder et al. 2003). Our data either support the hypothesis of a recent species divergence within Lubomirskiidae or question their specific status. The finding of undescribed species of Lubomirskiidae (voucher BK267) revealed that the systematic of this family is not completed.

Endemic sponges of the family Lubomirskiidae. a–d Sponge body, oscules, skeleton, and spicules of Swartschewskia papyracea. e–h Sponge body, oscules, skeleton, and spicules of Lubomirskia abietina
The lower divergence may be related to slower evolution of ITS in Lubomirskiidae than in Spongillidae, or may indicate that extant Lubomirskiidae are an evolutionarily young species. Such rapid evolution was noted for many other benthic organisms in Lake Baikal, e.g., worms and mollusks (Zubakov et al. 1997; Sherbakov 1999; Kaygorodova et al. 2007). Some early authors considered Lubomirskiidae to be relics of ancient freshwater or marine fauna (Berg 1910; Vereshchagin 1940; Rezvoi 1936). Martinson described the discovery of spicules of Lubomirskiidae in sediments of the Late Oligocene Age (Martinson 1936). Analyses of ITS sequences clearly indicated that divergence within Lubomirskiidae took place relatively recently and the evolutionary age of these species apparently does not exceed the geological age of Lake Baikal, which is considered to be about 30 million years (Mats 1993). Thus data from the ITS sequences analyses support claims of an autochthonous radiation of sponge species in Lake Baikal (Makushok 1925; Efremova and Goureeva 1989). Divergence of the Lubomirskiidae species flock took place after Baikal had already become a deep-water basin, and probably it is the result of adaptation to a variety of ecological niches which came into existence within the lake. Another reason for the low level of genetic divergence between Lubomirskiidae species may be a relatively recent bottleneck event in the evolutionary history of this family. This could explain the long distance between Lubomirskiidae and Spongillidae and short distances between extant Lubomirskiidae species. This assumption is also supported by the results of paleontological studies. According to the results of spicule analyses, in the Late Pliocene there was wider species diversity, including fossil species, than exists in Lake Baikal at the present time (Weinberg 2001).
Our results revealed the ITS region to be useful for study of the taxonomy and phylogeny of freshwater sponges, and we suppose that future studies of this gene region will help to clarify their phylogenetic history. Wider analyses including other species and families of the freshwater sponges and analyses of population structures are in progress.
References
Addis JS, Peterson KJ (2005) Phylogenetic relationships of freshwater sponges (Porifera, Spongillina) inferred from analyses of 18S rDNA, COI mtDNA, and ITS2 rDNA sequences. Zool Scr 34:549–557
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bargues MD, Vigo M, Horak P, Dvorak J, Patzner RA, Pointier JP, Jackiewicz M, Meier-Brook C, Mas-Coma S (2001) European Lymnaeidae (Mollusca: Gastropoda), intermediate hosts of trematodiases, based on nuclear ribosomal DNA ITS-2 sequences. Infect Genet Evol 1(2):85–107
Bargues MD, Horak P, Patzner RA, Pointier JP, Jackiewicz M, Meier-Brook C, Mas-Coma S (2003) Insights into the relationships of Palearctic and Nearctic lymnaeids (Mollusca: Gastropoda) by rDNA ITS-2 sequencing and phylogeny of stagnicoline intermediate host species of Fasciola hepatica. Parasite 10(3):243–255
Berg LS (1910) Baikalian fauna and its origin. Biol J 1(1):10–45 (in Russian)
Borchiellini C, Chombard C, Manuel M, Alivon E, Vacelet J, Boury-Esnault N (2004) Molecular phylogeny of Demospongiae: implications for classification and scenarios of character evolution. Mol Phylogenet Evol 32:823–837
Brien P (1970) Les Potamolepides africaines nouvelles du Luapula et du Lac Moero. Symp Zool Soc Lond 25:163–186
Coleman AW (2003) ITS2 is a double-edged tool for eukaryote evolutionary comparisons. Trends Genet 19(7):370–375
Coleman AW (2007) Paneukaryote ITS2 homologies revealed by RNA secondary structure. Nucleic Acids Res 35:3322–3329
Coleman AW, Vacquier VD (2002) Exploring the phylogenetic utility of ITS sequences for animals: a test case for abalone (haliotis). J Mol Evol 54(2):246–257
Coleman AW, Preparata RM, Mehrotra B, Mai JC (1998) Derivation of the secondary structure of the ITS-1 transcript in Volvocales and its taxonomic correlations. Protist 149:135–146
Duran S, Giribet G, Turon X (2004) Phylogeographical history of the sponge Crambe crambe (Porifera, Poecilosclerida): range expansion and recent invasion of the Macaronesian islands from the Mediterranean Sea. Mol Ecol 13:109–122
Efremova SM (2001) Sponges (Porifera). In: Timoshkin OA (ed) Index of animal species inhabiting lake Baikal and its catchment area, vol. 1. Lake Baikal, Book 1. Nauka, Novosibirsk, pp 182–192 (in Russian)
Efremova SM (2004) New genus and new species of sponges from family Lubomirskiidae Rezvoj, 1936. In: Timoshkin OA (ed) Index of animal species inhabiting lake Baikal and its catchement area, vol. 1, Lake Baikal, Book 2. Nauka, Novosibirsk, pp 1261–1278
Efremova SM, Goureeva MA (1989) The problem of the origin and evolution of Baikalian sponges. 1st Vereshchagin Baikal Int. Conf. Abstracts, Irkutsk, pp 21–22
Efremova SM, Itskovich VB, Parfenova V, Drucker VV, Müller WEG, Schröder HC (2002) Lake Baikal: a unique place to study evolution of sponges and their stress response in an environment nearly unimpaired by anthropogenic perturbation. Cell Mol Biol 48(4):359–371
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Galtier N, Gouy M, Gautier C (1996) SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comp Appl Biosci 12:543–548
Goertzen LR, Cannone JJ, Gutell RR, Jansen RK (2003) ITS secondary structure derived from comparative analysis: implications for sequence alignment and phylogeny of the Asteraceae. Mol Phylogenet Evol 29:216–234
Gustincich SG, Manfioletti G, Del Sal C, Schneider Carnincy P (1991) A fast method for high-quality genomic DNA extraction from whole human blood. Biotechniques 11:298–300
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hooper JNA, van Soest RWM (2002) Class Demospongiae Sollas, 1885. In: Hooper JNA, van Soest RWM (eds) Systema Porifera: a guide to the classification of sponges, vol 1. Kluwer Academic/Plenum, New York, pp 15–18
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogeny. Bioinformatics 17:754–755
Itskovich VB, Belikov SI, Efremova SM, Masuda Y (1999) Phylogenetic relationship between Lubomirskiidae, Spongillidae and some marine sponges according partial sequences of 18S rDNA. Mem Queensland Mus 44:275–280
Itskovich VB, Belikov SI, Efremova SM, Masuda Y, Krasko A, Schroder HC, Muller WEG (2006) Monophyletic origin of freshwater sponges in ancient lakes based on partial structures of COXI gene. Hydrobiologia 568(1):155–159
Itskovich VB, Belikov SI, Efremova SM, Masuda Y, Perez T, Alivon E, Borchiellini C, Boury-Esnault N (2007) Phylogenetic relationships between freshwater and marine Haplosclerida (Porifera, Demospongiae) based on the full length 18S rRNA and partial COXI gene sequences. In: Custódio MR, Hajdu E, Lôbo-Hajdu G, Muricy G (eds) Porifera research: biodiversity, innovation and sustainability. Museu Nacional, Rio de Janeiro, pp 383–391
Joseph N, Krauskopf E, Vera MI, Michot B (1999) Ribosomal internal transcribed spacer 2 (ITS2) exhibits a common core of secondary structure in vertebrates and yeast. Nucleic Acids Res 27:4533–4540
Kaygorodova IA, DYu Sherbakov, Martin P (2007) Molecular phylogeny of Baikalian Lumbriculidae (Oligochaeta): evidence for recent explosive speciation. Comp Cytogenet 1(1):71–84
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17(12):1244–1245
Mai JC, Coleman AW (1997) The internal transcribed spacer 2 exhibits a common secondary structure in green algae and flowering plants. J Mol Evol 44:258–271
Makushok ME (1925) To the question of origin of Lake Baikal spongiofauna. Russ Zool J 5(4):50–73 (in Russian)
Manconi R, Pronzato R (2002) Suborder Spongillina subord. nov.: freshwater sponges. In: Hooper JNA, van Soest RWM (eds) Systema Porifera: a guide to the classification of sponges, vol 1. Kluwer Academic/Plenum, New York, pp 921–1019
Marshall W (1885) On some new siliceous sponges collected by M. Pechuel-Losche in the Kongo. Ann Mag Nat Hist 12:391–412
Martinson GG (1940) Materials for the study of the fossil fauna of the Baikal area. Trudy Baikalskoi Limnolicheskoi Stantsii Acad Sci USSR 10:425–451 (in Russian)
Masuda Y, Satoh K (1992) Scanning electron microscopic observations on spicules, gemmule coats, and micropyles of the freshwater sponge, Eunapius sinensis (Annandale). Kawasaki Igakkai Shi Liberal Arts Sci Course 18:75–82
Masuda Y, Itskovich V, Veinberg EV, Efremova SM (1999) Perspective studies of freshwater sponges in Lake Baikal. Berliner Geowiss Abh 30:329–332
Mats VD (1993) The structure and development of the Baikal rift depression. Earth Sci Rev 34:81–118
Meixner MJ, Luter C, Eckert C, Itskovich V, Janussen D, von Rintelen T, Bohne AV, Meixner JM, Hess WR (2007) Phylogenetic analysis of freshwater sponges provide evidence for endemism and radiation in ancient lakes. Mol Phylogenet Evol 45(3):875–886
Nichols SA (2005) An evaluation of support for order-level monophyly and interrelationships within the class Demospongiae using partial data from the large subunit rDNA and cytochrome oxidase subunit I. Mol Phylogenet Evol 34:81–96
Nielsen C (2001) Animal evolution. Interrelationships of the living phyla, 2nd edn. Oxford University Press, Oxford
Penney JT, Racek AA (1968) Comprehensive revision of a worldwide collection of freshwater sponges (Porifera: Spongillidae). Bull US Natl Mus 272:1–184
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14(9):817–818
Pronzato R, Manconi R (2001) Atlas of European freshwater sponges. Ann Mus Civ St Nat Ferrara 4(3):64
Racek AA, Harrison FW (1974) The systematic and phylogenetic position of Palaeospongilla chubutensis (Porifera, Spongillidae). Proc Linn Soc New South Wales 99(3):157–165
Redmond NE, van Soest RW, Kelly M, Raleigh J, Travers SA, McCormack GP (2007) Reassessment of the classification of the order Haplosclerida (class Demospongiae, phylum Porifera) using 18S rRNA gene sequence data. Mol Phylogenet Evol 43:344–352
Resenchuk SM (1991) Gene Tools (preparation and processing of text files containing the nucleic or amino acids sequences). Version 1.0
Rezvoi PD (1936) Freshwater sponges of the USSR. In: Rezvoi PD (ed) The fauna of the USSR, vol 2. AS USSR, Moskow, pp 1–42 (in Russian)
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19(12):1572–1574
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
Schmidt HA, Strimmer K, Vingron M, von Haeseler A (2000) TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18(3):502–504
Schmitt S, Hentschel U, Zea S, Dandekar T, Wolf M (2005) ITS-2 and 18S rRNA gene phylogeny of Aplysinidae (Verongida, Demospongiae). J Mol Evol 60(3):327–336
Schröder HC, Efremova SM, Itskovich VB, Belikov SI, Masuda Y, Krasko A, Müller IM, Müller WEG (2003) Molecular phylogeny of the freshwater sponges in Lake Baikal. J Zool Syst Evol Res 41:80–86
Sherbakov DY (1999) Molecular phylogenetic studies on the origin of biodiversity in Lake Baikal. Trends Ecol Evol 14:92–94
Sperling EA, Pisani D, Peterson KJ (2007) Poriferan paraphyly and its implications for Precambrian paleobiology. Geol Soc Lond Spec Publ 286:355–368
Swofford DL (2002) PAUP*. Phylogenetic Analysis Using Parsimony (*and other methods). Sinauer Associates, Sunderland MA
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Timoshkin OA (ed) (1995) Index of animal species inhabiting Lake Baikal and its catchement area. Vol. 1. Lake Baikal, Book 2. Nauka, Novosibirsk, pp 1261–1278
van Nues RW, Rientjes JMJ, van Dersande CAFM, Zerp SF, Sluiter C, Venema J, Planta RJ, Raue HA (1994) Separate structural elements within internal transcribed spacer-1 of Saccharomyces cerevisiae precursor ribosomal RNA direct the formation of 17S and 26S ribosomal RNA. Nucleic Acids Res 22:912–919
Vereshchagin GY (1940) The origin and the history of Baikal, its fauna and flora. Trudi Baikalskoy Limnol Stanzcii AS USSR 10:73–239 (in Russian)
Volkmeir-Ribeiro C (1990) A new insight into the systematics, evolution and taxonomy of freshwater sponges. In: Rutzler K (ed) New perspectives in sponge biology. Smithsonian Institution Press, Washington, DC, pp 323–331
Volkmer-Ribeiro C, de Rosa-Barbosa R (1978) Neotropical freshwater sponges of the family Potamolepidae Brien, 1967. In: Levi C, Boury Esnault N (eds) Biologie des Spongiaires. Colloques Internationaux No. 291. Centre National de la Recherche Scientifique, Paris, pp 503–511
Volkmer-Ribeiro C, Watanabe Y (1983) Sanidastra yokotonensis, n. gen. and n. sp. of freshwater sponge from Japan. Bull Natl Sci Mus Tokyo Zool 9:151–159
Weinberg EV (2001) The sponge fauna of Lake Baikal in the Late Pliocene (according to studies of core samples from deep borehole BDP-96–1). Russ Geol Geophys 42(1–2):130–137
Wilkinson CR (1978) Microbial associations in sponges. II. Numerical analysis of sponge and water bacterial populations. Mar Biol 49:169–176
Willmer P (1990) Invertebrate relationships. Patterns in animal evolution. Cambridge University Press, Cambridge
Wolf M, Achtziger M, Schultz J, Dandekar T, Muller T (2005) Homology modelling revealed more than 20, 000 rRNA internal transcribed spacer 2 (ITS2) secondary structures. RNA 11:1616–1623
Wörheide G, Hooper JN, Degnan BM (2002) Phylogeography of western Pacific Leucetta ‘chagosensis’ (Porifera: Calcarea) from ribosomal DNA sequences: implications for population history and conservation of the Great Barrier Reef World Heritage Area (Australia). Mol Ecol 11(9):1753–1768
Wörheide G, Nichols SA, Goldberg J (2004) Intragenomic variation of the rDNA internal transcribed spacers in sponges (Phylum Porifera): implications for phylogenetic studies. Mol Phylogenet Evol 33(3):816–830
Yoon HS, Lee JY, Boo SM, Bhattacharya D (2001) Phylogeny of Alariaceae, Laminariaceae, and Lessoniaceae (Phaeophyceae) based on plastid-encoded RuBisCo spacer and nuclear-encoded ITS sequence comparisons. Mol Phylogenet Evol 21(2):231–243
Young I, Coleman AW (2004) The advantages of the ITS2 region of the nuclear rDNA cistron for analysis of phylogenetic relationships of insects: a Drosophila example. Mol Phylogenet Evol 30(1):236–242
Zubakov DI, Sherbakov DY, Sitnikova TI (1997) Analysis of phylogeny of endemic mollusca of family Baicaliidae, Clessin 1878 (Gastropoda, Pectinibranchia) from Baikal lake using fragments of nucleotide sequences of the mitochondrial gene CO1. Mol Biol (Mosk) 31(6):1092–1097
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31(13):3406–3415
Acknowledgments
We thank E. Weinberg and Y. Takemon for help with the collection of samples. This work was supported by an Obuchi fellowship, the Deutsche Forschungsgemeinschaft (DFG 436 RUS/17/20/01), and Federal Agency for Science and Innovations (FASI) Grant MK-4167.2007.4. We also thank G. McCormack for her help with the first version of the text. Two anonymous reviewers are acknowledged for critical comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Itskovich, V., Gontcharov, A., Masuda, Y. et al. Ribosomal ITS Sequences Allow Resolution of Freshwater Sponge Phylogeny with Alignments Guided by Secondary Structure Prediction. J Mol Evol 67, 608–620 (2008). https://doi.org/10.1007/s00239-008-9158-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-008-9158-5