
Abstract
It has been argued that the common ancestor of about 75% of all dicots possessed an S-RNase-based gametophytic self-incompatibility (GSI) system. S-RNase genes should thus be found in most plant families showing GSI. The S-RNase gene (or a duplicate) may also acquire a new function and thus genes belonging to the S-RNase lineage may also persist in plant families without GSI. Nevertheless, sequences that belong to the S-RNase lineage have been found in the Solanaceae, Scrophulariaceae, Rosaceae, Cucurbitaceae, and Fabaceae plant families only. Here we search for new sequences that may belong to the S-RNase lineage, using both a phylogenetic and a much faster and simpler amino acid pattern-based approach. We show that the two methods have an apparently similar false-negative rate of discovery (~10%). The amino acid pattern-based approach produces about 15% false positives. Genes belonging to the S-RNase lineage are found in three new plant families, namely, the Rubiaceae, Euphorbiaceae, and Malvaceae. Acquisition of a new function by genes belonging to the S-RNase lineage is shown to be a frequent event. A putative S-RNase sequence is identified in Lotus, a plant genus for which molecular studies on GSI are lacking. The hypothesis of a single origin for S-RNase-based GSI (before the split of the Asteridae and Rosidae) is further supported by the finding of genes belonging to the S-RNase lineage in some of the oldest lineages of the Asteridae and Rosidae, and by Baysean constrained tree analyses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In angiosperms, when looking at the plant family level, genetically determined self-incompatibility (SI) is estimated to have evolved independently 21 times (Weller et al. 1995). Detailed macrophylogenetic studies on the evolution of SI in Polemoniaceae (Barrett et al. 1996) and Asteraceae (Ferrer and Good-Avila 2007) suggest that this number could be higher since within each of the plant families multiple origins of SI must be argued in order to explain SI species distribution. Gametophytic self-incompatibility (GSI) is found in many plant families (de Nettancourt 1997) and evolved independently at least three times. In grasses GSI is governed by the complementary action of two gametophytically expressed genes, S and Z. An SI response occurs if both the S and the Z allele of a haploid pollen grain are expressed in the diploid stigmatic tissue. The molecular mechanism of the bifactorial grass SI system is still unknown. Nevertheless, in rye (Secale cereale L.) a molecular marker (called TC116908, which shows a high similarity to ubiquitin-specific protease genes) that is only expressed in rye pistils, shows high polymorphism among a random sample of rye plants, and cosegregates with Z has been identified (Hackauf and Wehling 2005). In Papaver rhoeas, S proteins encoded by the stigma interact with incompatible pollen, triggering a Ca2+-dependent signaling network resulting in pollen tube inhibition and programmed cell death (see review by McClure and Franklin-Tong 2006). S-RNase-based GSI has been described in Solanaceae, Rosaceae, and Scrophulariaceae (see references in Igic and Kohn 2001). The pollen component has been identified as being an F-box protein in all three plant families. The Prunus and Antirrhinum/Petunia pollen F-box genes are, however, apparently not orthologous (Wang et al. 2004; Ushijima et al. 2004). Nevertheless, it has been argued that the S-RNase-mediated GSI system evolved only once before the separation of the Asteridae and Rosidae (Igic and Kohn 2001; Steinbachs and Holsinger 2002). Therefore, it is inferred that the common ancestor of about 75% of all dicot plants possessed an S-RNase-based GSI system (Igic and Kohn 2001). Many higher dicot plant families do not present GSI, and thus, multiple GSI losses have occurred (Igic and Kohn 2001; Steinbachs and Holsinger 2002). The most likely fate of a nonfunctional S-RNase gene is to be mutated beyond recognition (Igic and Kohn 2001). Therefore, in plant groups where an old GSI loss is evident no S-RNase sequence is expected to be found. However, in a few cases, the S-RNase sequence may have acquired a new function. The LC1 and LC2 sequences from Luffa cylindrica (Cucurbitaceae), the MC sequence from Momordica charantia (Cucurbitaceae), and the HRGP sequence from Pisum sativum (Fabaceae) are thought to represent such rare changes of function (Igic and Kohn 2001; Steinbachs and Holsinger 2002).
Most of our knowledge on S-RNase-based GSI comes from three plant families only, the Solanaceae, Plantaginaceae, and Rosaceae. Thus, molecular studies on other plant families are needed in order to further test the hypothesis that the S-RNase-mediated GSI system evolved only once (Steinbachs and Holsinger 2002).
Large-scale plant sequence databases containing hundreds of T2 RNase sequences are available. Phylogenetic methods are very time-consuming when dealing with hundreds of sequences, thus they cannot be efficiently used to search for sequences belonging to the S-RNase lineage. Nevertheless, all plant T2 RNases have been proposed to derive from a common ancestor (Taylor et al. 1993; Sassa et al. 1996; Ushijima et al. 1998). Therefore, it is conceivable that due to functional constraints amino acid patterns that allow the distinction of S-RNase sequences from other T2 RNases exist. That the S-RNase gene and the other T2 RNase sequences may have such characteristic amino acid patterns has been suggested by Green (1994) based on the reduced data set then available (6 S-like RNases and 22 Solanaceae S-RNase sequences). It remains to be tested whether such characteristic amino acid patterns are observed in the larger data sets now available containing a diversity of S-RNases (from species of the Solanaceae, Plantaginaceae, and Rosaceae families) and S-like RNases (from monocot, dicot, and green algae species). Finding such amino acid patterns could greatly improve our capability to efficiently search for sequences that may belong to the S-RNase lineage. Traditional phylogenetic methods could then be used to test the hypothesis that the sequences retrieved belong to the S-RNase lineage.
In this work we report four amino acid patterns that allow the distinction of Solanaceae, Plantaginaceae, and Rosaceae functional S-RNase sequences from S-like RNase sequences from green algae, monocot, and dicot species. These amino acid patterns were used to identify a small number of new T2 RNase sequences found in large sequence databases that may belong to the S-RNase lineage. These results are compared with those obtained using a fast maximum likelihood method (GARLI; Zwickl 2006) using more than 350 T2 RNase sequences. We show that, when using the presence of typical amino acid patterns as a first screening method, high-quality results can be obtained with much less effort. Using a Bayesian phylogenetic framework, we show that our results are compatible with the hypothesis of a single origin for the S-RNase-based GSI system in eudicots. One sequence from Lotus japonicus (Fabaceae) may represent a functional S allele. Although GSI is known to exist in the Lotus genus, no molecular studies have been yet performed in the Fabaceae family.
Materials and Methods
Identification of Amino Acid Patterns
In order to find amino acid patterns that distinguish S-RNase from S-like sequences we consider amino acid patterns with ambiguous characters. More specifically, the pattern language is a subset of regular expressions. Every position in the pattern can only be composed by classes of characters belonging to the set of amino acid residues. A class is represented within brackets. For compactness of representation, it is also possible to negate the class. In this case, all characters belonging to the alphabet not present are the ones that compose the class. The negation is denoted by “^.”
A pattern is a regular expression that corresponds to the set of words that can be obtained by the pattern characters. A class of characters appearing in a pattern can be substituted by any element appearing within brackets. For instance, the pattern (A[GT]A) corresponds to the set of words {AGA, ATA}. A pattern is said to have a match in a sequence if the sequence contains at least one word that belongs to the pattern. For instance, the pattern with length 3 “[GT].A,” where “.” is any character, has two matches (underlined) in the sequence “TAAGTTA.”
Two data sets were used, namely, the set of 208 S-RNase sequences (64, 19, 37, and 88 sequences from Solanaceae, Plantaginaceae, Maloideae, and Prunoideae species, respectively) of Vieira et al. (2007) and a second set containing 69 plant S-like sequences from monocots, dicots, and green algae (see Supplementary Table 1). The latter sequences were obtained by BlastX using one Arabidopsis thaliana T2 RNase (gi21555182). It should be noted that a search of the protein database with the words “T2 RNase” and “Spermatophyta” does not produce any additional S-like gene entries. The 69 sequences encompass most of the T2 RNase gene and have been shown not to be S-RNases.
T2 RNase sequences that are not S alleles but that may belong to the S-RNase lineage were avoided (the LC1 and LC2 sequences from Luffa cylindrica, the MC sequence from Momordica charantia, and the HRGP sequence from Pisum sativum (Igic and Kohn 2001); the Prunus avium Pa1 gene (Yamane et al. 2003a); the Antirrhinum hispanicum S-like 29 sequence (Liang et al. 2003); one Nicotiana silvestris sequence (Golz et al. 1998); the Petunia inflata X2 sequence (Lee et al. 1992); and the Medicago truncatula (gi124365515) sequence, which is a gene of likely hybrid origin since, besides the T2 RNase domain, it also possesses a PDI (protein disulfil isomerase)/thioredoxin domain (see Discussion).
For both the S-RNase and the S-like data sets, a fast word discovery program (Pereira et al. 2006a) was used to find statistically interesting words. The minimum word length used was two (-m 2) and the minimum number of sequences where the word should occur (-e) was set to 98% and 40% of the number of sequences in the S-RNase and S-like data sets, respectively. A lower value was used for the S-like data set since it was a priori unknown whether two conserved consecutive amino acids would be found in most sequences from such a diverse origin as algae, monocot, and dicot species and from genes known to perform different functions. The words found were then filtered to discard those words that occurred more than three times in both data sets.
In order to find the largest and more general amino acid patterns that distinguish the two sets of sequences, the software Bioredx was used (Pereira et al. 2006b). This program accepts as input two sequence files and a word seed/word pattern and tries to find patterns (based on the seed given) that occur in one file (referred as the positive file) and not in the other (referred as the negative file). The software needed to perform the search for interesting words and the one needed to find amino acid patterns that can distinguish two sequence data sets can be found at http://evolution.ibmc.up.pt/~nf/projects/sigdis/. We have used this software to find patterns that occurred less than four times in the negative file and considered patterns up to 20 amino acid residues larger than the initial word seed/word pattern. It should be noted that the procedures used to find these patterns do not require the sequences to be aligned but care was taken to make sure that the pattern occurrences were located in the expected region of the molecule. Since the 208 S-RNases sequences represent the best available set of sequences for this gene (Vieira et al. 2007) and the 69 T2 RNases encompass most of the gene, this procedure was used to identify the most informative regions of the S-RNase gene. Nevertheless, many more S-RNase sequences are available. Therefore in order to use all the available information a much larger data set was collected, containing 757 sequences (237, 21, 211, and 288 from Solanaceae, Plantaginaceae, Maloideae, and Prunoideae, respectively; Supplementary Table 2) previously shown to be S-RNases. This data set was built by BlastX using a complete S-RNase sequence from each of the three plant families, and by performing searches using the combination of a specific word and “RNase.” The specific words used are “Solanaceae,” “Plantaginaceae,” “Rosaceae,” “Prunus,” “Malus,” “Pyrus,” “Sorbus,” “Crataegus,” “Antirrhinum,” and “Misopates.” GI accession duplicates were discarded. Only sequences annotated as S-RNases were used.
Sequence Alignments
Amino acid sequences were aligned using ClustalX version 1.64b (Thompson et al. 1997). For phylogenetic analyses, amino acid alignments were used as a guide to obtain the alignment of the corresponding nucleotide sequences.
Phylogenetic Methods
A fast maximum likelihood method of tree reconstruction as implemented in GARLI (Zwickl 2006) was used with the default options. The model implemented is the generalized time-reversible (GTR) model of sequence evolution, allowing for among-site rate variation and a proportion of invariable sites. This is the best-fit model of sequence evolution for the data set used, as indicated by the Akaike Information Criterion, as implemented in Modeltest (Posada and Crandall 1998).
For Bayesian tree estimation we use MrBayes (Huelsenbeck and Ronquist 2001) with the GTR model of sequence evolution, allowing for among-site rate variation and a proportion of invariable sites. This is the model indicated by the Akaike Information Criterion as being the one that best fits the data set used. Third codon positions were allowed to have a gamma-distributed rate with a different shape parameter than the first and second positions. Two simultaneous, completely independent analyses starting from different random trees were run for 500,000 generations (each with one cold and three heated chains). Samples were taken every 100th generation. The first 1250 samples were discarded (burn-in).
Results
S-RNase Amino Acid Patterns
In order to find amino acid patterns that distinguish S-RNases from other T2 RNases a data set that likely comprises all S-RNase sequences available in April 2007 (757 S-RNase sequences; Supplementary Table 2) was compiled, which includes 237, 21, 211, and 288 sequences from Solanaceae, Plantaginaceae, Maloideae, and Prunoideae species, respectively. This data set was compared with a set of 69 complete or almost-complete T2 RNase sequences that are not S-RNases (Supplementary Table 1). Although most S-RNase sequences are partial, hundreds of sequences encompass the regions that have been identified as most informative when a smaller S-RNase data set that represents a compromise between data set size and gene coverage is used (see Materials and Methods). The results are reported in Table 1, and the location of the amino acid patterns along the gene is shown in Fig. 1.

Relative location of the patterns for S-RNases (shaded regions in the S-RNase sequences) and for S-like sequences (shaded regions in the S-like sequences). Regions in boldface represent the two T2 RNase conserved active site (CAS) sequences. Stars indicate active site amino acid residues
Patterns 1 and 2 (Table 1) are exclusively found in S-RNases. Pattern 1 is found in 467 of 468 possible sequences (the exception being the Prunus cerasus S6 m RNase; giABD49100). The sour cherry stylar part mutant S6 m-haplotype has a 2.7-kb mutator-like element insertion in the putative promoter region of the S6 RNase, and this S-RNase is not expressed in this plant and thus it is nonfunctional (Yamane et al. 2003b). A mutation in the coding region of a nonfunctional S allele may be one possible justification for the absence of pattern 1 in the P. cerasus S6 m RNase sequence. The second pattern is found in 689 of 691 possible sequences (the exceptions being the Solanum chilense S3 RNase [Lycopersicon chilense; gi21623705] and the Antirrhinum hispanicum subsp. mollissimum S-RNase [gi20067963]).
In the first pattern the following amino acids are present at >1% frequency at a given site (frequencies are shown as subscripts):
This pattern invokes two invariant amino acid positions for all S-RNases (sites 5 and 7), plus five almost-invariant positions (sites 1, 2, 4, 6, and 8; at these positions the main amino acid variant (F, T, H, L, and P, respectively) is present at a frequency >98%). It should be noted that the amino acid pattern here identified overlaps (from position 1 to position 8) the first conserved active site sequence (CAS) of T2 RNases (Itagaki et al. 2006). According to the Conserved Domain Database (CDD; Marchler-Bauer et al. 2007), three of the residues (positions 4, 7, and 9) encompassed by the pattern derived here for S-RNases are part of the active site.
In the second pattern the following amino acids are present in more than 1% frequency at a given site (frequencies are shown as subscripts):
This pattern invokes only one almost-invariant position (at site 1, where a W is present at a frequency >98%).
S-Like Amino Acid Patterns
The amino acid pattern [HY]EW is found in 54 of 69 S-like RNases and in only 7 of 658 S-RNase sequences (the 7 sequences are all from Prunus). The finding of this amino acid pattern in S-allele sequences is not a spurious result since this pattern is found in the expected place when sequences are aligned. The words HEWE and HEWEKHGTC (possible words starting at position 1) appear 35 and 34 times, respectively, in the S-like RNase data set and never in the S-RNase data set.
It is surprising that almost all S-RNases and a subset of 17 S-like RNase sequences do not have the HEW amino acid pattern. This pattern partially overlaps the second conserved active site sequence (CAS) of T2 RNases (Itagaki et al. 2006). According to the CDD (Marchler-Bauer et al. 2007) the H and E residues at positions 1 and 2, respectively, are part of the active site of T2 RNases. Only 3 of the 17 S-like RNase sequences not having the HEW motif have the H residue at position 1 and only 9 have the E residue at position 2. All 17 sequences are from monocots (data not shown).
Amino acid pattern 4 (see Table 1) is not found in any of the 658 S-RNase sequences that encompass the region where the pattern is located. This pattern is found in 64 of 69 S-like sequences (3 Oryza sativa [gi125538782, gi23616976, and gi125603519], 1 Hordeum vulgare [gi2150002], and 1 Panax ginseng [gi51701931] sequence do not show this amino acid pattern). This pattern is found very close to the previously discussed amino acid pattern but does not encompass the second conserved active site sequence. The word CPS (a possible word for positions 1–3 of the pattern) occurs 40 times in the S-like RNase data set and not in the S-RNase data set.
Screening of EST Databases for T2 RNases Showing Amino Acid Motifs Characteristic of S-RNases
EST (expressed sequence tags) data are publicly available for 98 plant species (http://www.ncbi.nlm.nih.gov/genomes/PLANTS/PlantList.html). These EST collections were first screened for T2 RNase sequences by comparing each of the six possible open reading frames of each EST sequence against the Nicotiana alata S2, Antirrhinum hispanicum S2, Prunus avium S1, and Pyrus pyrifolia S4 S-RNases using BLASTP. Sequences showing significant (E value <0.05) homology with these T2 RNases (called the T2 RNAse EST collection) were then inspected for the presence of amino acid patterns 1 and 2 (Table 1). In total, 12 unique sequences (Malus domestica, CN579810; Malus domestica, CO414712; Lotus japonicus, CN825207; Malus domestica, CV629264; Marchantia polymorpha, BJ859209; Helianthus annuus, DY922217; Hedyotis terminalis, CB078110; Euphorbia esula, DV121487; Lactuca sativa, DY981169; Gossypium hirsutum, DT463266; Glycine max, BM520744; and Aquilegia formosa × A. pubescens, DR949132) were retrieved, showing the rarity of these patterns in T2 RNase sequences represented in EST collections. Two sequences are highly similar to already described S-RNase alleles (accession numbers CN579810 and CO414712) and are from Malus domestica (Rosaceae). In addition to these EST sequences, only the Lotus japonicus (Fabaceae; accession number CN825207) sequence presents the two amino acid patterns expected for an S-RNase sequence. This sequence shows 35% amino acid identity with S-RNase sequences from Rosaceae. This is the expected pattern for an S allele from a Fabaceae species since the latter group of species is more closely related to Rosaceae species than to Solanaceae/Plantaginaceae species (Wikström et al. 2001). For comparison, S alleles from Solanaceae share about 35% amino acid identity with Plantaginaceae S alleles (data not shown). These two plant families have been diverging for the last 82–86 million years, while Fabaceae and Rosaceae have been diverging for the last 89–91 million years (Wikstrom et al. 2001).
Since co-occurrence of patterns 1 and 2 is found for the vast majority of functional S-RNases, it is unlikely that the sequences showing pattern 1 or 2 only represent functional S alleles. Nevertheless, these sequences might represent rare cases of S-RNase sequences that acquired a new function.
The Marchantia polymorpha (a liverwort; BJ859209) and Aquilegia formosa × A. pubescens (a stem eudicotyledon; DR949132) sequences are likely false positives since the S-RNase lineage is tought to have appeared just before the separation of the Asteridae and Rosidae (Igic and Kohn 2001; Steinbachs and Holsinger 2002).
Testing the Inclusivness of the Approach Used
Looking for amino acid patterns in nonannotated EST sequences showing similarity to S-RNases is a simple and fast approach. Nevertheless, it could be argued that the patterns derived reflect our still limited knowledge on genes that are known to belong to the S-RNase lineage. Although much more time-consuming and computationally intensive, this caveat would not apply when using a phylogenetic approach.
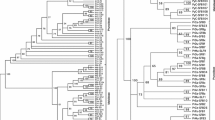
In order to compare the two approaches, we selected from the T2 RNase EST collection (see above), per species, the three (in order to limit computational burden) unique sequences that showed the highest similarity to the set of four S-RNases used to compile that data set. Then we added to this data set all EST sequences showing pattern 1 or 2 (see previous section), all known S-like sequences, sequences previously described as belonging to the S-RNase lineage, and the Solanaceae, Plantaginaceae, and Rosaceae data sets used by Vieira et al. (2007). At this stage, in order to reduce the computational burden we discarded sequences showing fewer than five nucleotide differences from other already included sequences. The final data set contains 376 aligned nucleotide sequences. The fast maximum likelihood algorithm implemented in GARLI (Zwickl 2006) was used to obtain 250 maximum likelihood trees using this data set. It should be noted that gapped positions are ignored. The consensus of the 250 trees is shown in Fig. 2.

Consensus of the 250 maximum likelihood trees generated with GARLI. 1, Prunus avium non-S RNase PA1 (AB096918); 2, Malus domestica Mdfrt3099j19 (CV629264); 3, Nicotiana sylvestris relic S-RNase (AJ002296); 4, Luffa cylindrica RNase LC2 (D64012); 5, Luffa cylindrica RNase LC1 (D64011); 6, Aquilegia formosa × A. pubescens (DT744263); 7, Medicago truncatula (AW776643); 8, Hedyotis terminalis (CB078110); 9, Lotus japonicus (CN825207); 10, Pisum sativum (Y11824); 11, Arachis hypogaea (EE124041); 12, Glycine max (BM520744); 13, Gossypium hirsutum (DT463266); 14, Euphorbia esula (DV121487); 15, Medicago truncatula (AC159124); 16, Malus domestica (CO414712); 17, Malus domestica (CN579810); 18, Petunia hybrida S x2 -RNase (M81685); 19, Antirrhinum hispanicum S-like 29 (AJ315592); 20, Marchantia polymorpha lwb14d22 (BJ859209), (see also Supplementary Table 3)
Of the sequences identified by the maximum likelihood method as likely belonging to the S-RNase lineage (excluding sequences known to be S-RNases), all but two were identified by simply looking at amino acid patterns typical of S-RNases (Table 2). On the other hand, six sequences showing amino acid patterns typical of S-RNases were not identified by the maximum likelihood phylogenetic method, including those from Luffa cylindrica previously reported as belonging to the S-RNase lineage (Steinbachs and Holsinger 2002).
Detailed Phylogenetic Studies Using a Set of 43 Sequences
A Bayesian phylogenetic approach was used in order to try to better define the relationship of the set of sequences listed in Table 2 and known S-RNase and S-like sequences. In order to avoid computational burden, sequences listed in Table 2 known to be relic S-RNases (the A. hispanicum S-like 29, P. inflata X2, and N. silvestris sequences [Lee et al. 1992; Golz et al. 1998; Liang et al. 2003]) were not used.
The unconstrained Bayesian tree presented in Fig. 3a has very little definition. Nevertheless, it is clear from this tree that the Lactuca sativa and Helianthus annus sequences do not belong to the S-RNase lineage since they cluster with strong support with the S-like type I and II sequences used as a reference. The observed lack of definition of the tree is not unusual since very divergent sequences are being used. For instance, the Bayesian phylogenetic analyses performed by Steinbachs and Holsinger (2002) show the Luffa cylindrica (Cucurbitaceae) LC1 and LC2 sequences as a sister group to Prunus S-RNases rather than a sister group to all Rosaceae S-RNases, as expected. Nevertheless, based on this tree, these authors still conclude that these sequences belong to the S-RNase lineage.

Cladograms built using a Bayesian approach. (a) Unconstrained tree; (b) tree constrained on having all S-like type I and II reference sequences plus the Lactuca sativa and Helianthus annus sequences as a monophyletic group, and all other sequences, with the exception of the outgroup, as monophyletic as well. Numbers represent posterior probability values. Both trees were rooted using the Aspergillus sequence used by Steinbachs and Holsinger (2002)
The Bayesian tree presented in Fig. 3b was constrained on having all S-like type I and II reference sequences plus the Lactuca sativa and Helianthus annus sequences as a monophyletic group. Furthermore, all other sequences, with the exception of the outgroup, were forced to be monophyletic as well. When the difference of Bayes factors between this tree and the unconstrained tree is calculated, a value of 2.06 is obtained. It has been suggested that the constrained tree should be considered as significantly worse only if twice this difference gives a number higher than 10 (Kass and Raftery 1995). Thus, the constrained tree is not significantly worse than the unconstrained one. Only three T2 RNase classes have been described based on phylogenetic and intron number patterns (Igic and Kohn 2001; Steinbachs and Holsinger 2002). Therefore, it seems reasonable to assume that all sequences that cluster in Fig. 3b with known S-RNases belong to the S-RNase lineage. It should be noted that at least two S-like genes (the Nicotiana alata U13256 (Dodds et al. 1996) and the Oryza sativa CM000133 genes) do present the same intron profile found in S-RNase genes.
The tree shown in Fig. 3b still shows little definition. In principle, it is possible to compare the Bayes factor of a tree constrained to a specific evolutionary hypothesis with that for the unconstrained tree. Gene duplications are nevertheless apparent since, for instance, two sequences that seem to belong to the S-RNase lineage were retrieved from M. truncatula. In order to test a specific evolutionary hypothesis we would need to specify all gene duplications and when they occurred. Too many, a priori, reasonable alternatives are possible, and at present it is impracticable to test all of them.
Discussion
The four amino acid patterns described here can be used to distinguish S-RNases from other T2 RNases. The finding that two of the patterns that distinguish S-like RNases from S-RNases overlap the conserved active site sequences suggests that such differences are functionally important, but this assumption must be confirmed experimentally. These patterns were used to identify putative sequences that belong to the S-RNase lineage among hundreds of T2 RNase sequences. It is clear that this method produces some false positives since, for instance, a T2 RNase sequence harboring amino acid pattern 1 (typical of S-RNases) is found in a liverwort (Table 2). Nevertheless, there is a very good overlap between those results and the ones obtained using a fast maximum likelihood phylogenetic approach (Table 2).
The Bayesian tree presented in Fig. 3b suggests that, when using the pattern-based approach, the rate of false positives is about 15%. Furthermore, the rate of false negatives seems to be about 10%. It should be noted that, under the conditions used here, the fast maximum likelihood method of tree reconstruction produces about 15% false negatives. Therefore, the much simpler method of pattern searching identifies most sequences that seem to belong to the S-RNase lineage without producing too many false negatives or positives. We thus suggest that in future studies the amino acid pattern-based approach is used to gather a small set of putative sequences that may belong to the S-RNase lineage, and that must then be tested using a proper phylogenetic approach. Further amino acid patterns exclusively found in genes belonging to the S-RNase lineage may be used, in order to try to decrease the false-negative rate of discovery.
The sequence from L. japonicus (Fabaceae) shows all the features expected for a functional S allele. The L. japonicus EST library was prepared using mRNA extracted from green seed pods at very early stages to fully developed green pods. Although there is no report in the literature showing whether the S-RNase gene is expressed at these stages, even at low levels, we have found two M. domestica S-RNase sequences in one EST library prepared from young fruitlets (accession numbers CN579810 and CO414712). Furthermore, the L. japonicus sequence presents the two amino acid patterns found in most S alleles and does not show the two amino acid patterns typical of S-like sequences. In principle, primers can now be designed for conserved regions between the L. japonicus sequence and sequences shown to be S alleles in other plant species. The use of such primers should greatly facilitate the isolation of other putative S alleles from species of the Lotus genus. By looking at variability patterns, further support for this sequence being a functional S allele can be found. It should be noted that L. japonicus is a diploid self-compatible species (Ross and Jones 1985), although other Lotus species are self-incompatible, including L. corniculatus (Ross and Jones 1985), a polyploid species that is difficult to distinguish morphologically from L. japonicus (Liston 2006; available at http://www.ncbi.nlm.nih.gov/genomes/PLANTS/PlantList.html). It is thus conceivable that self-compatibility was lost in L. japonicus due to mutations in the pollen component rather than in the pistil (S-RNase) component. In Prunus (Rosaceae) loss of GSI has been found to be mostly associated with mutations in the pollen gene (Tao et al. 2007).
The possibility that polyploid Lotus species may have a GSI S-RNase-based system is intriguing since this would imply that competitive interaction (i.e., in polyploid plants pollen carrying two different pollen S alleles fails to function in SI) described in Solanaceae (Sijacic et al. 2004; Tsukamoto et al. 2005) and Maloideae (Rosaceae; de Nettancourt 1997), but absent in Prunus (Rosaceae; Hauck et al. 2006; Nunes et al. 2006), is also absent in Lotus (Fabaceae). In Prunus, polyploidy does not causes self-compatibility unless two nonfunctional S-haplotypes are present in the same individual (Hauck et al. 2006).
It has been argued that upon loss of S-RNase-based GSI the S-RNase gene rarely acquires a new function (Igic and Kohn 2001). Sequences showing only one of the patterns for S-RNases could represent such cases. A case-by-case discussion is provided below.
The species Hedyotis terminalis belongs to the Rubiaceae family. This family is known to contain species showing GSI, although it is not known whether it is S-RNase based (Igic and Kohn 2001) (Fig. 4). The H. terminalis sequence comes from an EST library prepared from flowers at preanthesis stage 2. The S-RNase gene is known to be expressed at low levels in ovaries and petals (Sims 1993) and styles at this stage (Anderson et al. 1986). It is conceivable that we have identified an S-RNase sequence, although the lack of amino acid pattern 1, as well as the presence of pattern 3 (typical of S-like sequences), suggests otherwise. It is unclear whether this putative non-S-RNase gene arose by duplication or loss of function of an S-RNase gene.

Relationship of dicot plant families where evidence for sequences belonging to the S-RNase lineage has been found. G, presence of GSI; +, use of S-RNases. Divergence times are according to Wikström et al. (2001)
The two L. cylindrica (LC1 and LC2) and M. charantia (for M. charantia only the amino acid sequence is available, thus this sequence was not used in phylogenetic analyses) sequences come from species that belong to the Cucurbitaceae family, in which no species presenting GSI are known (Fig. 4). Among Cucurbitales, only the Begoniaceae family is known to contain species showing GSI (Igic and Kohn 2001), although it is unknown whether it is S-RNase based. Begoniaceae and Cucurbitaceae have been diverging for about 65–66 million years (Wikstrom et al. 2001). Therefore the change in function of the S-RNase gene likely occurred about 65 million years ago or earlier.
Our analyses also show that the previously reported non-S-allele P. avium Pa1 (Yamane et al. 2003a) and a gene from Malus domestica (CV629264) clearly belong to the S-RNase lineage since they cluster with high confidence with Prunus S-allele sequences. Although Yamane et al. (2003a), using a Southern blot approach, could not find the PA1 gene in species from Maloideae (Malus domestica and Pyrus pyrifolia), the M. domestica (CV629264) sequence could be orthologous to the Prunus PA1 sequence.
The P. sativum (Fabaceae) HRGP gene is of hybrid origin, is involved in the regulation of DNA replication in the chloroplast, and shows no RNase activity (Gaikwad et al. 1999; Igic and Kohn 2001). No domain besides that for T2 RNase (about 190 amino acids long) is detected in the P. sativum HRGP sequence (data not shown). The M. truncatula (Fabaceae) sequence (AC159124) reported here shows a PDI (protein disulfyl isomerase)/thioredoxin domain (~80 amino acids long) and a T2 RNase domain (~170 amino acids long). Since the PDI domain is not present in the P. sativum HRGP sequence, it is unlikely that they are orthologous. Under this hypothesis, the two genes were created as the result of two independent events involving an S-RNase gene. At present there is no evidence that such chimeric genes are present in non-Fabaceae species. Indeed, Gaikwad et al. (1999) failed to find an orthologue of the P. sativum HRGP gene in other angiosperms when performing a Northern blot survey. Based on this observation, Igic and Kohn (2001) have concluded that this copy is possibly unique to P. sativum and its relatives. The Fabaceae chimeric genes are thus likely less than 89–91 million years old (the time for the separation of Fabaceae from its sister taxa [Wikstrom et al. 2001]). Three other Fabaceae nonchimeric genes that seem to belong to the S-RNase lineage have been found in M. truncatula (AW776643), Arachis hypogaea (EE124041), and Glycine max (BM520744).
All species belonging to the Euphorbiaceae family show unisexual flowers (Flora Iberica; http://www.rjb.csic.es/floraiberica/PHP/cientificos.php). The split between Euphorbiaceae species and the other Rosidae plant families considered here happened about 94–98 million years ago (Fig. 4). Therefore, about 94–98 million years ago or earlier, when S-RNase GSI was likely lost in the Euphorbiaceae lineage, the S-RNase gene acquired a new function that is represented by the Euphorbia esula sequence. S-RNase-based GSI is found in Rosaceae (Sassa et al. 1996; Tao et al 1997) and Fabaceae (see above) species. Cucurbitaceae species are more closely related to species from the Rosaceae and Fabaceae families than to Euphorbiaceae species. Furthermore, no molecular homologues of the L. cylindrica genes have been found outside the Cucurbitaceae (Igic and Kohn 2001). Thus, it is unlikely that the sequences from L. cylindrica and M. charantia are orthologous to the Euphorbiaceae sequence reported here.
Gossypium hirsutum (Malvaceae) is a self-compatible allotetraploid species (Flora Iberica; http://www.rjb.csic.es/floraiberica/PHP/cientificos.php), but other species of the Malvaceae family present GSI, although it is not known whether it is S-RNase based (Igic and Kohn 2001) (Fig. 4). The G. hirsutum EST library was prepared using cotton ovules. It is known that the S-RNase gene is expressed at low levels in the whole ovary (Sims 1993). Therefore it is conceivable that the G. hirsutum sequence represents the S-RNase gene. Nevertheless, the absence of amino acid pattern 1 and presence of pattern 3 suggest otherwise (Table 2). Species of the Brassicaceae family, which includes Arabidopsis thaliana, have been diverging from species of the Malvaceae family for the last 85–90 million years (Wikstrom et al. 2001). No A. thaliana sequence belongs to the S-RNase lineage (Igic and Kohn 2001; Steinbachs and Holsinger 2002; data not shown). Thus no orthologous sequence of the G. hirsutum gene reported here is found in the fully sequenced A. thaliana genome. This suggests that the G. hirsutum gene is younger than 90 million years. Nevertheless, it is also conceivable that it may be older and that it was lost in the Brassicaceae lineage.
The above considerations suggest that the S-RNase gene acquired a new function after GSI loss on several independent occasions. In contrast, our data are compatible with a single origin for the S-RNase GSI system before the split of the Asteridae and Rosidae (about 120 million years ago [Wikstrom et al. 2001]) (Fig. 4). Within Asteridae the oldest plant family showing S-RNase-based GSI is the Campanulaceae (Stephenson et al. 1992), although no S-RNase sequence is yet available. Campanulaceae species have been diverging from Rubiaceae, Plantaginaceae, and Solanaceae species for the last 102–112 million years (Wikstrom et al. 2001). Within Rosidae we found a gene derived from an S allele in a species from the Malvaceae family. Malvaceae species have been diverging from Cucurbitaceae, Rosaceae, Fabaceae, and Euphorbiaceae species for the last 100–109 million years (Wikstrom et al. 2001). Finding evidence for genes belonging to the S-RNase lineage in such old plant families offers support for the single-origin hypothesis. Under the alternative hypothesis that S-RNase-based GSI appeared independently in Asteridae and Rosidae, two S-RNase-based GSI systems would have evolved in the time space of about 10 million years, and this seems unlikely. Moreover, the Bayesian tree shown in Fig. 3b, which is constrained in having all S-like type I and II reference sequences plus the Lactuca sativa and Helianthus annus sequences as a monophyletic group, and all other sequences, with the exception of the outgroup, as monophyletic as well, is not statistically worse than the unconstrained tree. This finding also suggests a single common origin for S-RNase-based GSI.
References
Anderson MA, Cornish EC, Mau SL et al (1986) Cloning of cDNA for a stylar glycoprotein associated with expression of self-incompatibility in Nicotiana alata. Nature 321:38–44
Barrett SCH, Harder LD, Worley AC (1996) The comparative biology of pollination and mating in flowering plants. Philos Trans Roy Soc B 351:1271–1280
de Nettancourt D (1997) Incompatibility in angiosperms. Springer-Verlag, Berlin
Dodds PN, Clarke AE, Newbigin E (1996) Molecular characterisation of an S-like RNase of Nicotiana alata that is induced by phosphate starvation. Plant Mol Biol 31:227–238
Ferrer MM, Good-Avila SV (2007) Macrophylogenetic analyses of the gain and loss of self-incompatibility in the Asteraceae. New Phytol 173:401–414
Gaikwad A, Tewari KK, Kumar D, Chen W, Mukherjee SK (1999) Isolation and characterisation of the cDNA encoding a glycosylated accessory protein of pea chloroplast DNA polymerase. Nucleic Acids Res 27:3120–3129
Golz JF, Clarke AE, Newbigin E, Anderson M (1998) A relic S-RNase is expressed in the styles of self-compatible Nicotiana sylvestris. Plant J 16:591–599
Green PJ (1994) The ribonucleases of higher plants. Annu Rev Plant Physiol Plant Molec Biol 45:421–445
Hackauf B, Wehling P (2005) Approaching the self-incompatibility locus Z in rye (Secale cereale L.) via comparative genetics. Theor Appl Genet 110:832–845
Hauck NR, Yamane H, Tao R, Iezzoni AF (2006) Accumulation of nonfunctional S-haplotypes results in the breakdown of gametophytic self-incompatibility in tetraploid Prunus. Genetics 172:1191–1198
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Igic B, Kohn JR (2001) Evolutionary relationships among self-incompatibility RNases. Proc Natl Acad Sci USA 98:13167–13171
Itagaki T, Koyama H, Daigo S, Kobayashi H, Koyama T, Iwama M, Ohgi K, Irie M, Inokuchi N (2006) Primary structure and properties of ribonuclease Bm2 (RNase Bm2) from Bryopsis maxima. Biol Pharm Bull 29:875–883
Kass RE, Raftery AE (1995) Bayes factors. J Am Stat Assoc 90:773–795
Lee HS, Singh A, Kao T-H (1992) RNase X2, a pistil-specific ribonuclease from Petunia inflata, shares sequence similarity with solanaceous S proteins. Plant Mol Biol 20:1131–1141
Liang L, Huang J, Xue Y (2003) Identification and evolutionary analysis of a relic S-RNase in Antirrhinum. Sex Plant Reprod 16:17–22
Marchler-Bauer A, Anderson JB, Derbyshire MK et al (2007) CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res 35:D237–D240
McClure BA, Franklin-Tong V (2006) Gametophytic self-incompatibility: understanding the cellular mechanisms involved in “self” pollen tube inhibition. Planta 224:233–245
Nunes MDS, Santos RAM, Ferreira SM, Vieira J, Vieira CP (2006) Variability patterns and positively selected sites at the gametophytic self-incompatibility pollen SFB gene in a wild self-incompatible Prunus spinosa (Rosaceae) population. New Phytol 172:577–587
Pereira P, Fonseca NA, Silva F (2006a) Fast discovery of statistically interesting words. Technical Report DCC-2007-01. DCC-FC & LIACC, Universidade do Porto, Porto, Portugal
Pereira P, Fonseca NA, Silva F (2006b) A high performance distributed tool for mining patterns in biological sequences. Technical Report DCC-2006-08. DCC-FC & LIACC. Universidade do Porto, Porto, Portugal
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Ross MD, Jones WT (1985) The origin of Lotus corniculatus. Theor Appl Genet 71:284–288
Sassa H, Nishio T, Kowyama Y, Hirano H, Koba T, Ikehashi H (1996) Self-incompatibility (S) alleles of the Rosaceae encode members of a distinct class of the T2/S ribonuclease superfamily. Mol Gen Genet 250:547–557
Sijacic P, Wang X, Skirpan AL, Wang Y, Dowd PE, McCubbin AG, Huang S, Kao TH (2004) Identification of the pollen determinant of S-RNase-mediated self-incompatibility. Nature 429:302–305
Sims TL (1993) Genetic regulation of self-incompatibility. Crit Rev Plant Sci 12:129–167
Steinbachs JE, Holsinger KE (2002) S-RNase-mediated gametophytic self-incompatibility is ancestral in eudicots. Mol Biol Evol 19:825–829
Stephenson AG, Winsor JA, Richardson TE, Singh A, Kao TH (1992) Effects of style age on the performance of self and cross pollen in Campanula rapunculoides. In: Ottaviano E, Mulcahy DL, Ms Gorla, Mulcahy GB (eds) Angiosperm pollen and ovules. Springer-Verlag, New York, pp 117–121
Tao R, Yamane H, Sassa H, Mori H, Gradziel TM, Dandekar AM, Sugiura A (1997) Identification of stylar RNases associated with gametophytic self-incompatibility in almond (Prunus dulcis). Plant Cell Physiol 38:304–311
Tao R, Watari A, Hanada T, Habu T, Yaegaki H, Yamaguchi M, Yamane H (2007) Self-compatible peach (Prunus persica) has mutant versions of the S haplotypes found in self-incompatible Prunus species. Plant Mol Biol 63:109–123
Taylor CB, Bariola PA, delCardayre SB, Raines RT, Green PJ (1993) RNS2: a senescence-associated RNase of Arabidopsis that diverged from the S-RNases before speciation. Proc Natl Acad Sci USA 90:5118–5122
Thompson J, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX window interface: flexible stategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tsukamoto T, Ando T, Watanabe H, Marchesi E, Kao TH (2005) Duplication of the S-locus F-box gene is associated with breakdown of pollen function in an S-haplotype identified in a natural population of self-incompatible Petunia axillaris. Plant Mol Biol 57:141–153
Ushijima K, Sassa H, Tao R, Yamane H, Dandekar AM, Gradziel TM, Hirano H (1998) Cloning and characterization of cDNAs encoding S-RNases from almond (Prunus dulcis): primary structural features and sequence diversity of the S-RNases in Rosaceae. Mol Gen Genet 260:261–268
Ushijima K, Yamane H, Watari A, Kakehi E, Ikeda K, Hauck NR, Iezzoni AF, Tao R (2004) The S haplotype-specific F-box protein gene, SFB, is defective in self-compatible haplotypes of Prunus avium and P. mume. Plant J 39:573–586
Vieira J, Morales-Hojas R, Santos RA, Vieira CP (2007) Different positively selected sites at the gametophytic self-incompatibility pistil S-RNase gene in the Solanaceae and Rosaceae (Prunus, Pyrus, and Malus). J Mol Evol 65:175–185
Wang L, Dong L, Zhang Y, Zhang Y, Wu W, Deng X, Xue Y (2004) Genome-wide analysis of S-Locus F-box-like genes in Arabidopsis thaliana. Plant Mol Biol 56:929–945
Weller SG, Donoghue MJ, Charlesworth D (1995) The evolution of self-incompatibility in flowering plants: a phylogenetic approach. In: Hoch PC, Stephenson AG (eds) Experimental and molecular approaches to plant biosystematics. Missouri Botanical Garden, St Louis, MO, pp 355–382
Wikstrom N, Savolainen V, Chase MW (2001) Evolution of the angiosperms: calibrating the family tree. Proc Biol Sci 268:2211–2220
Yamane H, Tao R, Mori H, Sugiura A (2003a) Identification of a non-S RNase, a possible ancestral form of S-RNases, in Prunus. Mol Genet Genomics 269:90–100
Yamane H, Ikeda K, Hauck NR, Iezzoni AF, Tao R (2003b) Self-incompatibility (S) locus region of the mutated S6-haplotype of sour cherry (Prunus cerasus) contains a functional pollen S allele and a non-functional pistil S allele. J Exp Bot 54:2431–2437
Zwickl DJ (2006) Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Ph.D. dissertation. The University of Texas at Austin, Austin
Acknowledgments
This work was partially funded by Fundação para a Ciência e Tecnologia (FCT), research projects POCTI/AGG/44800/2002 and POCI/BIA-BDE/59887/2004 (funded by POCI 2010, cofunded by FEDER funds). Nuno A. Fonseca is the recipient of postdoctoral grant SFRH/BPD/26737/2006 from the FCT.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Vieira, J., Fonseca, N.A. & Vieira, C.P. An S-RNase-Based Gametophytic Self-Incompatibility System Evolved Only Once in Eudicots. J Mol Evol 67, 179–190 (2008). https://doi.org/10.1007/s00239-008-9137-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-008-9137-x