
Abstract
Dominant mutations in the LRRK2 gene, a member of the Roco family, cause both familial and sporadic Parkinson disease. LRRK genes had so far been detected only in bilaterian animals. In deuterostomes, including humans, two LRRK genes (LRRK1 and LRRK2) exist, while in protostomes a single LRRK gene has been found. In this study, I combine structural and phylogenetic analyses to show that the cnidarian Nematostella vectensis has four LRRK genes. One of them is a bona fide orthologue of the human LRRK2 gene, demonstrating that this gene has an ancient origin. Two others are, respectively, orthologues of the deuterostome LRRK1 and the protostome LRRK genes. The fourth gene is probably cnidarian-specific. This precise characterization of the early evolution of LRRK genes in animals has important implications, because it indicates that the Drosophila and Caenorhabditis LRRK genes, which are studied to gain an understanding of LRRK2 function, are not true orthologues of the human Parkinson disease gene. Novel functional insights are also gained by comparison of the structures of LRRK2 genes in distantly related species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The discovery of genes involved in the genesis of familial forms of Parkinson disease (PD) has opened new ways to understand the molecular basis of this pathology. LRRK2 is the first gene whose mutations have been consistently associated with both the familial and the much more frequent sporadic cases of PD. Dominant mutations in LRRK2 may explain 13% of the familial and up to 5% of sporadic PD cases (Berg et al. 2005; Mata et al. 2006; Taylor et al. 2006). This general involvement of the LRRK2 gene in both types of PD suggests a central role in dopaminergic cell death and the onset of the disease (Mata et al. 2006; Taylor et al. 2006). In addition, mutations in LRRK2 may be involved in other neurodegenerative diseases (Chen-Plotkin et al. 2008). All these results have led to a great interest in understanding the biochemical and cellular functions of this gene.
LRRK2 protein has a complex structure, which includes several types of repeats and three large domains, called the Roc, COR, and kinase domains (Bosgraaf and Van Hastert 2003; Marín 2006; Mata et al. 2006). Proteins with a Roc domain, which is a Ras-like GTPase domain, plus a COR domain, a characteristic domain of unknown function, belong to the Roco family (Bosgraaf and Van Hastert 2003). Roco family genes are present in both prokaryotes and eukaryotes. Vertebrates typically contain four Roco genes: LRRK1, LRRK2, MFHAS1, and DAPK1 (Bosgraaf and Van Hastert 2003; Marín 2006). Phylogenetic analyses showed that LRRK1 is the closest relative of LRRK2. This is confirmed by structural similarity, because they both encode a kinase domain of the TKL group that is missing from the other two vertebrate genes. However, LRRK2 proteins have a 650-amino acid-long N-terminal region, formed by repeats, which is absent in LRRK1 proteins (Marín 2006). LRRK1 and LRRK2 genes are present in all deuterostomes for which we have complete genomic data, including not only vertebrates, but also echinoderms. However, protostomes like insects and nematodes have a single LRRK gene that is structurally very similar to LRRK1 (Bosgraaf and Van Hastert 2003; Marín 2006). This led to the hypothesis that LRRK2 is a deuterostome-specific gene, emerged by duplication after the protostome–deuterostome split (Marín 2006).
The recent release of the first draft of the genome sequence of a cnidarian, the sea anemone Nematostella vectensis, has challenged our views of how complex the genomes of archaic animals were. N. vectensis has a very elaborate genome, more similar to the human one than to the genomes of protostome model species (Putnam et al. 2007). The presence in the anemone of many genes found in vertebrates but absent in Drosophila or Caenorhabditis suggests that these two model organisms have lost many genes that existed in the ancestor of all animals. This unexpected finding has important implications. It means that N. vectensis is a key organism to trace the origin of any human gene. Particularly, we can now test whether a gene present in humans but absent in protostome models (which would thus in principle be considered deuterostome-specific) is, on the contrary, ancient and it has been secondarily lost in protostomes. Moreover, the finding of cnidarian orthologues of human genes may also contribute to the understanding of the functions of our genes by showing what is essential and what may change in them.
With all this in mind, I decided to search for LRRK genes in N. vectensis. Surprisingly, this organism has the most complex set of LRRK genes described so far for any animal, with four paralogues. Phylogenetic and structural analyses show that a bona fide LRRK2 gene is present in cnidarians. Moreover, new evidence indicates that the protostome LRRK genes are paralogues of the human LRRK2 genes. Finally, I show that the comparison of the cnidarian and vertebrate LRRK2 genes refines our understanding of the structure and function of these genes. I discuss the important implications that these findings have for research, relevant to PD, on LRRK2.
Methods
Sequence Retrieval and Reconstruction of Nematostella LRRK Genes
The protein sequence of the COR domain, which is exclusive of the Roco family, may be used to find all members of this family present in the databases. The protein sequences of the human LRRK1 and LRRK2 COR domains were used to perform TblastN searches against all databases available at the National Center for Biotechnology Information (NCBI). These searches were finished in September 2007. Many novel sequences, not available at the time of my previous study (Marín 2006) were found. After excluding some primate proteins which were almost identical to the human ones, I generated a database containing 55 COR domain LRRK sequences. Only 26 of them were included in my previous analyses. Among them, four distinct N. vectensis sequences with strong similarity to LRRK1 and LRRK2 were detected. Similar searches were performed to detect the Roco family-specific kinase and Roc GTPase domains. Again, after excluding some very similar primate sequences, I detected 42 complete kinase sequences, instead of the 17 found in my previous work. Three of them derived from the N. vectensis genome. For Roc GTPases, I found 52 instead of the 23 detected in my previous work. Again, three of the new sequences came from N. vectensis.
I then performed searches with the full-length protein sequences derived from human, sea urchin (Strongylocentrotus purpuratus), and Drosophila melanogaster LRRK genes to detect all LRRK-related regions in the N. vectensis genome. Combining the results of those searches with automatic gene structure predictions using GenScan (Burge and Karlin 1997), I built models of the four N. vectensis LRRK genes. Although none of these models can be considered definitive, and two are obviously incomplete (see details below), the structures defined were sufficiently precise as to test for congruence with the results of the phylogenetic analyses described next.
Phylogenetic Analyses
Analyses of their COR or kinase domains generate consistent phylogenetic reconstructions of the relationships among LRRK genes, while the shorter Roc domain is less informative (Marín 2006). I first built phylogenetic trees with the 55 animal LRRK COR domains detected plus the domains of the proteins encoded by three Dictyostelium discoideum Roco genes (GbpC, Pats, and Roco10 [see Bosgraaf and Van Hastert 2003; Marín 2006]), which were used as outgroups. These 58 protein sequences were aligned using ClustalX 1.83 (Thompson et al. 1997) and manually corrected using GeneDoc (Nicholas and Nicholas 1997). A few regions which contained gaps in most sequences were eliminated to obtain a final alignment of 257 amino acidic sites. Trees were then built using that alignment, with three different procedures, namely, neighbor joining (NJ), maximum parsimony (MP), and maximum likelihood (ML). The NJ tree was obtained using the routine in MEGA 4 (Tamura et al. 2007), MP was performed using PAUP* beta 10 version (Swofford 2003), and ML reconstructions were established using PhyML (Guindon and Gascuel 2003). For NJ, sites with gaps were included and Kimura's correction was implemented. Parameters for MP were as follows: (1) all sites included; (2) randomly generated trees used as seeds; (3) maximum number of trees saved equal to 100; and (4) heuristic search using the Tree Bisection and Reconnection (TBR) algorithm. This method is more exhaustive, and therefore better, than the one that I used in my previous work. It was possible to use it here because the number of sequences was smaller. Finally, for ML analyses, the BioNJ tree was used to start the iterative searches and the Blosum62 matrix was chosen to model amino acidic transitions. Reliability of the topologies was tested in all cases by bootstrap analyses. One thousand bootstrap replicates were performed for the NJ and MP analyses and 500 for the ML analyses.
Identical analyses were performed with the kinase and Roc domains. The LRRK kinase and Roc domain sequences detected in the searches detailed above plus three D. discoideum outgroups were aligned and dendrograms built following the same methods described for the COR domains. In this case, the final length of the sequences aligned was 198 amino acidic sites for the kinases and 111 for the Roc GTPases. Dendrograms showing results for the COR, kinase, and Roc domains were drawn using the tree editor of MEGA 4.
Structural Analyses
Domains in LRRK proteins were characterized using InterProScan (Zbodnov and Apweiler 2001), which includes in a single search comparisons with the patterns of several structure databases, such as SMART and Pfam. Repeats in the N-terminal region of N. vectensis LRRK2 protein were determined by combining two methods: (1) aligning that region with the sequences of the other nine LRRK2 proteins (from vertebrates and echinoderms), using ClustalX 1.83; and (2) automatic detection of repeats with HHrep (Söding et al. 2006).
Results
As already indicated, four Nematostella vectensis COR domain sequences which had very strong similarity to those encoded by LRRK genes were detected in the databases (expect values ranged from 1 × 10−16 to 8 × 10−41). I built models for the four corresponding proteins using comparisons of the LRRK genes in vertebrates and invertebrates and the genomic Nematostella sequences plus ab initio analyses of the gene structures using GeneScan. Two of these models may correspond to full-length proteins. The third, which corresponds to the LRRK2 gene of N. vectensis (see next paragraph), lacks a small part of the Roc domain, unavailable so far in the databases, but seems otherwise complete. The fourth, which I have called Nv LRRK3 for reasons that will soon become clear, lacks its C-terminal end, including part of the kinase domain characteristic of LRRK proteins, and may also lack its N-terminal end. The structures deduced for these four models is compared with representative human, S. purpuratus, and D. melanogaster LRRK proteins in Fig. 1.

Structures of selected LRRK proteins. The three main domains (Roc, COR, kinase) plus the repeats characteristic of these proteins (LRRK2-specific, Leucine-rich repeats [LRR], WD40, Ankyrin) are indicated. Species abbreviations are as follows: Hs, Homo sapiens; Sp, Strongylocentrotus purpuratus; Nv, Nematostella vectensis; Dm, Drosophila melanogaster. Broken lines refer to domains not fully characterized due to lack of data. The arrows in Nv LRRK3 indicate that the full-length protein cannot be reconstructed with the available information and both ends of the protein are missing
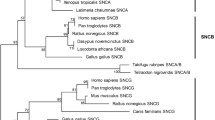
One of the proteins found in N. vectensis showed a very high sequence similarity to LRRK2 proteins in TblastN analyses. Structural analyses demonstrated that it also has the characteristic domains that I described for deuterostome LRRK2 proteins (Marín 2006). Particularly, it has a set of 11 N-terminal repeats (schematized in Fig. 1) that are very similar to those found in other LRRK2 proteins (Fig. 2). Phylogenetic analyses based on COR domain sequences and kinase sequences, shown in Figs. 3 and 4, confirmed the close relationship of the LRRK2-like protein in N. vectensis with deuterostome LRRK2 proteins. They appear together in a monophyletic group which is supported by the three methods of phylogenetic reconstruction in the independent analyses using both domains. Thus, I consider the gene encoding that protein as the bona fide orthologue of LRRK2 in Nematostella vectensis, hence the name Nv LRRK2 is used for it from now on. The analyses of the shorter Roc domain sequences did not provide any additional information (no significant bootstrap support was found for the critical internal branches of the tree; see Supplementary Fig. 1) and were not further considered.



N-terminal repeats specific to LRRK2 proteins in five mammals (Homo sapiens, Mus musculus, Canis familiaris, Bos taurus, Monodelphis domestica), a bird (Gallus gallus), two fishes (Danio rerio, Tetraodon nigroviridis), an echinoderm (Strongylocentrotus purpuratus), and a cnidarian (Nematostella vectensis). The locations of the 14 repeats detected in humans (Marín 2006) are indicated. N. vectensis LRRK2 lacks repeats 11–13

Dendrogram obtained when COR domain sequences are analyzed. The topology shown is the one deduced using NJ. Those generated with MP and ML are, however, so similar to this one that all results can be shown in a single tree. Numbers refer to the percentages of bootstrap replicates that support the corresponding branches. The three numbers correspond, respectively, to NJ/MP/ML analyses (see Methods for details). Only branches supported by the three methods and in which bootstrap support was higher than 50% in at least two of them are shown. Significant values for some outer branches, which group vertebrates or insects, have been omitted for simplicity

Dendrogram obtained analyzing kinase domain sequences. Conventions are as in Fig. 3. Again, some significant results have been omitted for simplicity
There is some controversy in the literature regarding the repeats found in the N terminus of LRRK2. I indicated in my previous study that they are gene specific. I used the patterns deduced from the human and S. purpuratus LRRK2 proteins to search for similarities in other proteins but did not detect any significant match. On the contrary, Mata et al. (2006) suggested, without any indication of how they obtained the evidence, that the N-terminal region of human LRRK2 contains a set of armadillo repeats. I decided to check for this possibility using the set of 10 sequences shown in Fig. 2. InterProScan analyses of those sequences failed to detect any armadillo repeat in nine of them. The exception was the S. purpuratus sequence, in which InterProScan detected, albeit with a low score, two armadillo repeats, in positions 99–141 and 506–549. This corresponds approximately to repeats number 3 and 12 in the S. purpuratus protein. Thus, only 2 of the 135 repeats shown in Fig. 2 are detected in the structural analyses performed against multiple databases by the InterProScan tool as canonical armadillo repeats. These results refute the hypothesis of the LRRK2-specific repeats being armadillo repeats. The exceptional detection of those repeats in automatic analyses is explained by the fact that both the LRRK2-specific repeats and the armadillo repeats contain multiple hydrophobic residues in somewhat similar positions. However, armadillo repeats are just one among several different types of repeats which are able to generate long alpha-helical structures known as “Armadillo-like helical domains” (InterPro domain no. IPR011989 [Groves and Barford 1999]). Most significantly, InterProScan detected these domains in 8 of the 10 sequences shown in Fig. 2. The exceptions were the two fish sequences, which I reconstructed from genomic data and may be incomplete (see gaps in Fig. 2). Thus, we may conclude that LRRK2 proteins contain a peculiar Armadillo-like helical domain, absent from the rest of LRRK proteins. This kind of structure is considered to be a protein-protein interaction surface (Groves and Barford 1999).
The structures predicted for two other N. vectensis proteins were very similar to those characteristic of LRRK1 proteins in deuterostomes or LRRK proteins in protostomes. Going from the N terminus to the C-terminal region, they have ankyrin repeats, leucine-rich repeats, and the typical Roc, COR, and kinase domains (Fig. 1; Nv LRRK1, Nv LRRK4). The fourth gene was, as already indicated, only partially characterized (Fig. 1; Nv LRRK3). The reconstructed region is uninformative with respect to its possible relationships to other LRRK genes. Phylogenetic analyses place two of these genes in positions that strongly suggest that they are either the orthologue of deuterostome LRRK1 genes (hence the name Nv LRRK1) or the orthologue of protostome LRRK genes (Nv LRRK3). The fourth gene (Nv LRRK4) appears in an intermediate position between LRRK1 and LRRK2 (Figs. 3 and 4). Apart from these cnidarian genes, the rest of new LRRK sequences found in my searches and detailed in Figs. 3 and 4 fit very well with the picture of LRRK gene evolution described in Marín (2006). All new genes in vertebrates, from mammals, reptiles and fishes, were obvious orthologues of either LRRK1 or LRRK2 and additional genes in insect and nematode species were also very similar to the protostome LRRK genes that I described before.
The trees in Figs. 3 and 4 can be interpreted as a whole considering the phylogenetic relationships among species. The simplest hypothesis to explain the topology of those trees is that there were at least three genes prior to the cnidarian-bilaterian split. These three genes gave rise to the branches annotated as LRRK1, LRRK2, and LRRK3 in Figs. 3 and 4. Thus, two of these genes correspond to the LRRK1 and LRRK2 genes in vertebrates, which are found also in echinoderms and cnidarians. The third gene would correspond to the one that was hitherto defined only for protostomes and called LRRK (Marín 2006). The results shown in Fig. 3, however, strongly suggest that a gene, which I think it is appropriate to call LRRK3, existed in the ancestor of all eumetazoans. It has been conserved in cnidarians, echinoderms, and protostomes, but lost in vertebrates. Figure 4 confirms that a monophyletic branch, including protostome and S. purpuratus LRRK genes, exists. In this case, however, the current lack of an alignable N. vectensis LRRK3 kinase domain precludes further confirmation that this gene is older than the protostome/deuterostome split. Note that the only small incongruence in the LRRK3 branch is the atypical position of the Caenorhabditis proteins. However, it is well known that Caenorhabditis genes evolve rapidly (Mushegian et al. 1998; Stein et al. 2003), so this result can be simply explained by acceleration in the rate of evolution of LRRK3 genes in nematodes. Finally, the Nv LRRK4 gene, which is the only one found outside the three main branches which define the LRRK1–LRRK3 genes (Figs. 3 and 4), may be a Nematostella-specific paralogue.
These results mean that protostomes and vertebrates do not contain orthologous LRRK genes. Protostomes have kept LRRK3, at the same time losing the LRRK1 and LRRK2 genes, while vertebrates have kept LRRK1 and LRRK2 but lost LRRK3. Among the groups for which we have data, the three genes are present only in cnidarians and echinoderms. Therefore, these results demonstrate that LRRK2 is an ancient gene, emerging in ancestral animals prior to the cnidarian-bilaterian split. They also indicate that no true orthologue of LRRK2 exists in model organisms such as Drosophila melanogaster and Caenorhabditis elegans.
Discussion
Tracing the origin of a human gene may greatly contribute to our understanding of its functions. In a series of works, my group has described the origin and evolution of several Parkinson disease genes such as parkin, DJ-1 and LRRK2 (Marín and Ferrús, 2002; Marín et al. 2004; Lucas et al. 2006; Marín 2006; Lucas and Marín 2007). These studies have contributed to delineate the best simple model organisms in which to study the functions of the human genes. In the case of LRRK2, I warned against the potential problems of using Drosophila melanogaster or Caenorhabditis elegans as models, given that it was unclear that those species had true LRRK2 orthologues (Marín 2006). The only LRRK gene present in protostomes encodes for a protein that not only is very dissimilar in sequence but also is structurally different from LRRK2. Thus, I suggested concentrating on deuterostome species as models (Marín 2006). Recent papers, however, described apparently promising results using protostome models. Sakaguchi-Nakashima et al. (2007) showed that mutations in the C. elegans LRK-1 gene, the only LRRK gene present in this species, generate anomalies in the localization of synaptic vesicle proteins. This result, which involves LRRK gene products in the regulation of vesicular transport, agrees with functional data for human LRRK2, which implicates it in synaptic vesicle recycling and neurite outgrowth (MacLeod et al. 2006; Hatano et al. 2007). Even more striking were the results obtained by Lee et al. (2007), showing that loss of function of the LRKK gene of D. melanogaster leads to the death of dopaminergic neurons in the fly brain and anomalies in locomotor activity.
The results reported here further support the idea that LRRK genes in Drosophila and Caenorhabditis and LRRK2 genes in humans are paralogous. Therefore, these recent results should be reevaluated, taking this fact into account. In my opinion, all the results in invertebrates and the results obtained for LRRK1 (Korr et al. 2006; Greggio et al. 2007; Taylor et al. 2007) and LRRK2 (summarized in Marín 2006; Thomas and Beal 2007) in vertebrates are compatible with all the LRRK gene products in animals being involved in neuronal functions. This is consistent with their presence only in animals with a developed nervous system, from cnidarians to vertebrates. However, that these neuronal-specific functions are the same for all LRRK genes in different animal species is not supported by the available data. For example, the Drosophila results indicated above—which are so far the only indication that LRRK genes in protostomes might be useful models to understand why LRRK2 mutations are involved in PD—are in obvious contradiction with the evidence obtained in our species. In the fly, overexpression of the LRRK gene does not lead to any obvious phenotype, whereas loss of function causes dopaminergic cell death, among other problems (Lee et al. 2007). In humans, however, it is unknown whether loss-of-function mutations in LRRK2 lead to dopaminergic cell death. On the contrary, it seems that it is increased/constitutive activity of the LRRK2 protein (probably through increased kinase function) what leads to the loss of dopaminergic cells in patients with PD (West et al. 2005; Gloeckner et al. 2006; Smith et al. 2006; Guo et al. 2007; Li et al. 2007). In summary, researchers in this field should be very careful before extrapolating results obtained for protostome LRRK genes to humans.
I suggested in my previous study that LRRK2 originated recently, by a duplication which occurred after the protostome–deuterostome split. This hypothesis has been refuted by the new findings in Nematostella. I now favor a more complex explanation: first, two duplications led to the presence of three genes before the protostome–deuterostome split; second, after that split two of those genes were lost in protostomes and the third one was lost in deuterostomes. This was obviously impossible to foresee, not knowing the cnidarian data. It is a good example of the radical change in our interpretations that genomic analyses of basal organisms may provide. As I commented in the Introduction, so far the scientific community has been working under the impression that we should expect a progressive increase in genome complexity when we get closer to humans, perhaps with some anomalies here and there caused by particular features in the ways of life of some species, by species-specific genome duplications, etc. Thus, a typical reasoning was that, whenever single genes are found in invertebrates and multiple genes are detected in vertebrates, the most parsimonious explanation was the occurrence of vertebrate-specific duplications. However, the unexpected complexity of the genome of this simple anemone may lead to a paradigmatic change, in which Drosophila or Caenorhabditis are viewed as possessing genomes in which many of the genes of their animal ancestors were lost. This may lead to a reevaluation of the evolution of many gene families in the light of cnidarian data, as I have done in this study.
The characterization of orthologous genes in very distant animals may also significantly contribute to our understanding of the biochemical potential of their products. Here, by comparing the N-terminal region of the LRRK2 orthologues, I have shown that they contain an Arm-like surface. Mutations in this surface have been involved in familial PD (Nichols et al. 2007). Given that this is considered to be a protein-protein interaction surface, as well as its absence in LRRK1 proteins, it is a good candidate for use in experiments aimed at identifying cellular partners specific to LRRK2.
References
Berg D, Schweitzer K, Leitner P, Zimprich A, Lichtner P, Belcredi P, Brussel T, Schulte C, Maass S, Nagele T (2005) Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson’s disease. Brain 128:3000–3011
Bosgraaf L, Van Haastert PJ (2003) Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta 1643:5–10
Burge C, Karlin S (1997) Prediction of complete gene structures in human genomic DNA. J Mol Biol 268:78–94
Chen-Plotkin AS, Yuan W, Anderson C, Wood EM, Hurtig HI, Clark CM, Miller BL, Lee VM, Trojanowski JQ, Grossman M, Van Deerlin VM (2008) Corticobasal syndrome and primary progressive aphasia as manifestations of LRRK2 gene mutations. Neurology 70:521–527
Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O’Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M (2006) The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet 15:223–232
Greggio E, Lewis PA, van der Brug MP, Ahmad R, Kaganovich A, Ding J, Beilina A, Baker AK, Cookson MR (2007) Mutations in LRRK2/dardarin associated with Parkinson disease are more toxic than equivalent mutations in the homologous kinase LRRK1. J Neurochem 102:93–102
Groves MR, Barford D (1999) Topological characteristics of helical repeat proteins. Curr Opin Struct Biol 9:383–389
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG (2007) The Parkinson’s disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res 313:3658–3670
Hatano T, Kubo S, Imai S, Maeda M, Ishikawa K, Mizuno Y, Hattori N (2007) Leucine-rich repeat kinase 2 associates with lipid rafts. Hum Mol Genet 16:678–690
Korr D, Toschi L, Donner P, Pohlenz HD, Kreft B, Weiss B (2006) LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal 18:910–920
Lee SB, Kim W, Lee S, Chung J (2007) Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun 358:534–539
Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z (2007) Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J Neurochem 103:238–247
Lucas JI, Arnau V, Marín I (2006) Comparative genomics and protein domain graph analyses link ubiquitination and RNA metabolism. J Mol Biol 357:9–17
Lucas JI, Marín I (2007) A new evolutionary paradigm for the Parkinson disease gene DJ-1. Mol Biol Evol 24:551–561
MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A (2006) The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52:587–593
Marín I (2006) The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol Biol Evol 23:2423–2433
Marín I, Ferrús A (2002) Comparative genomics of the RBR family, including the Parkinson’s disease-related gene parkin and the genes of the ariadne subfamily. Mol Biol Evol 19:2039–2050
Marín I, Lucas JI, Gradilla AC, Ferrús A (2004) Parkin and relatives: the RBR family of ubiquitin ligases. Physiol Genomics 17:253–263
Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA (2006) LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci 29:286–293
Mushegian AR, Garey JR, Martin J, Liu LX (1998) Large-scale taxonomic profiling of eukaryotic model organisms: a comparison of orthologous proteins encoded by the human, fly, nematode, and yeast genomes. Genome Res 8:590–598
Nicholas KB, Nicholas HB Jr (1997) GeneDoc: a tool for editing and annotating multiple sequence alignments. Distributed by the author
Nichols WC, Elsaesser VE, Pankratz N, Pauciulo MW, Marek DK, Halter CA, Rudolph A, Shults CW, Foroud T; for the Parkinson Study Group-PROGENI Investigators (2007) LRRK2 mutation analysis in Parkinson disease families with evidence of linkage to PARK8. Neurology (in press)
Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV, Jurka J, Genikhovich G, Grigoriev IV, Lucas SM, Steele RE, Finnerty JR, Technau U, Martindale MQ, Rokhsar DS (2007) Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317:86–94
Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N (2007) LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol 17:592–598
Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA (2006) Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 9:1231–1233
Söding J, Remmert M, Biegert A (2006) HHrep: de novo protein repeat detection and the origin of TIM barrels. Nucleic Acids Res 34:W137–W142
Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, Coulson A, D’Eustachio P, Fitch DH, Fulton LA, Fulton RE, Griffiths-Jones S, Harris TW, Hillier LW, Kamath R, Kuwabara PE, Mardis ER, Marra MA, Miner TL, Minx P, Mullikin JC, Plumb RW, Rogers J, Schein JE, Sohrmann M, Spieth J, Stajich JE, Wei C, Willey D, Wilson RK, Durbin R, Waterston RH (2003) The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol 1:E45
Swofford DL (2003) PAUP*. Phylogenetic Analysis Using Parsimony (*and other methods). Version 4. Sinauer Associates, Sunderland, MA
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Taylor JP, Mata IF, Farrer MJ (2006) LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol Med 12:76–82
Taylor JP, Hulihan MM, Kachergus JM, Melrose HL, Lincoln SJ, Hinkle KM, Stone JT, Ross OA, Hauser R, Aasly J, Gasser T, Payami H, Wszolek ZK, Farrer MJ (2007) Leucine-rich repeat kinase 1: a paralog of LRRK2 and a candidate gene for Parkinson’s disease. Neurogenetics 8:95–102
Thomas B, Beal MF (2007) Parkinson’s disease. Hum Mol Genet Spec No 2:R183–R194
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24:4876–4882
West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM (2005) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA 102:16842–16847
Zdobnov EM, Apweiler R (2001) InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17:847–848
Acknowledgments
This research was supported by Grant SAF2006-08977 (Programa Nacional de Biomedicina. Ministerio de Educación y Ciencia, Spain) and Grant 200720I021 (Proyectos intramurales especiales, CSIC, Spain).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Marín, I. Ancient Origin of the Parkinson Disease Gene LRRK2 . J Mol Evol 67, 41–50 (2008). https://doi.org/10.1007/s00239-008-9122-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-008-9122-4