
Abstract
The GTPase center of the large ribosomal subunit, being a landing platform for translation factors, and regarded as one of the oldest structures in the ribosome, is a universally conserved structure in all domains of life. It is thought that this structure could be responsible for the major breakthrough on the way to the RNA/protein world, because its appearance would have dramatically increased the rate and accuracy of protein synthesis. The major part of this center is recognized as a distinct structural entity, called the stalk. The main functional part of the stalk in all domains of life is composed of small L12/P proteins, which are believed to form an evolutionarily conserved group. However, some data indicate that the bacterial and archaeo/eukaryal proteins are not related to each other structurally, and only a functional relationship may be clearly recognized. To clarify this point, we performed a comprehensive comparative analysis of the L12/P proteins from the three domains of life. The results show that bacterial and archaeo/eukaryal L12/P-proteins are not structurally related and, therefore, might not be linked evolutionarily either. Consequently, these proteins should be regarded as analogous rather than homologous systems and probably appeared on the ribosomal particle in two independent events in the course of evolution.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ribosome is the core of the translation machinery of all organisms and assures two key functions: the decoding of the genetic information and the formation of peptide bonds. The ribosome, probably being a reminiscence of the ancient RNA world, is perceived as a ribozyme because its two primary key functions are dependent on rRNA (Nissen et al. 2000). However, as (Noller 2004) stated, “The invention of protein synthesis, probably instructed and catalyzed by RNA, was not to create a protein world, but to extend the structural, and therefore the functional, capabilities of the RNA world, and initially was the crowning achievement of the RNA world, but also began its demise.” It is thought that at a very early stage of life everything turned on the evolution of translation (Woese 1998). Initially, only small proteins could evolve—along with any larger, imprecisely translated ones (called “statistical proteins”) that the primitive cell was able to produce and use (Woese and Fox 1977). Each slight improvement in that process would have permitted a new generation of proteins to emerge; these new proteins, in turn, refined and developed the metabolic pathways and generally improved the performance of the cell, which then set the stage for a further round of improvement in translation (Woese 1998). According to Woese (1998), “This iterative, bootstrapping evolution continued until the accuracy of translation reached a level where it no longer prevented the evolution of the types of proteins we see today.” Therefore, translation was among the first, if not the very first, of the cellular subsystems to crystallize, and moreover, the improvement of accurate protein synthesis paved the way from progenotes to the modern organisms called genotes (Woese 1998). One of the ribosomal structures responsible for a major breakthrough on the way from RNA to protein world could have been the GTPase center, whose appearance, together with translation factors, would have dramatically increased the rate and accuracy of protein synthesis, leading to today’s richness of life forms (Frank et al. 2007; Hury et al. 2006). This ribosomal center, being an interacting place for translation factors, is regarded as one of the oldest in the ribosome, and occurs in all living organisms (Mears et al. 2002). The major part of this center is recognized on the large ribosomal subunit as a distinct structural entity, called the stalk. It is composed of conserved proteins which form an oligomeric structure, and represents an isolated entity without extensive contacts with other ribosomal components (Diaconu et al. 2005; Gonzalo and Reboud 2003; Wahl and Moller 2002). These proteins exist in two main classes—bacterial and archaeo/eukaryal. In Bacteria the stalk is composed of two types of proteins. The first is represented by the L10 protein, which forms the base of the stalk (Wahl and Moller 2002). L10 proteins from all organisms were found to be similar (Shimmin et al. 1989) and are classified among the 29 universally conserved proteins constituting the genetic core of the hypothetical universal ancestor (Harris et al. 2003). The L12 protein represents the second group (also denoted L7/L12 in the case of E. coli) and occurs on the ribosome in multiple copies, attached to it through the L10 protein. A species-dependent variability in stoichiometry has been documented for the L12 protein, showing a lack of uniformity of bacterial ribosomes in this respect. In thermophiles, there are three, whereas mesophiles have two L12 dimers, forming heptameric L10(L12)6 or pentameric L10(L12)4 complexes, respectively (Diaconu et al. 2005; Ilag et al. 2005). The archaeo/eukaryal stalk is also composed of two types of proteins, P0 and P1/P2 (Ballesta and Remacha 1996; Tchorzewski 2002). P0 is homologous to the bacterial L10 protein and, forming the base of the stalk, constitutes the anchor for the two P1/P2 protein dimers (Wahl and Moller 2002). The P1/P2 group is regarded as a functional counterpart of the bacterial L12 protein, however, it seems that they have no significant similarity in the primary structure to the bacterial protein (Liao and Dennis 1994). Alignment of P1 and P2 with the archaeal homologue, the aL12 protein, indicated that P1 and P2 form a distinct monophyletic group and arose from duplication and subsequent divergence of an ancestral form of archaeal aL12 that occurred very early in the eukaryotic lineage (Liao and Dennis 1994). These proteins form P1-P2 heterodimers (Gonzalo et al. 2001; Shimizu et al. 2002; Tchorzewski et al. 2000b), which in some eukaryotes, such as the yeast S. cerevisiae, form two distinct P1A-P2B and P1B-P2A complexes (Guarinos et al. 2001; Tchorzewski et al. 2003). In all organisms, the stalk is directly involved in ensuring the speed and accuracy of protein synthesis and constitutes part of the GTPase-associated center, with the L12/P proteins being its main functional elements (Rodnina and Wintermeyer 2001). The whole stalk is indispensable for cell survival (Krokowski et al. 2006), but for proper functioning of the ribosome only one dimer is sufficient in bacteria (Griaznova and Traut 2000), while in Eukaryotes the P1/P2 proteins are not absolutely required for the ribosome activity or cell viability (Remacha et al. 1995). Despite the apparent identical function of the L12/P proteins on the ribosome, they are not interchangeable between Bacteria and Archaea/Eukaryotes, and moreover, the P proteins confer factor binding specificity on the ribosome (Nomura et al. 2006; Uchiumi et al. 1999). What is more, in Eukaryotes, the P proteins from distinct species are not fully complementary in terms of function, and it seems that they have diverged during ribosome evolution and, apart from their primary function, might also represent regulatory elements enabling distinct organisms to accommodate their metabolism to changing environmental conditions (Rodriguez-Gabriel et al. 2000).
A structural model of the whole stalk has been proposed for the bacterial ribosome (Diaconu et al. 2005), but the intact archaeo/eukaryal stalk has not been described yet. The only structure available today is a low-resolution model of the S. cerevisiae P1A-P2B dimer, and preliminary analysis showed that the yeast complex does not resemble, in its shape, the structure of the bacterial counterpart (Grela et al. 2007). Moreover, the evolutionary relationship among the L12/P proteins from diverse organisms is unclear (Liao and Dennis 1994). Bacterial L12 proteins were classified as universally conserved in the three phylogenetic domains (Mears et al. 2002), but it seems that they are different in terms of structural organization from their archaeo/eukaryal counterparts. Therefore the question arises into which pathway of evolution the L12/P proteins may fall: divergent, where all proteins are coming from a common ancestor but the rate of evolution was so high that they have diverged to the point that no similarity can be detected anymore; or convergent, where bacterial and archaeo/eukaryal proteins were recruited independently for the same function.
Therefore, to shed more light on the evolutionary relationship among the stalk L12/P proteins, we undertook structural characterization of these proteins from the three phylogenetic domains: Bacteria, Escherichia coli; Archaea, Sulfolobus solfataricus; and Eukaryotes, Homo sapiens. The analyses showed that, considering all structural aspects, the bacterial and archaeo/eukaryal proteins are not structurally related, raising the question of the evolutionary origins of these proteins. In our view the structural data are more consistent with convergent evolution.
Materials and Methods
Protein Expression and Purification
The human P1-P2 heterodimer and P2-P2 homodimer were prepared according to the procedure described earlier (Grela et al. 2008; Tchorzewski et al. 2000a). Recombinant Sulfolobus solfataricus aL12 protein was prepared as follows. Appropriate sequence was PCR amplified from genomic DNA (a kind gift from Dr. Emmanuele De Vendittis, Universita di Napoli Federico II, Italy) with the aid of specific primers. The amplified fragment was introduced into expression vector pLM1 (Sodeoka et al. 1993). The aL12 protein was expressed with a 6xHis-tag on its N-terminus in E. coli BL21(DE3) cells and purified using affinity chromatography on an Ni column, followed by size-exclusion chromatography. The bacterial Escherichia coli L7/L12 protein was expressed using plasmid pET24b (a kind gift from Dr. Suparna Sanyal, Biomedical Center, Uppsala, Sweden), and the recombinant protein was prepared according to the procedure published earlier (Mulder et al. 2004).
Phylogenetic Analysis
The alignment of amino acid sequences from representative species, obtained using ClustalX1.8 with default options (Thompson et al. 1997), was manually checked and edited. For phylogenetic analysis sites of uncertain homology, which could not be unambiguously aligned, were removed. After removal, 75 positions (of 105) were left in the alignment. Bayesian phylogenetic analysis was performed, and its substitution model with fixed rate matrices and rate parameters (Prset aamodelpr=mixed, LSET rates=invgamma) were estimated by the MrBayes 3.0B4 program (Huelsenbeck and Ronquist 2001). Gamma distribution was approximated using four categories. Four Markov chains were run, with 1 million generations per chain, and the 50% majority rule consensus tree was built, with the first 2000 trees discarded. The tree was drawn with Tree View, Version 1.6.1, for Microsoft Windows (Page 1996). The neighbor-joining (NJ) and minimal evolution (ME) phylogenetic trees were constructed with the MEGA program, version 3.1 (Kumar et al. 2004). For both methods, the parameters p-distance model and pairwise deletion were selected, and the interior branch test and bootstrap test were performed with 1000 replications each.
Analysis of Secondary Structure Propensity
The whole amino acid sequences of the acidic ribosomal aL12/P proteins were analyzed using secondary structure prediction tools: Jpred (Cuff et al. 1998) and Sspro (Cheng et al. 2005).
Small-Angle X-Ray Scattering (SAXS) Experiment
The SAXS method was used to determine the three-dimensional (3D) structure of the studied proteins. This method has proven to be a very useful technique for obtaining low-resolution molecular shape of proteins and their complexes in solution. The main advantage of solution scattering is its ability to study the structure of native particles under nearly physiological conditions. Recently, there has been significant progress in the development of ab initio methods for low-resolution shape restoration, placing SAXS as a very fast and reliable structural approach (Svergun et al. 2001).
The synchrotron radiation X-ray scattering data were collected in two experimental sessions on the X33 camera (Boulin et al. 1988; Koch and Bordas 1983) at the European Molecular Biology Laboratory Hamburg (EMBL) on the storage ring DORIS III of the Deutsches Elektronen Synchrotron (DESY) using multiwire proportional chambers with delay line readout (Gabriel and Dauvergne 1982). The scattering patterns from all proteins were recorded at multiple solute concentrations ranging from 2 to 15 mg/ml (in the case of L7/L12 protein, from 2 to 50 mg/ml) at a sample-detector distance of 2.3 m, covering the range of momentum transfer 0.15 < s < 3.5 nm-1 (s = 4π sin(θ)/λ, where 2θ is the scattering angle and λ = 0.15 nm is the X-ray wavelength). The data collected in 10 successive 1-min frames were analyzed for the absence of radiation damage, averaged after normalization to the intensity of the incident beam, corrected for the detector response, and the scattering of the buffer was subtracted using the program PRIMUS (Konarev et al. 2003). The difference curves were scaled for the protein concentrations and extrapolated to infinite dilution following standard procedures (Feigin and Svergun 1987). The maximum dimensions of all proteins D max were estimated using the orthogonal expansion program ORTOGNOM (Svergun 1993). The forward scattering values I(0) and the radii of gyration R g were evaluated using the Guinier approximation (Guinier 1939). These parameters were also computed from the entire scattering patterns using the indirect transform package GNOM (Svergun 1992), which also provides the distance distribution functions p(r) of the particles.
Low-resolution models of the proteins were generated ab initio by the program GASBOR (Svergun et al. 2001) as previously described (Grela et al. 2007). The models of each protein were reconstructed using appropriate numbers of residues, according to the protein sequence (P1-P2, 229 residues; P2-P2, 230 residues; L7/L12, 242 residues; aL12, 222 residues). For the homodimeric P2-P2 complex, reconstructions without and also with symmetry restrictions (i.e., assuming P2 point symmetry) were performed.
For each solute, results from a dozen separate GASBOR runs were averaged to determine common structural features using the programs DAMAVER (Volkov and Svergun 2003) and SUPCOMB (Kozin and Svergun 2001). The latter program aligns two arbitrary low- or high-resolution models represented by ensembles of points by minimizing a dissimilarity measure called normalized spatial discrepancy (NSD). For every point (in this case, coordinate of DRs) in the first model, the minimum value among the distances between this point and all points in the second model is found, and the same is done for the points in the second model. These distances are added and normalized against the average distances between the neighboring points for the two models. Generally, NSD values close to unity indicate that the two models are similar. The program DAMAVER generates the average model of the set of superimposed structures and also specifies the most typical model (i.e., that having the lowest average NSD with all the other models of the set).
Rigid Body Modeling
Rigid body modeling represents the method where it is possible to build models of protein complexes from high-resolution structures of their subunits or domains against low-resolution models of the whole complexes derived from SAXS data (Petoukhov and Svergun 2005). Molecular modeling was done with the available atomic models of both domains of the L7/L12 protein. The NMR (Bocharov et al. 2004) (1RQU; fragment 1-31) and the crystallographic (Leijonmarck et al. 1980) (1CTF) structures were used as models of the N- and the C-terminal folded domains, respectively. The program BUNCH (Petoukhov and Svergun 2005) was employed to model the protein structure fitting the SAXS merged profile covering an angular range from 0.014 to 0.34 Å−1. A P2 internal symmetry, found for the N-terminal domain in crystallographic studies (Wahl et al. 2000), was imposed. BUNCH combines rigid-body with ab initio modeling for proteins consisting of folded domains, for which high-resolution models are available, joined by flexible linkers of unknown structure. A simulated annealing optimization procedure is employed to find the optimal positions and orientations of the domains that are moved as rigid bodies attached to the linkers, modeled as a chain of dummy residues separated by 3.8 Å (Petoukhov et al. 2002). Starting from an arbitrary conformation of domains and linkers, BUNCH performs random modifications of the model maintaining the structures of the domains and the connectivity of the linkers. At each step the scattering profile is computed and compared with the experimental one, and in a simulated annealing manner the model converges toward a configuration that minimizes a merit function. The agreement with the experimental scattering profile and some geometrical parameters of the models are included in this merit function (Petoukhov and Svergun 2005). The structural discrepancy between the resulting models of 20 independent BUNCH runs was evaluated with the program SUPCOMB (Kozin and Svergun 2001) through the NSD parameter. NSD values close to unity indicate that two models are similar, and when this parameter departs from 1.0, the models become more and more dissimilar.
Results
Initially, primary structures were compared to assess the similarity among the L12/P proteins. Alignment of L12/P protein sequences from all domains of life was attempted by the authors of the previous analysis (Liao and Dennis 1994), who found some similarities between them, but the assumption of their common ancestry was less than certain. However, we found no considerable similarity between bacterial and archaeal/eukaryal L12/P proteins in our multiple sequence alignment of those proteins (Fig. 1). This suggests that these two groups of proteins may not be evolutionarily related, or the relation, if any, could only be preserved at a higher structural level. Therefore, the phylogenetic analysis was only applied to the archaeal and eukaryal proteins. The phylogenetic trees of archaeal aL12 and eukaryal P proteins generated by the Bayesian, NJ, and ME methods showed essentially the same topology (data not show), and the tree obtained by the Bayesian inference is shown in Fig. 2. The protein sequences were divided into three well-supported clades, corresponding to P1, P2, and aL12 proteins (Fig. 2). Within these three clades some groups of sequences corresponded to known taxa, while some of the other groupings did not reflect recognized phylogenetic associations. Our results thus confirm earlier findings of Liao and Dennis (1994), but only with respect to relationships between archaeal and eukaryal proteins.

Alignment of representative ribosomal stalk L12/P proteins from three domains of life. Black boxes indicate fragments of sequences used for phylogenetic analysis, which also correspond roughly to the most conserved N- and C-terminal domains (NTD and CTD) in eukaryotic proteins. The red boxes indicate the NTD and CTD in bacterial proteins

Phylogenetic tree of aL12/P proteins obtained by Bayesian inference. Numbers at nodes represent Bayesian posterior probabilities/NJ bootstrap support of particular clades. (–) That clade does not exist in NJ analysis
Since evolutionary relationships are not always well recognized at the level of primary structure, we performed a set of structural analyses to evaluate the secondary and tertiary structure of the L12/P proteins. First, a secondary structure analysis was performed using three representative proteins, the H. sapiens, S. cerevisiae and S. solfataricus, with the aid of the Jpred and Sspro programs. The secondary structure (2D) of E. coli L7/L12 protein was extracted from known 3D models (Bocharov et al. 2004; Leijonmarck et al. 1980). Once again, the aL12/P proteins could be easily aligned, with the 2D organization of aL12 closely resembling that found in the P-protein group (Fig. 3). The bacterial L7/L12 did not show significant similarities in the 2D structure to the archaeal/eukaryal aL12/P proteins, except for the first 30 amino acids, which form two α-helices responsible for dimerization.

Secondary structure of the whole proteins as determined by the Jpred and Sspro programs. The secondary structure of L7/L12 was extracted from known 3D structures (PDB nos: 1RQT, 1RQS). H, α-helical structure; E, β-sheet
To provide a more exhaustive insight into the issue, tertiary structures of proteins were analyzed with the aid of SAXS. The structures were solved for human P1-P2 and P2-P2 dimers, S. solfataricus aL12 dimer, and E. coli L7/L12 complex. The experimental scattering pattern of all complexes is presented in Fig. 4, and the structural parameters computed from these data are listed in Table 1.

Experimental X-ray scattering patterns of L12/P proteins and scattering computed from their models. Dots with error bars depict experimental data, and solid lines are fits obtained from the ab initio models. The insets show the distance distribution functions [p(r)] computed from experimental data by GNOM. (A, B) Human P1-P2 and P2-P2 dimers, respectively; (C) aL12 archaeal S. solfataricus dimer; (D) L7/L12 bacterial E. coli dimer
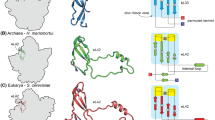
A low-resolution SAXS model of the human P1-P2 heterodimer was reconstructed ab initio as described under Materials and Methods. Twelve independent reconstructions using the program GASBOR yielded superimposable results providing reasonable fits to the experimental data. The ab initio model computed by the program DAMAVER is presented in Fig. 5. The model displays an elongated bent shape consisting of two domains with a maximum diameter of about 12 nm and a cross section of about 3 nm (Fig. 4A). However, the estimated molecular mass (MM) is somewhat larger than the value expected from the sequence, which indicates that a fraction of the protein may undergo aggregation in solution. This information is supported by our observation that the human heterocomplex has a tendency to aggregate (Grela et al. 2008). But there was no significant increase in the apparent MM of the solute at higher protein concentrations, which should have been the case if unspecific protein aggregation or extensive formation of oligomers were taking place. Instead, the Guinier plots of the experimental data displayed a linear behavior characteristic of monodisperse solutions. It could thus be concluded that the protein is largely dimeric in solution, however, a small portion of the dimer may exist in higher oligomeric forms which may slightly increase the experimental MM value. Additionally, since the human P2-P2 homodimer may exist in solution as a stable entity and may represent a transient form during the exchangeability of the P proteins (Grela et al. 2008), we used SAXS to determine the overall shape of the homodimer as well. Low-resolution shape reconstructions of the homodimer, performed assuming P2 symmetry and without symmetry restrictions, lead to superimposable shapes. The distance distribution function of the homodimer (Fig. 4B, inset) indicates an elongated particle with a maximum diameter of 11 nm and a cross section of about 2.5 nm. A shoulder at higher intraparticle distances observed in the p(r) function suggests that the average separation between the centers of two domains is about 6 nm. The ab initio model is displayed in Fig. 5. The shape of the homodimer clearly displays two linearly positioned domains with a waist in the middle. In the case of human P2-P2 dimer the estimated MM of the solute agrees very well with the value expected from the sequence, indicating that the protein is monodisperse in solution. As the next step, we analyzed the structure for the orthologous archaeal protein aL12. The structural parameters are almost identical to those obtained for the eukaryotic counterparts (Table 1). Analysis of the aL12 protein showed a good agreement between theoretical and estimated MM, underscoring the fact that the protein exists as a single species in solution, which was also verified by size exclusion chromatography and native PAGE analyses (data not shown). The distance distribution function of the aL12 dimer (Fig. 4C, inset) indicates an elongated particle with a maximum diameter of 12 nm and a cross section of about 2.5 nm. Also in this case, a shoulder at higher intraparticle distances was observed in the p(r) function, suggesting that the average separation between the centers of two domains is about 6 nm. Following the procedure presented above, an ab initio model was constructed (Fig. 5). The shape of the aL12 dimer clearly resembles the structure of the eukaryotic complexes, with two linearly positioned domains with a waist in the middle.

Ab initio models of the L12/P proteins. P1-P2 and P2-P2, human hetero- and homodimers, respectively; the complexes were resolved at 1.9-nm resolution. P1A-P2B, S. cerevisiae dimer (Grela et al. 2007); resolution, 0.7 nm. aL12, S. solfataricus dimer; resolution, 1.8 nm. L7/L12 E. coli dimer; resolution, 1.8. All models are shown in the front view (A) and the view rotated by 90° around the Y-axis (B). The models were generated with the VMD program (Humphrey et al. 1996)
There is no available SAXS model for bacterial L7/L12 protein, and available structural data for the bacterial dimer are not suitable for direct comparison with the archaeal/eukaryal SAXS models. Therefore, in order to obtain compatible structural model of bacterial proteins, the SAXS analysis was performed with the E. coli L7/L12 dimer. The structure of the individual domains for that protein has been solved by crystallography (Leijonmarck et al. 1980; Wahl et al. 2000) and NMR spectroscopy (Bocharov et al. 2004), but the overall shape of the whole dimer in solution or of the stalk has not been solved, except for a provisional proposition presented recently (Diaconu et al. 2005). Consequently, we were able to obtain the ab initio low-resolution model of L7/L12, as depicted in Fig. 5. Analysis of the E. coli L7/L12 dimer showed that the protein exists in solution as a monodisperse entity, even at very high protein concentrations, and the estimated MM of the solute agrees well with the value computed from the L7/L12 sequence (Table 1). The p(r) function (Fig. 4D, inset) indicates an elongated particle with the maximum diameter of 12 nm.
The L7/L12 dimer displays three distinct and well-separated domains. The two C-terminal domainss (CTDs) are very well recognizable and one centrally located N-terminal domain (NTD) can also be identified confidently. The angle between the two CTDs is of about 120o between the directions of their long axes. It should be emphasized that this is the first insight into the molecular organization of individual domains of the L7/L12 dimer under native conditions and it fully agrees with the propositions made previously (Bocharov et al. 2004; Mulder et al. 2004). Moreover, to support our L7/L12 model, the experimental scattering profile was fitted with the rigid-body modeling program BUNCH in order to find the spatial arrangement of the available high-resolution structures representing the folded domains (see Materials and Methods). The program was run 20 times from random starting configurations, and an excellent agreement with the SAXS curve was obtained in all cases, with χ values between 1.04 and 1.33, as shown in Fig. 6A. The average NSD obtained between the 20 structural models derived was 0.88 ± 0.07, indicating that all structures present an almost-equivalent domain distribution. Derived structural models present an open arm shape, where the NTD is located at the elbow and the two CTDs are at the extremes. The average interdomain distances between both CTDs and between the NTD and the CTDs are 85.0 ± 2.2 and 48.2 ± 0.4 Å, respectively, and the angle formed between the three domains is 124.0 ± 5.8°. Therefore, the measured SAXS profile tightly constrains the relative domain positions in L7/L12, although their orientations and linker conformations varied from model to model. This result produced a remarkable fit, at the level of resolution available between SAXS and BUNCH models, and fully confirmed the domain orientation in the SAXS model (Figs. 6B and C).

Rigid-body modeling of the L7/L12 protein dimer. (A) Experimental X-ray scattering pattern of the L7/L12 dimer. Dots with error bars depict experimental data; the red line represents the fit from the rigid-body model. (B) Rigid-body models of the L7/L12 protein. The N-terminal domain and C-terminal domain are shown in green and blue, respectively; the linker is marked in red. Models are shown in the top view (I) and front view (II), rotated by 90° around the X-axis. (C) Ab initio SAXS model of L7/L12 proteins (front view). The models were generated by the VMD program
Summing up, the obtained archaeal/eukaryal SAXS models, which can be described as two linearly positioned domains with a waist in the middle, depart significantly from the bacterial one, which showed a typical three-domain organization, and moreover, this structure suggests the presence of limited amount of dynamics in the linkers connecting the NTD and the CTD in L7/L12. Therefore, considering 1D, 2D, and 3D analyses, there is no similarity between bacterial and archaeal/eukaryal proteins.
Discussion
Phylogenetic studies of ribosomal RNA have revolutionized our understanding of biological diversity by revealing that modern organisms fall into three phylogenetic domains: Bacteria, Archaea, and Eukarya (Woese et al. 1990). In principle, the rRNA sequence is well suited for determining deep phylogenetic relationships because rRNA is present in all organisms, and has evolved at a sufficiently slow rate to retain phylogenetic information between distantly related organisms. On the other hand, phylogenetic analysis of amino acid sequences does not always determine clear evolutionary relationships among the proteins of interest. Systematic phylogenetic analyses of universally conserved proteins have revealed a small set of genes that can be traced back to the universal ancestor and have coevolved since that time with the ribosomal RNA since their divergence from a last universal common ancestor (LUCA). As found, most of the core three-domain genes/proteins belong to the nucleic acid-based central information pathway (ribosomal proteins, DNA/RNA polymerase subunits, ribosomal elongation factors) and more than half are ribosomal proteins (29 of 50) (Harris et al. 2003), supporting the conclusion that the divergence of the three types of ribosomes (bacterial, archaeal, and eukaryal) occurred after a relatively efficient ribosome structure had already been formed (Olsen and Woese 1997). The majority of the universally conserved ribosomal proteins showing a three-domain phylogeny is represented by small-subunit proteins. The proteins of the large ribosomal subunit are a more complex group, and a smaller fraction of these was universally conserved (Harris et al. 2003). In total, 32 ribosomal proteins are strictly conserved in all bacterial, archaeal, and eukaryotic ribosomes which ensure preservation of the core and global shape of the ribosome (Lecompte et al. 2002). The majority of orthologous ribosomal proteins show a clear sequence-based relationship reflected by functional link. However, the proteins that form the ribosomal stalk do not fully obey this rule. The base of the prokaryotic/eukaryotic stalk is formed by a very conserved protein, L10/P0, which shares the “three-domain” phylogenetic rRNA topology (Liao and Dennis 1994). Having such properties, these proteins have also been classified as universally conserved genes, belonging at the same time to the genetic core that traces its ancestry back to the LUCA of life (Harris et al. 2003). While the evolutionary connection between L10 and P0 from the three domains is unambiguous, the relationships among the L12/P proteins are less obvious. Several evolutionary analyses have linked those proteins, implying that they might have a common evolutionary origin (Liao and Dennis 1994; Ramirez et al. 1989), however, the path of evolution is still obscure. There is no doubt that the L12/P proteins play identical roles on the ribosome, being part of the GTPase-associated center which is directly responsible for the stimulation of translation-factor-dependent GTP hydrolysis (Rodnina et al. 2000). However, our present analyses, considering all structural levels, provide evidence that these proteins have different structural organization and in fact may not be orthologous.
In our initial analyses, inspection of a multiple sequence alignment did not indicate a close relationship between the bacterial and the archaeal/eukaryal L12/P proteins, implying that they may represent two unrelated sequence types. Phylogenetic analysis confirms close relationships between archaeal and eukaryal proteins. The placement of the root of the tree of life is still an open question (Gribaldo and Philippe 2002; Philippe and Forterre 1999; Walsh and Doolittle 2005). Even though we follow herein the view on the original position of the LUCA (Woese 1998; Woese et al. 1990), it must be pointed out that the postulated replacement event on the ribosomal stalk has taken place on the branch between Bacteria and Archaea plus Eukarya, irrespective of the actual position of the root. In fact, the structural similarity of L12/P ribosomal proteins in Archaea and Eukarya is yet another synapomorphy, linking the two groups together.
A secondary structure comparison confirmed this conjecture by showing that there are significant differences between the analyzed proteins. The NTD of L7/L12, which is responsible for dimerization, is composed of 30 amino acids and has two α-helices (Bocharov et al. 2004). On the other hand, the NTD of aL12/P proteins, which is also responsible for protein-protein interactions, contains about 65 amino acids and has four α-helices (Jose et al. 1995; Tchorzewski et al. 2003). At first glance, one may find some resemblance in the 2D structural organization, but it should be noted that these two domains are of profoundly different sizes. Even more pronounced differences are seen in the C-terminal parts. The C-terminal segment of L7/L12 proteins, consisting of 68 amino acids (residues 53–120), is considered a functional element and adopts a compact globular α/β-fold (Leijonmarck et al. 1980). On the other hand, the CTD of the L12/P proteins is also regarded as a functional part (Santos and Ballesta 1995) and, in principle, should be conserved throughout evolution, but that is not the case here. Our prediction indicates that the C-terminal fragment of the P proteins, comprising only some 20 residues, does not form a compact domain, although the first half of this region has a propensity to form an α-helix and the second half folds into a turn-like conformation at low temperatures, as shown before (Soares et al. 2004). Therefore, it seems that these two groups of proteins have a totally different 2D organization in the C-terminal part, which in turn may reflect a very distinct 3D organization.
The results of the 2D analysis are fully supported by the structural study using SAXS, which provided convincing evidence that the bacterial and archaeal/eukaryal L12/P proteins are not structurally related. The archaeal/eukaryal aL12/P proteins exhibit a very similar spatial organization. Both aL12 and P dimers have a very elongated, slightly dumbbell-shaped structure with a typical waist separation. In this structure the CTDs are present as separate domains, but as we have already suggested the movement of the C-terminal segments relative to the central NTD dimer makes them appear to have wider cross sections than the central portions (Grela et al. 2007). On the other hand, the SAXS model for the bacterial L7/L12 dimer showed a structural organization entirely different from that of their archaeal/eukaryal counterparts. In the dimer, the three structural domains are very well recognized, with the NTD located centrally and the two CTDs placed at the two termini, which is in excellent agreement with the available 3D structures (Bocharov et al. 2004) and also as shown by rigid-body modeling. The hinge region requires special attention; in this model the two hinges have unequal lengths, which might suggest that one is structured, probably adopting an α-helical form, and the second one is in a relaxed conformation. This observation may provide support for models presented previously, where it was suggested that the hinge regions of the L7/L12 protein dimer might undergo an α-helix coil transition in aqueous solution, whereas in a nonpolar environment, it prefers to fold into long α-helices, which would close the hydrophobic cleft of the NTD (Bocharov et al. 2004; Chandra Sanyal and Liljas 2000; Wahl et al. 2000). It should be emphasized that this is the first insight into the spatial organization of individual NTDs and CTDs of the whole L7/L12 complex in solution. All these experiments suggest that the bacterial and archaeal/eukaryal L12/P proteins belong to different groups that are not structurally related. This observation is in line with the earlier data showing that they are not interchangeable between Bacteria and Archaea/Eukaryotes, despite their identical functions (Uchiumi et al. 1999).
It seems that a rudimentary GTPase-associated center was present in the LUCA in the form of L10/P0 protein and some additional rRNA elements such as a sarsin-ricin loop, which could support the basic activity of this center and probably enabled the transition from progenotes to genotes, but then much of the stalk structure evolved in a very dynamic way in the bacterial and the archaeal/eukaryotic lines of descent, with a significant further refinement occurring in the eukaryotic line once it separated from the Archaea, as was the case for other ribosomal processes (Kyrpides and Woese 1998). On the basis of our results the two main evolutionary paths could be considered for L12/P proteins. The first path, convergent evolution, represents, in our view, the most likely scenario, where the bacterial and the archaeal/eukaryal L12/P proteins probably appeared on the ribosomal particle as two independent events in the evolution of the GTPase center, to fulfill the special needs for further adjustment of speed and accuracy of the ribosomal machinery and, also, as has been postulated (Krokowski et al. 2007; Remacha et al. 1995), to bring a regulatory aspect into protein synthesis. The second path, where the L12/P proteins come from an ancestral protein in the LUCA genome, but would have diverged to the point that no similarity can be detected, cannot be ruled out. However, the first scenario seems more likely. First, the structural differences on all levels of protein structure are so dramatic that most likely they are related to two autonomous evolutionary incidents leading to independent creation of two protein structures with the same function. Second, the ribosomes deprived of those proteins have residual activity toward stimulation of translation factor-dependent GTP hydrolysis (Mohr et al. 2002), which may indicate that addition of the stalk proteins was a relatively late event, realized after all basic elements of the GTPase-associated center in the LUCA was put in place. Third, the P0 stalk protein is homologous to L10 and they show sequence similarity (Maki et al. 2007). If L12 has been coevolving with the P1/P2 protein, it should have been showing the same pattern of sequence and structural change. Consequently, we postulate that these proteins should be regarded as analogous rather than homologous.
References
Ballesta JP, Remacha M (1996) The large ribosomal subunit stalk as a regulatory element of the eukaryotic translational machinery. Prog Nucleic Acid Res Mol Biol 55:157–193
Bocharov EV, Sobol AG, Pavlov KV, Korzhnev DM, Jaravine VA, Gudkov AT, Arseniev AS (2004) From structure and dynamics of protein L7/L12 to molecular switching in ribosome. J Biol Chem 279:17697–17706
Boulin CJ, Kempf R, Gabriel A, Koch MHJ (1988) Data acquisition systems for linear and area X-ray detectors using delay line readout. Nucl Instrum Meth 269:312–320
Chandra Sanyal S, Liljas A (2000) The end of the beginning: structural studies of ribosomal proteins. Curr Opin Struct Biol 10:633–636
Cheng J, Randall AZ, Sweredoski MJ, Baldi P (2005) SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res 33:W72–W76
Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ (1998) JPred: a consensus secondary structure prediction server. Bioinformatics 14:892–893
Diaconu M, Kothe U, Schlunzen F, Fischer N, Harms JM, Tonevitsky AG, Stark H, Rodnina MV, Wahl MC (2005) Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell 121:991–1004
Feigin LA, Svergun DI (1987) Structure analysis by small-angle X-ray and neutron scattering. Plenum Press, New York
Frank J, Gao H, Sengupta J, Gao N, Taylor DJ (2007) The process of mRNA–tRNA translocation. Proc Natl Acad Sci USA 104:19671–19678
Gabriel A, Dauvergne F (1982) The localization method used at EMBL. Nucl Instrum Method 201:223–224
Gonzalo P, Reboud JP (2003) The puzzling lateral flexible stalk of the ribosome. Biol Cell 95:179–193
Gonzalo P, Lavergne JP, Reboud JP (2001) Pivotal role of the P1 N-terminal domain in the assembly of the mammalian ribosomal stalk and in the proteosynthetic activity. J Biol Chem 276:19762–19769
Grela P, Helgstrand M, Krokowski D, Boguszewska A, Svergun D, Liljas A, Bernado P, Grankowski N, Akke M, Tchorzewski M (2007) Structural characterization of the ribosomal P1A-P2B protein dimer by small-angle X-ray scattering and NMR spectroscopy. Biochemistry 46:1988–1998
Grela P, Sawa-Makarska J, Gordiyenko Y, Robinson CV, Grankowski N, Tchorzewski M (2008) Structural properties of the human acidic ribosomal p proteins forming the p1-p2 heterocomplex. J Biochem 143:169–177
Griaznova O, Traut RR (2000) Deletion of C-terminal residues of Escherichia coli ribosomal protein L10 causes the loss of binding of one L7/L12 dimer: ribosomes with one L7/L12 dimer are active. Biochemistry 39:4075–4081
Gribaldo S, Philippe H (2002) Ancient phylogenetic relationships. Theor Popul Biol 61:391–408
Guarinos E, Remacha M, Ballesta JP (2001) Asymmetric interactions between the acidic P1 and P2 proteins in the Saccharomyces cerevisiae ribosomal stalk. J Biol Chem 276:32474–32479
Guinier A (1939) La diffraction des rayons X aux tres petits angles; application a l’etude de phenomenes ultramicroscopiques. Ann Phys (Paris) 12:161–237
Harris JK, Kelley ST, Spiegelman GB, Pace NR (2003) The genetic core of the universal ancestor. Genome Res 13:407–412
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33-8, 27–28
Hury J, Nagaswamy U, Larios-Sanz M, Fox GE (2006) Ribosome origins: the relative age of 23S rRNA Domains. Orig Life Evol Biosph 36:421–429
Ilag LL, Videler H, McKay AR, Sobott F, Fucini P, Nierhaus KH, Robinson CV (2005) Heptameric (L12)6/L10 rather than canonical pentameric complexes are found by tandem MS of intact ribosomes from thermophilic bacteria. Proc Natl Acad Sci USA 102:8192–8197
Jose MP, Santana-Roman H, Remacha M, Ballesta JP, Zinker S (1995) Eukaryotic acidic phosphoproteins interact with the ribosome through their amino-terminal domain. Biochemistry 34:7941–7948
Koch MHJ, Bordas J (1983) X-ray diffraction and scattering on disordered systems using synchrotron radiation. Nucl Instrum Meth 208:461–469
Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI (2003) PRIMUS—a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36:1277–1282
Kozin MB, Svergun DI (2001) Automated matching of high- and low-resolution structural models. J Appl Crystallogr 34:33–41
Krokowski D, Boguszewska A, Abramczyk D, Liljas A, Tchorzewski M, Grankowski N (2006) Yeast ribosomal P0 protein has two separate binding sites for P1/P2 proteins. Mol Microbiol 60:386–400
Krokowski D, Tchorzewski M, Boguszewska A, McKay AR, Maslen SL, Robinson CV, Grankowski N (2007) Elevated copy number of L-A virus in yeast mutant strains defective in ribosomal stalk. Biochem Biophys Res Commun 355:575–580
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform 5:150–163
Kyrpides NC, Woese CR (1998) Archaeal translation initiation revisited: the initiation factor 2 and eukaryotic initiation factor 2B alpha-beta-delta subunit families. Proc Natl Acad Sci USA 95:3726–3730
Lecompte O, Ripp R, Thierry JC, Moras D, Poch O (2002) Comparative analysis of ribosomal proteins in complete genomes: an example of reductive evolution at the domain scale. Nucleic Acids Res 30:5382–5390
Leijonmarck M, Eriksson S, Liljas A (1980) Crystal structure of a ribosomal component at 2.6 A resolution. Nature 286:824–826
Liao D, Dennis PP (1994) Molecular phylogenies based on ribosomal protein L11, L1, L10, and L12 sequences. J Mol Evol 38:405–419
Maki Y, Hashimoto T, Zhou M, Naganuma T, Ohta J, Nomura T, Robinson CV, Uchiumi T (2007) Three binding sites for stalk protein dimers are generally present in ribosomes from archaeal organism. J Biol Chem 282:32827–32833
Mears JA, Cannone JJ, Stagg SM, Gutell RR, Agrawal RK, Harvey SC (2002) Modeling a minimal ribosome based on comparative sequence analysis. J Mol Biol 321:215–234
Mohr D, Wintermeyer W, Rodnina MV (2002) GTPase activation of elongation factors Tu and G on the ribosome. Biochemistry 41:12520–12528
Mulder FA, Bouakaz L, Lundell A, Venkataramana M, Liljas A, Akke M, Sanyal S (2004) Conformation and dynamics of ribosomal stalk protein L12 in solution and on the ribosome. Biochemistry 43:5930–5936
Nissen P, Hansen J, Ban N, Moore PB, Steitz TA (2000) The structural basis of ribosome activity in peptide bond synthesis. Science 289:920–930
Noller HF (2004) The driving force for molecular evolution of translation. RNA 10:1833–1837
Nomura T, Nakano K, Maki Y, Naganuma T, Nakashima T, Tanaka I, Kimura M, Hachimori A, Uchiumi T (2006) In vitro reconstitution of the GTPase-associated centre of the archaebacterial ribosome: the functional features observed in a hybrid form with Escherichia coli 50S subunits. Biochem J 396:565–571
Olsen GJ, Woese CR (1997) Archaeal genomics: an overview. Cell 89:991–994
Page RD (1996) TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358
Petoukhov MV, Svergun DI (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J 89:1237–1250
Petoukhov MV, Eady NA, Brown KA, Svergun DI (2002) Addition of missing loops and domains to protein models by X-ray solution scattering. Biophys J 83:3113–3125
Philippe H, Forterre P (1999) The rooting of the universal tree of life is not reliable. J Mol Evol 49:509–523
Ramirez C, Shimmin LC, Newton CH, Matheson AT, Dennis PP (1989) Structure and evolution of the L11, L1, L10, and L12 equivalent ribosomal proteins in eubacteria, archaebacteria, and eucaryotes. Can J Microbiol 35:234–244
Remacha M, Jimenez-Diaz A, Bermejo B, Rodriguez-Gabriel MA, Guarinos E, Ballesta JP (1995) Ribosomal acidic phosphoproteins P1 and P2 are not required for cell viability but regulate the pattern of protein expression in Saccharomyces cerevisiae. Mol Cell Biol 15:4754–4762
Rodnina MV, Wintermeyer W (2001) Fidelity of aminoacyl-tRNA selection on the ribosome: kinetic and structural mechanisms. Annu Rev Biochem 70:415–435
Rodnina MV, Stark H, Savelsbergh A, Wieden HJ, Mohr D, Matassova NB, Peske F, Daviter T, Gualerzi CO, Wintermeyer W (2000) GTPases mechanisms and functions of translation factors on the ribosome. Biol Chem 381:377–387
Rodriguez-Gabriel MA, Remacha M, Ballesta JP (2000) The RNA interacting domain but not the protein interacting domain is highly conserved in ribosomal protein P0. J Biol Chem 275:2130–2136
Santos C, Ballesta JP (1995) The highly conserved protein P0 carboxyl end is essential for ribosome activity only in the absence of proteins P1 and P2. J Biol Chem 270:20608–20614
Shimizu T, Nakagaki M, Nishi Y, Kobayashi Y, Hachimori A, Uchiumi T (2002) Interaction among silkworm ribosomal proteins P1, P2 and P0 required for functional protein binding to the GTPase-associated domain of 28S rRNA. Nucleic Acids Res 30:2620–2627
Shimmin LC, Ramirez C, Matheson AT, Dennis PP (1989) Sequence alignment and evolutionary comparison of the L10 equivalent and L12 equivalent ribosomal proteins from archaebacteria, eubacteria, and eucaryotes. J Mol Evol 29:448–462
Soares MR, Bisch PM, Campos de Carvalho AC, Valente AP, Almeida FC (2004) Correlation between conformation and antibody binding: NMR structure of cross-reactive peptides from T. cruzi, human and L. braziliensis. FEBS Lett 560:134–140
Sodeoka M, Larson CJ, Chen L, LeClaira KP, Verdine GL (1993) A multifunctional plasmid for protein expression by ECPCR: overproduction of the p50 subunit of NF-κB Bioorg. Med Chem Lett 3:1089–1095
Svergun DI (1992) Determination of the regularization parameter in indirect transform methods using perceptual criteria. J Appl Crystallogr 25:495–503
Svergun DI (1993) A direct indirect method of small-angle scattering data treatment. J Appl Crystallogr 26:258–267
Svergun DI, Petoukhov MV, Koch MH (2001) Determination of domain structure of proteins from X-ray solution scattering. Biophys J 80:2946–2953
Tchorzewski M (2002) The acidic ribosomal P proteins. Int J Biochem Cell Biol 34:911–915
Tchorzewski M, Boguszewska A, Dukowski P, Grankowski N (2000a) Oligomerization properties of the acidic ribosomal P-proteins from Saccharomyces cerevisiae: effect of P1A protein phosphorylation on the formation of the P1A-P2B hetero-complex. Biochim Biophys Acta 1499:63–73
Tchorzewski M, Boldyreff B, Issinger O, Grankowski N (2000b) Analysis of the protein-protein interactions between the human acidic ribosomal P-proteins: evaluation by the two hybrid system. Int J Biochem Cell Biol 32:737–746
Tchorzewski M, Krokowski D, Boguszewska A, Liljas A, Grankowski N (2003) Structural characterization of yeast acidic ribosomal P proteins forming the P1A-P2B heterocomplex. Biochemistry 42:3399–3408
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Uchiumi T, Hori K, Nomura T, Hachimori A (1999) Replacement of L7/L12.L10 protein complex in Escherichia coli ribosomes with the eukaryotic counterpart changes the specificity of elongation factor binding. J Biol Chem 274:27578–27582
Volkov VV, Svergun DI (2003) Uniqueness of ab initio shape determination in small angle scattering. J Appl Crystallogr 36:860–864
Wahl MC, Moller W (2002) Structure and function of the acidic ribosomal stalk proteins. Curr Protein Pept Sci 3:93–106
Wahl MC, Bourenkov GP, Bartunik HD, Huber R (2000) Flexibility, conformational diversity and two dimerization modes in complexes of ribosomal protein L12. EMBO J 19:174–186
Walsh DA, Doolittle WF (2005) The real ‘domains’ of life. Curr Biol 15:R237–R240
Woese C (1998) The universal ancestor. Proc Natl Acad Sci USA 95:6854–6859
Woese CR, Fox GE (1977) The concept of cellular evolution. J Mol Evol 10:1–6
Woese CR, Kandler O, Wheelis ML (1990) Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 87:4576–4579
Acknowledgments
This work was supported by the Ministry of Science and Higher Education grant 2 P04A 004 29, 2005/2008. The SAXS measurements were supported by the European Community-Research Infrastructure Action under the FP6 “Structuring the European Research Area Programme,” contract number RII3/CT/2004/5060008. P.B. was supported by funds from the Ramóny Cajal research program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Grela, P., Bernadó, P., Svergun, D. et al. Structural Relationships Among the Ribosomal Stalk Proteins from the Three Domains of Life. J Mol Evol 67, 154–167 (2008). https://doi.org/10.1007/s00239-008-9132-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-008-9132-2