
Abstract
Hippidions are past members of the equid lineage which appeared in the South American fossil record around 2.5 Ma but then became extinct during the great late Pleistocene megafaunal extinction. According to fossil records and numerous dental, cranial, and postcranial characters, Hippidion and Equus lineages were expected to cluster in two distinct phylogenetic groups that diverged at least 10 MY, long before the emergence of the first Equus. However, the first DNA sequence information retrieved from Hippidion fossils supported a striking different phylogeny, with hippidions nesting inside a paraphyletic group of Equus. This result indicated either that the currently accepted phylogenetic tree of equids was incorrect regarding the timing of the evolutionary split between Hippidion and Equus or that the taxonomic identification of the hippidion fossils used for DNA analysis needed to be reexamined (and attributed to another extinct South American member of the equid lineage). The most likely candidate for the latter explanation is Equus (Amerhippus) neogeus. Here, we show by retrieving new ancient mtDNA sequences that hippidions and Equus (Amerhippus) neogeus were members of two distinct lineages. Furthermore, using a rigorous phylogenetic approach, we demonstrate that while formerly the largest equid from Southern America, Equus (Amerhippus) was just a member of the species Equus caballus. This new data increases the known phenotypic plasticity of horses and consequently casts doubt on the taxonomic validity of the subgenus Equus (Amerhippus).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Two groups of equids were present in Late Pleistocene South America, Hippidion and Equus. Hippidion appeared in the South American fossil record around 2.5 Ma, shortly after the Great American Interchange, and is clearly different from Equus, which appeared only around 1.0 million years ago (MA). Cladistic analysis of dental, cranial, and postcranial characters separate Hippidion and Equus into two different clades, which share the North American late-Miocene Pliohippus as a common ancestor around 10 MA (Prado and Alberdi 1996). The first clade includes Equus, Astrohippus, and Dinohippus, while the second consists of Hippidion. Alternatively, MacFadden (1997) has suggested that Equus is derived from Dinohippus, and Hippidion from Pliohippus sensu lato (including Astrohippus), implying that the divergence between Dinohippus and Pliohippus occurred prior to 10 MA.
A molecular phylogeny proposed by Orlando et al. (2003) raised a completely different picture, with Hippidion nesting within a paraphyletic Equus group. These authors analyzed two mitochondrial DNA sequences of the hypervariable region I (HVR-I) from two Chilean fossils attributed to Hippidion saldiasi. The results raised questions about the generic classification (Hippidion versus Equus) of several fossil equids. This misidentification was potentially due to the pronounced shortening of the distal limbs in both hippidiform and Equus (Amerhippus) horses, a likely convergent adaptation to life in sloped terrains.
Alberdi et al. (2005) raised concerns about this interpretation and called for further genetic analysis. Central to their comment was an extensive morphological survey of almost all available collections of fossils from South America, which confirmed that the teeth samples used for the ancient DNA analyses undoubtedly belonged to Hippidion. They concluded that the phylogenetic DNA clustering obtained by Orlando et al. (2003) required deep taxonomic revision and should be checked using molecular data from remains that indisputably belong to Equus (Amerhippus) species.
Seven additional Hippidion fossils were independently analyzed (Weinstock et al. 2005) and resulted in virtually identical HVR-I sequences to Orlando et al. (2003). Additional sequence data from the second hypervariable region (HVR-II) confirmed that Hippidion formed part of a paraphyletic Equus, and postdated the emergence of the genus Equus (Weinstock et al. 2005). Most of these seven new specimens consisted of phalanges and showed morphological characteristics typical of Hippidion (e.g., two short tuberosities in the first phalanges, in contrast to the single, long, and V-shaped forms found in Equus).
Consequently, it seems unlikely that the nine Hippidion samples that have been genetically analyzed so far could all be misattributed, supporting the new phylogenetic arrangement where Hippidion is not a separate lineage dating back more than 10 Ma. However, to fully clarify this issue, it is necessary to obtain sequences from an Equus (Amerhippus) specimen. In this study, eight samples unambiguously related to Equus (Amerhippus) (Table 1) were subjected to ancient DNA extraction. A new Hippidion saldiasi specimen from Patagonia was also analyzed to further check the homogeneity of the mtDNA gene pool of the species (Table 1). DNA was extracted as previously described (Orlando et al. 2003, 2006; Weinstock et al. 2005) using appropriate ancient DNA methods and controls. A 183-bp fragment of the cytochrome b gene was targeted by PCR using the previously described cytb2L/cytb2H primers (Orlando et al. 2003). Furthermore, we designed a new set of primers to recover six short overlapping DNA fragments encompassing 546 bp of the horse mtDNA HVR-I (Table 2). All PCR reactions were conducted in a total volume of 25 μl using either Taq Gold (2.5 units; Perkin-Elmer), 1 × buffer, 2 mM MgCl2, 1 mg/ml BSA, 250 μM of each dNTP, and 0.5–1 μM of the different primers (France), or Taq Hifi (1.5 units; Invitrogen), 1 × buffer, 2 mM MgSO4, 2 mg/ml BSA, 250 μM of each , and 1 μM of each primer (ACAD). PCR conditions were the following: for Taq Gold, activation (92°C, 10 min), 50 cycles of DNA denaturation (92°C, 40 s), primer annealing (50°C, 40 s), and extension (72°C, 40 s), with a final elongation step of 10 min; and for Taq HiFi, activation (94°C, 1 min), 50 cycles of DNA denaturation (94°C, 20 s), primer annealing (51°C, 20 s), and extension (68°C, 40 s), with a final 10-min elongation step.
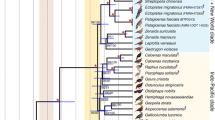
We successfully recovered DNA sequences for two Equus (Amerhippus) specimens (CH423 and CH425) and for the additional Hippidion saldiasi sample (ACAD1652) (Table 2). Notably, the two samples that gave maximal sequence length information (ACAD1652 and CH423) both originated from arid cave deposits (Table 1). For each DNA fragment, the final sequence was determined from the consensus of clones from at least two independent PCR products to minimize the impact of artifactual substitutions induced by DNA damage (Hofreiter et al. 2001). A total of 48 PCR products and 381 clones were analyzed in Lyon (Table 2). In addition, the mtDNA HVR-I sequences of fragments 15492F-15625R, 15668F-15847R, and 15950F-16083R of specimen CH423 were independently replicated with complete sequence identity at the Australian Centre for Ancient DNA (Adelaide). The mtDNA HVR-I sequence of the new Hippidion specimen from Patagonia was highly similar to previously reported Hippidion sequences (Orlando et al. 2003; Weinstock et al. 2005) (see branch lengths of the phylogenetic tree reported in Fig. 1). The sequences were deposited in GenBank under accession numbers EU030679–EU030682.

Phylogenetic relationships as shown by the mtDNA HVR-I sequence data. (A) Midpoint-rooted phylogenetic tree. GTR+G4 + I, 488 sites, 105 sequences; ML α = 0.417, I = 0.432, loglk = –3350.39608; Bayes α = 0.237, I = 0.504. (B) Two rhinos (Ceratotherium simum and Rhinoceros unicornis; accession nos. NC001808-NC001779) were used as outgroups as by Weinstock et al. (2005): GTR+G4+I, 475 sites, 107 sequences; ML α = 0.292, I = 0.163, loglk = –4246.20573; Bayes α = 0.217, I = 0.237. Bootstrap values (%) and posterior probabilities are indicated above and below the principal nodes of the tree, respectively. As the data sets include an extensive number of taxa, some parts of the tree have been compressed (black triangles) using the MEGA software. The number of sequences included in each of these groups is reported. Accession numbers are as follows: Equus asinus—NC_001788; Equus burchelli—AF220916–AF220924; Equus caballus—AF354425, AF354426, AF354427, AF354428, AF354429, AF354431, AF354432, AF354433, AF354434, AF354436, AF354437, AF354438, AF354439, AF354440, AF354441, AF169009, AF169010, AF014406, AF014407, AF014408, AF014409, AF014411, AF014412, AF014413, AF014414, AF014415, AF014416, AF064627, AF064628, AF064629, AF064630, AF064631, AF064632, AY049718, AY049719, AY049720, AY246174, AY246175, AY246176, AY246177, AY246178, AY246179, AY246180, AY246181, AY246184, AY246186, AY246187, AY246190, AY246192, AY246195, AY246196, AY246197, AY246198, AY246211, AY246212, AY246214, AY246219, AY246220, AY246221, AY246222, AY246225, AY246226, AY246229, AY246231, Y246234 , AY246235, AY246236, AY246240, AY246241, AY246242, AY246243, AY246248, AY246253, AY246254, AY246256, AY246257, AY246259, AY246261, AY246266, AY246267, AY246271, DQ297634, DQ297635, DQ297637, DQ297638, DQ327893, DQ327897, DQ327900, DQ327903, DQ327904, DQ327905, DQ327908, DQ327915, DQ327916, DQ327918, DQ327919, DQ327920, Q327921 , DQ327923, DQ327924, DQ327925, DQ327926, DQ327927, DQ327928, DQ327929, DQ327934, DQ327935, DQ327936, DQ327937, DQ327942, DQ327944, DQ327945, DQ327946, DQ327948, DQ327950, DQ327951, DQ327956, DQ327957, DQ327958, DQ327959, DQ327960, DQ327968, DQ327969, DQ327970, DQ327971, DQ327973, DQ327974, DQ327976, DQ327981, DQ327982, DQ327983, DQ327984, DQ327986, DQ327989, DQ327990, DQ328002, DQ328005, DQ328007, DQ328012, DQ328015, DQ328018, DQ328020, DQ328021, DQ328023, DQ328025, DQ328034, DQ328035, DQ328037, DQ328039, DQ328040, DQ328042, DQ328043, DQ328044, DQ328045, DQ328050, DQ328052, DQ328053, DQ328054, DQ328056, HRSMTTRNAA, HRSMTTRNAB, HRSMTTRNAC; Equus grevyi—AF220928–AF220930; Equus hemionus—AF220934–AF220937; Equus kiang—AF220933, AY569539; Equus quagga—AY914318–AY914323; Equus zebra—AF22025–AF22027, AF220931; Hippidion saldiasi—DQ007560, DQ007562–DQ007564; “stilt-legged” horses—DQ007568, DQ007621
Importantly, the Equus (Amerhippus) sequences are highly divergent from both the existing and the new Hippidion sequences (Fig. 1). This convincingly demonstrates that the genetically analyzed Hippidion samples were not misattributed Equus (Amerhippus) specimens, in agreement with Alberdi et al. (2005) and Weinstock et al. (2005). On the contrary, the Equus (Amerhippus) sequence reveals similarities with several caballine horse haplotypes including members of Thoroughbred, Quarter, and Shire breeds. Over 546 bp, the CH423 haplotype shows complete identity to previously reported horse mtDNA HVR-I sequences (accession nos. AF072976, AF072980, AF072991, AY246192, AY246193, AY246216, AY246217, AY246230, DQ297636, DQ327891, DQ327917). The same holds true for the CH425 specimen, though in this case, the sequence information retrieved is rather short (i.e., 89 bp), and consequently the list of putatively identical horse haplotypes is longer.
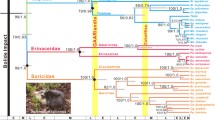
To examine the phylogenetic position of the Equus (Amerhippus) sequences in relation to horses and hippidions, we performed analyses using a mtDNA data set of HVR-I sequences for a range of equids available in GenBank. Sequences shorter than 500 bp, or exhibiting large stretches of undetermined nucleotide positions (e.g., Hippidion under accession no. DQ007561), were discarded, leaving a final data set of 348 sequences. Redundant or very similar haplotypes (pairwise Kimura2 distances <0.005) as well as two highly divergent caballine horse haplotypes (accession nos. AY049718–AY049719) were eliminated. In total, the final data comprised 105 sequences of extant and extinct equids (available upon request; Fig. 1). The sequence information was used to construct maximum-likelihood (PHYML online; Guindon and Gascuel 2005) and Bayesian (MrBayes 3.1.2; Huelsenbeck and Ronquist 2001) trees using the best model of molecular evolution according to the AIC criterion of Modeltest (Posada and Crandall 1998). The strength of the phylogenetic signal was assessed via nonparametric bootstrapping (1000 pseudo-replicates) and posterior probabilities (20 million generations, sampling frequency = 1 every 1000 generations, burn-in value = 1000). Regardless of whether the tree was midpoint rooted (Fig. 1A) or rooted with two rhino sequences (Fig. 1B), the Equus (Amerhippus) sequences fall inside the caballine horse cluster with maximum bootstrap support and posterior probabilities. The Equus (Amerhippus) haplotype appears among typical caballine horse haplotypes in median-spanning network analyses (NETWORK 4.2 software available at http://www.fluxus-engineering.com; Bandelt et al. 1999) but four steps from the nearest Hippidion relative (Fig. 2). Similarly, the HVR-I sequence from the ACAD1652 specimen also unambiguously clusters (maximal bootstrap support and posterior probabilities) with previously reported Hippidion sequences, confirming the homogeneity of the Hippidion mtDNA gene pool (Fig. 1).

Median spanning network showing the proximity of the 137-bp cytochrome b in horses and Equus (Amerhippus). All the available cytochrome b sequences of equids were retrieved from GenBank and redundant haplotypes were eliminated using the DNAsp software (Rozas et al. 2003). Additionally, three highly divergent horse sequences were discarded (accession nos. DQ236094, AY819736–AY819737). The arrow indicates the location of the cytochrome b sequence from the CH423 specimen. Accession numbers are as follows: Equus asinus (ASI1- to -4)—AF380135, AF380133, AF380132, AF380130; Equus burchelli (BUR1, -2)—AY534349, DQ470804; Equus caballus (CAB1- to 4)—DQ223537, DQ223533, DQ297658, DQ297640; Equus grevyi (GRE1)—X56282; Equus hemionus (HEM1)—DQ464015; Equus hydruntinus (HYD1)—DQ464013; Hippidion saldiasi (HIP1)—AY152859
Our analysis provides the first genetic characterization of Equus (Amerhippus) fossils. This new sequence information definitively removes the possibility that previously examined Hippidion specimens were all misattributed Equus (Amerhippus) specimens. At the same time it casts doubt on the current taxonomic status of Equus (Amerhippus), as they were found to be members of the caballine horse lineage (rather than members of a distinct subgenus inside equids). Given that modern caballine horse breeds exhibit mitochondrial haplotypes found in Equus (Amerhippus), the two taxa should presumably be recognized as conspecific caballine equids. This finding supports calls for a taxonomic revision of the more than 50 recognized species of American Pleistocene equids (Azzaroli 1998) and the suggestion that only a few authentic species might have been present in Late Pleistocene America (Weinstock et al. 2005). The taxonomy of the genus Equus has been in disorder for several decades (Winans 1989), mainly because the interspecific variation in skeletal morphology is generally not much greater than the intraspecific variation. Consequently, none of the qualitative and quantitative differences that have been used to separate species the genus Equus are great enough to assign species unambiguously, and the majority of palaeontologists who have worked with this genus would agree that at least some, if not most, of the nominal species are of dubious validity.
According to currently available ancient DNA data, at least three equid lineages were present in America during the late Pleistocene, namely, caballine horses (sensu stricto), “stilt legged horses” (a group of caballine horses sensu lato named by Weinstock et al. [2005]) and hippidions (Orlando et al. 2003; Weinstock et al. 2005) (Fig. 1). These data suggest that temporal and regional variation in body size and morphological and anatomic features should be considered a sign of extraordinary plasticity within each of these lineages. Such environment-driven adaptative changes would explain why the taxonomic diversity of equids has been overestimated on morphoanatomical grounds.
References
Alberdi MT, Cartelle C, Prado JL (2003) El registro Pleistoceno de Equus (Amerhippus) e Hippidion (Mammalia, Perissodactyla) de Brasil. Consideraciones paleoecológicas y biogeográficas. Ameghiniana 40:173–196
Alberdi MT, Prado JL, Prieto A (2005) Considerations on the paper “Morphological Convergence in Hippidion and Equus (Amerhippus) South American Equids Elucidated by Ancient DNA Analysis,” by Ludovic Orlando, Véra Eisenmann, Frédéric Reynier, Paul Sondaar, and Catherine Hänni. J Mol Evol 61:145–147
Azzaroli A (1998) The genus Equus in North America. PalaeontographItal 85:1–60
Bandelt H-J, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Guindon S, Lethiec F, Duroux P, Gascuel O (2005) PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res 33:W557–W559
Hofreiter M, Jaenicke V, Serre D, Haeseler Av A, Pääbo S (2001) DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res 29:4793–4799
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
MacFadden BJ (1997) Pleistocene horses from Tarija, Bolivia, and validity of the genus Onohippidium (Mammalia: Equidae). J Vertebr Paleontol 17:199–218
Orlando L, Eisenmann V, Reynier F, Sondaar P, Hänni C (2003) Morphological convergence in Hippidion and Equus (Amerhippus) southern-american equids elucidated by ancient DNA analysis. J Mol Evol 57 (Suppl 1):S29–S40
Orlando L, Mashkour M, Burke A, Douady CJ, Eisenmann V, Hänni C (2006) Geographic distribution of an extinct equid (Equus hydruntinus: Mammalia, Equidae) revealed by morphological and genetical analyses of fossils. Mol Ecol 15:2083–2093
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Prado JL, Alberdi MT (1994) A quantitative review of the horse genus Equus from South America. Palaeontology 37:459–481
Prado JL, Alberdi MT (1996) A cladistic analysis of the horses of the tribe Equini. Palaeontology 39:663–680
Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Weinstock J, Willerslev E, Sher A, Tong W, Ho SY, Rubenstein D, Storer J, Burns J, Martin L, Bravi C, Prieto A, Froese D, Scott E, Xulong L, Cooper A (2005) Evolution, systematics, and phylogeography of pleistocene horses in the new world: a molecular perspective. PLoS Biol 3:e241
Winans MC (1989) A quantitative study of the North American fossil species of the genus Equus. In: Prothero DR, RM RM Schoch (eds) The evolution of Perissodactyls. Oxford University Press, New York, pp 262–297
Author information
Authors and Affiliations
Corresponding author
Additional information
This is a reply to the Comment published by Alberdi et al. (2005).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Orlando, L., Male, D., Alberdi, M.T. et al. Ancient DNA Clarifies the Evolutionary History of American Late Pleistocene Equids. J Mol Evol 66, 533–538 (2008). https://doi.org/10.1007/s00239-008-9100-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-008-9100-x