
Abstract
Recently there has been a lively debate about a new class of rare genomic characters, RGC_CAMs, and their implications for deep bilaterian phylogeny. Most recently, nine bilaterian species were analyzed along with subsets of six outgroups (Rogozin et al. 2007b), and support for a coelomate clade reported. The authors suggested that our previously reported support for an ecdysozoan clade (Irimia et al. 2007) reflected (i) one outgroup, Nematostella vectensis, being too closely related to bilaterians and (ii) lack of “rigorous statistical analysis.” Here, we report further studies of these characters. First, we discuss general issues of outgroup choice. Second, we point out that an argument used by Rogozin et al. against backmutation is not statistically significant. Third, we point out that the statistical method of Rogozin et al. fails to incorporate backmutations, leading to systematic placement of the long-branch taxon as the outgroup. A simple modification of the method yields very different results: 51 of 63 outgroup combinations favor Ecdysozoa, inlcuding 51 of 52 with at least eight phylogenetically informative characters, and all 19 with statistically significant signal. These results indicate that the Coelomata signal is a long-branch artifact.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The relationship of major bilaterian groups continues to be debated, with much of the debate between those who support grouping molting animals together (the Ecdysozoa hypothesis [Aguinaldo et al. 1997]) and those who support grouping animals with a true coelom (the Coelomata hypothesis [Field et al. 1988]). Traditional sequence analysis as well as various classes of rare genomic characters have repeatedly been interpreted as support for both sides (Aguinaldo et al. 1997; Mushegian et al. 1998; Blair et al. 2002; Stuart and Berry 2004; Wolf et al. 2004; Dopazo and Dopazo 2005; Nguyen et al. 2005; Philip et al. 2005; Philippe et al. 2005; Roy and Gilbert 2005; Delsuc et al. 2006; Zheng et al. 2007).
Recently, Rogozin and coauthors (2007a) proposed a new class of rare genomic characters, RGC_CAMs (Rare Genomic Characters–Conserved Amino acids Multiple Changes), potentially useful to resolve difficult trifurcations such as the Ecdysozoa-Coelomata conundrum. These characters are defined as protein sequence positions for which (i) all members of one of the three groups share an amino acid with all outgroups; (ii) all members of the other two groups share a different amino acid, consistent with change in a putative ancestor; and (iii) the two amino acids differ by multiple nucleotide changes. First, Rogozin et al. (2007a) analyzed 694 sets of orthologues from six bilaterian species and four distant outgroups (fungi, plants, and apicomplexans), finding slightly more characters (34) grouping arthropods with deuterostomes (consistent with Coelomata) than with nematodes (26; consistent with Ecdysozoa), which they interpreted as strong support for Coelomata based on a novel statistical method. We then reanalyzed their data including a much more closely related outgroup (the sea anenome Nematostella vectensis) and additional key bilaterian species in order to break the long branches (Irimia et al. 2007). The inclusion of these four species eliminated the vast majority of Coelomata but not Ecdysozoa characters: 13 remaining characters supported Ecdysozoa, versus only 1 for Coelomata.
Most recently, the original authors (Rogozin et al. 2007b) provided a new analysis using the augmented set of taxa plus the choanoflagellate Monosiga brevicolis. Using all possible subsets of outgroups, they found an excess of Ecdysozoa characters whenever N. vectensis was included and, usually, an excess of Coelomata excess when N. vectensis was excluded (Table 1; “RGC_CAMs supporting”). They attributed the lack of Coelomata signal to N. vectensis being too closely related to bilaterians, leading to too great a reduction in informative characters.
It is not at all clear how this could be so: use of more closely related outgroups is expected to reduce artifactual characters but not true synapomorphies. In this case, detecting true Coelomata/Ecdysozoa synapomorphies requires that the outgroups reflect the ancestral bilaterian state and, so, requires (i) no change along the internode from the outgroup-bilaterian ancestor to the bilaterian ancestor and (ii) no change along external outgroup branches. Thus usage of more closely related and short branch outgroups is not expected to decrease the number of true synapomorphies. By contrast, closely related outgroups eliminate artifactual characters due to backmutations, since such characters require (i) change along the internode or (ii) change in the outgroup. N. vectensis is thus a much better choice that the next closest studied outgroup Monosiga brevicolis, due to (i) a much shorter internal branch (3 versus 35 RGC_CAM units; Fig. 1; see Rogozin et al. [2007b] for details) and (ii) much shorter external branch (66 vs 213; Fig. 1).

Branch lengths leading to N. vectensis and M. brevicolis (as calculated by Rogozin et al. 2007b)
This yields a specific prediction: if one topology is primarily supported by artifactual backmutation characters, its support will be disproportionately reduced by addition of a close ingroup. Exactly such a difference across topologies is seen in the data (Table 1; “Effect of close outgroup”). The table is grouped so that each pair of lines differs only by exclusion/inclusion of N. vectensis. For each pair, one can compare the reduction in Coelomata and Ecdysozoa characters when N. vectensis is added (Table 1). In total, while the average number of positions supporting Ecdysozoa is reduced from 9.2 to 6.2 (by 33%), the number of characters supporting Coelomata is dramatically reduced, from 21.6 to 2.8 (87%). In all 31 pairs, Coelomata characters suffer more of a reduction than for Ecdysozoa, in each case leading to a larger number of total characters supporting Ecdysozoa.
Thus: (i) N. vectensis is the best available outgroup; (ii) for all sets of outgroups that include N. vectensis, more characters support Ecdysozoa; and (iii) comparison of sets of outgroups including/excluding N. vectensis suggest that many of the Coelomata characters are attributable to backmutation. These findings suggest that the data support Ecdysozoa.
Rogozin et al. (2007b) reject this conclusion for two reasons. First, they argue that despite equal branch lengths, the number of backmutations in the Caenorhabditis ancestor (“Reversions” in Table 1) is smaller than the number of Coelomata characters, thus backmutations cannot explain all of the Coelomata characters (please see the Rogozin et al. [2007b] for details of this argument). However, this claim is not statistically supported: the numbers are only ever statistically different if only one or two outgroups are used, which defies the central logic of RGC_CAMs—that they are widely conserved among outgroups. Moreover, Rogozin et al.’s use of a t-test across sets of outgroups is inappropriate since tests using different outgroup sets are far from independent: in fact, use of a superset of outgroups implies a subset of RGC_CAMs.
Second, Rogozin et al. (2007b) rightly point out that the numbers of parallel changes supporting the Ecdysozoa versus Coelomata topologies are expected to be different. Since the nematode branch is much longer than the deuterostome branch, more parallel changes are expected in nematodes and arthropods (artifactually supporting Ecdysozoa) than in deuterostomes and arthropods (supporting Coelomata). To correct for this discrepancy they deploy a novel statistic: they compare the total number of RGC_CAMs supporting Coelomata (N AD , for shared between Arthropods and Deuterostomes) and Ecdysozoa (N AN ) to the deuterostome (N D ) and nematode (N N ) branch lengths. They interpret findings of \( \frac{{N_{{AD}} }} {{N_{D} }} > \frac{{N_{{AN}} }} {{N_{N} }} \) as evidence for Coelomata. This often leads them to find statistically significant support for Coelomata even in cases where there is a clear excess of Ecdysozoa characters (7:1, 10:3,17:6, 40:19, etc.), in itself troubling given that a central appeal of rare genomic characters is their hoped intuitiveness and simplicity of interpretation (Rokas and Holland 2000). Moreover, in correcting for homoplasy the analysis essentially moves from the realm of maximum parsimony to that of maximum likelihood, at which point it is not clear why characters should be chosen a priori (rather than implementing a more general model incorporating differences in rates across sites).
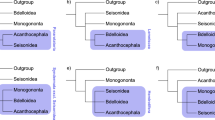
However, since their test ignores backmutation, it is expected to systematically place the long-branch group as the outgroup (in this case the nematodes, favoring the Coelomata topology). While greater branch length in nematodes increases parallel changes with arthropods (supporting Ecdysozoa), it also presumably increases backmutations (forward mutations in the bilaterian ancestor and backmutations in nematodes) supporting Coelomata (as well as more complex scenarios such as forward mutation in the metazoan ancestor and backmutations in both nematodes and N. vectensis) (Fig. 2). If both parallel changes and backmutations are proportional to branch length, and there are a total of P parallel changes (in arthropods and another group) and B backmutations (in either deuterostomes or nematodes), the expected relative numbers of artifactual Coelomata and Ecdysozoa are PN D + BN N and PN N + BN D , respectively. Thus, if there are any backmutations (i.e., B > 0), we expect \( \frac{{N_{{AD}} }} {{N_{D} }} > \frac{{N_{{AN}} }} {{N_{N} }} \) (since this is equivalent to P + BN N /N D > P + BN D /N N , which is so since N N >> N D [Table 1]), exactly the finding taken by Rogozin et al. as evidence for Coelomata. Thus in the absence of sufficiently large numbers of true synapomorphies, Rogozin et al.’s method is expected to systematically place the long nematode branch as the outgroup.

Major sources of homoplasy in analysis of RGC_CAMs. (i) Parallel mutations in groups 2 and 3 lead to apparent support for a clade including groups 2 and 3. (ii) Forward mutation in the ancestor and backmutation in group 1 leads to apparent support for a clade including groups 2 and 3
A more complete method would incorporate both parallel changes and backmutations. Assuming (i) that the probability of change along a given branch is proportional to the branch length as measured by Rogozin et al. and (ii) equal rates of forward and backmutation, the expected relative numbers of characters artifactually supporting Ecdysozoa and Coelomata are N A N N + N STEM N D and N A N D + N STEM N N , respectively, where N STEM is the number of changes along the internode from the outgroup to the bilaterian ancestor.
Using this reasoning, we calculated the fraction of Ecdysozoa + Coelomata characters that support Ecdysozoa [that is, E/(C + E), and compared it to the observed values (Table 1; “New test of E versus C”)]. A positive/negative value in the “excess” column indicates a larger/smaller than expected fraction of Ecdysozoa characters. Using this improved metric shows a much different picture. Ecdysozoa is supported over Coelomata for 51 of 63 sets of outgroups, including 51 of 52 for which there are at least eight phylogenetically informative characters, and for all 19 that give statistically significant results.
In total, then, analysis of RGC_CAMs from 694 sets of eukaryotic orthologues indicates that previously reported support for Coelomata was a long-branch artifact: (i) support for Coelomata (but not Ecdysozoa) disappears when a more appropriate outgroup is used; and (ii) support for Coelomata disappears when a statistical method also accounting for backmutation is utilized. In our opinion, it is less clear what to finally conclude from the data. While on its face the data support Ecdysozoa, the shortcomings and complexities of RGC_CAMs and their clear potential for homoplasy reduce our confidence in the utility of these characters overall. On balance, we suspect that more traditional treatments of sequence that take into account differences in rates across sites are likely to be more reliable (e.g., Philippe et al. 2005). Nonetheless, whatever confidence is placed in the methods themselves, these results indicate that RGC_CAMs do not provide support for Coelomata.
References
Aguinaldo AMA, Turbeville JM, Linford LS, Rivera MC, Garey JR, Raff RA, Lake JA (1997) Evidence for a clade of nematodes, arthropods and other moulting animals. Nature 387:489–493
Blair J, Ikeo K, Gojobori T, Hedges S (2002) The evolutionary position of nematodes. BMC Evol Biol 2:7
Delsuc F, Brinkmann H, Chourrout D, Philippe H (2006) Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 439:965–968
Dopazo H, Dopazo J (2005) Genome-scale evidence of the nematode-arthropod clade. Genome Biol 6:R41
Field K, Olsen G, Lane D, Giovannoni S, Ghiselin M, Raff E, Pace N, Raff R (1988) Molecular phylogeny of the animal kingdom. Science 239:748–753
Irimia M, Maeso I, Penny D, Garcia-Fernandez J, Roy SW (2007) Rare coding sequence changes are consistent with Ecdysozoa, not Coelomata. Mol Biol Evol 24:1604–1607
Mushegian A, Garey J, Martin J, Liu L (1998) Large-scale taxonomic profiling of eukaryotic model organisms: a comparison of orthologous proteins encoded by the human, fly, nematode, and yeast genomes. Genome Res 8:590–598
Nguyen H, Yoshihama M, Kenmochi N (2005) New maximum likelihood estimators for eukaryotic intron evolution. PLoS Comput Biol 1:e79
Philip GK, Creevey CJ, McInerney JO (2005) The Opisthokonta and the Ecdysozoa may not be clades: stronger support for the grouping of plant and animal than for animal and fungi and stronger support for the Coelomata than Ecdysozoa. Mol Biol Evol 22:1175–1184
Philippe H, Lartillot N, Brinkmann H (2005) Multigene analyses of bilaterian animals corroborate the monophyly of Ecdysozoa, Lophotrochozoa, and Protostomia. Mol Biol Evol 22:1246–1253
Rogozin IB, Wolf YI, Carmel L, Koonin EV (2007a) Ecdysozoan clade rejected by genome-wide analysis of rare amino acid replacements. Mol Biol Evol 24:1080–1090
Rogozin IB, Wolf YI, Carmel L, Koonin EV (2007b) Analysis of rare amino acid replacements supports the Coelomata clade. Mol Biol Evol 24(12): 2594–2597
Rokas A, Holland PWH (2000) Rare genomic changes as a tool for phylogenetics. Trends Ecol Evol 15:454–459
Roy SW, Gilbert W (2005) Resolution of a deep animal divergence by the pattern of intron conservation. Proc Natl Acad Sci USA 102:4403–4408
Stuart G, Berry M (2004) An SVD-based comparison of nine whole eukaryotic genomes supports a coelomate rather than ecdysozoan lineage. BMC Bioinformatics 5:204
Wolf Y, Rogozin I, Koonin E (2004) Coelomata and not Ecdysozoa: evidence from genome-wide phylogenetic analysis. Genome Res 14:29–36
Zheng J, Rogozin IB, Koonin EV, Przytycka TM (2007) Support for the Coelomata clade of animals from a rigorous analysis of the pattern of intron conservation. Mol Biol Evol 24(11): 2583–2592
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Roy, S.W., Irimia, M. Rare Genomic Characters Do Not Support Coelomata: RGC_CAMs. J Mol Evol 66, 308–315 (2008). https://doi.org/10.1007/s00239-008-9077-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-008-9077-5