
Abstract
A genomic PCR fragment of 4581 bp, referred to as PyRE10G, was isolated from the red alga Porphyra yezoensis. PyRE10G contained a putative open reading frame encoding gag, protease, integrase, reverse transcriptase (RT), and RNase H, but one stop codon was present in the integrase region. Southern blot analysis revealed that PyRE10G exists as a single copy in the genome. From the order of gene arrangement of polyproteins, PyRE10G appears to be a copia-like retrotransposon. Amino acid sequences of PyRE10G RT and RNase H were closely related to those of copia-like retrotransposons. In contrast, the phylogenetic tree suggested that PyRE10G integrase stands within the gypsy elements and outside the copia group. PyRE10G is the first example of a chimeric composition of copia- and gypsy-like polyprotein genes in a single element, supporting the hypothesis that long terminal repeat-containing retrotransposons have evolved by fusion of ancestral RT/RNase H and other polyprotein genes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Eukaryotic transposable elements are divided into two major classes according to whether their transposition intermediate is RNA or DNA (Bennetzen 2000; Kumar and Bennetzen 1999). These RNA- and DNA-dependent transposable elements are called retrotransposons and transposons, respectively. Each group of two elements contains autonomous and nonautonomous elements. Autonomous elements have either one or multiple open reading frames (ORFs) encoding the products required for transposition. Nonautonomous elements have no ORF and their transposition mechanism remains unclear. Autonomous retrotransposons are classified into long terminal repeat (LTR)-containing retrotransposons and LTR-lacking elements known as LINEs (long interspersed nuclear elements). LTR retrotransposons encode gag, protease, integrase, reverse transcriptase (RT), and RNase H, while LINEs encode gag, RT, and RNase H (Bennetzen 2000; Kumar and Bennetzen 1999). The full-length elements of these groups have been reported in a wide variety of plant species of seed plants (Kumar and Bennetzen 1999).
Although the gene composition varies between different types of retroelements, all autonomous retrotransposons possess an RT gene that is usually followed by an RNase H gene (Kumar and Bennetzen 1999). The RT gene is absent in the nuclear genome of eukaryotes but found in some bacterial RT elements such as bacterial retrons and mitochondrial retroplasmids (Xiong and Eickbush 1990). Phylogenetic studies of RT and RNase H sequences suggest that retrotransposons are derived from bacterial RT elements and that LINEs are older than LTR retrotransposons (Xiong and Eickbush 1990; Malik and Eickbush 2001).
LTR-containing retroelements are further divided into Ty1/copia and Ty3/gypsy elements, which are abbreviated hereafter as copia and gypsy, respectively. The two types can be distinguished by nucleotide sequence differences in their catalytic enzymes and an inversed gene arrangement order of integrase and RT/RNase H. Though RTs of LTR retrotransposons are believed to be derived from bacterial elements, there is no information about when or how the different orders of gene arrangement were constructed. To understand the evolutionary history of LTR retrotransposons, characterization of LTR retrotransposons in primitive eukaryotes is necessary. So far, Osser in the green alga Volvox carteri is the only example of the full-length copia retrotransposon among plant species other than vascular plants.
Red algae are known to be derived from a primitive plant lineage. Recently, the whole-genome sequence of a small unicellular red alga, Cyanidioschyzon merolae, was announced (Matsuzaki et al. 2004). We investigated the genome data and reported that C. merolae possesses RT sequences related to LINEs but lacking LTR retrotransposons (Nozaki et al. 2007), whereas a macro red alga, Porphyra yezoensis, possesses RT sequences of copia- and gypsy-like retrotransposons (Zhang et al. 2006). Further, a cDNA encoding RT sequences isolated from P. yezoensis showed a unique structural feature in that it possesses RT and RNase H genes closely related to those of Volvox copia retrotransposons but lacks the rest of the genes present in typical copia-retrotransposons (Zhang et al. 2006). We therefore tentatively speculated that the red algal cDNA could be a single RT/RNase H gene and a progenitor of copia retrotransposons. Subsequently, we thought that isolation and characterization of different families of LTR retrotransposons from Porphyra could provide valuable information about the evolution of LTR retrotransposons in plants. In this study, a copia-like element PyRE10G encoding the same proteins as typical LTR retrotransposons was isolated from the genome of P. yezoensis, and its sequence data suggest that the RT/RNase H sequences are closely related to those of copia-like retrotransposons but the integrase sequences are related to gypsy-like elements. This finding permits us to discuss the evolutionary history of copia-like retrotransposons in plants.
Materials and Methods
Plant Material and Culture Conditions
Gametophytic blades of Porphyra yezoensis Ueda (strain TU-1; Kuwano et al. 1996) were kindly provided by Professor N. Saga (Hokkaido University, Japan). The cultures were grown in a medium containing 3.5% Sealife powder (Marintech Co., Ltd., Japan) and 1% (v/v) ESS2 stock solution (pH 8.0) (Nikaido et al. 2000). The concentration of nitrate in the medium was changed to 2.8 mM. The culture for gametophytes of P. yezoensis was maintained at 15°C on a photoregime of 10 h light:14 h dark, with illumination from cool white fluorescent lamps (4500 lux) with constant air bubbling. Young thalli (<3 cm in length) germinated from monospores were used as the sample.
Isolation of a Retrotransposon-like Gene Fragment
Our preliminary experiment showed that the P. yezoensis EST clone AV430372 (Nikaido et al. 2000) was a 5′-end truncated gene fragment (3540 bp) encoding RT and RNase H related to those copia retrotransposons. Then, by inverse PCR and LA-PCR, a full-length gene was isolated from the genomic DNA of P. yezoensis as follows.
Total DNA was extracted from young blades with hexadecyltrimethyl ammonium bromide (CTAB) and chloroform according to the method of Apt and Grossman (1993). For inverse PCR, genomic DNA was digested with selected restriction enzymes, and the restriction fragments were treated with chloroform and isoamyl alcohol and then purified by ethanol precipitation. The purified restriction fragments were self-ligated using solution I from DNA ligation kit version 2.1 (Takara Bio, Shiga, Japan) overnight at 14°C. After the incubation period, the samples were purified by ethanol precipitation and used for inverse PCR with outward primers specific to the known region of the PyRE10G element. The resulting PCR fragments were gel purified, cloned into a pT7Blue vector, and identified by DNA sequencing.
LA PCR was carried out using a LA PCR kit (Takara Bio) in a 50-μl reaction mixture containing 100 ng of genomic DNA, 200 μM dNTPs, 5 μl LA buffer (Mg2+ free), 2 mM MgCl2, 10 pmol of each forward (LA1: 5′-TGCCTACGGCACGCTCAAGAC-3′) and reverse (LA2: 5′-GCTACCGTTCCGACATCTTG-3′) primer, and 0.2 μl of LA Taq polymerase (5 U/μl). The PCR conditions were initial denaturation at 94°C for 2 min followed by 40 cycles at 96°C for 20 sec and 66°C for 6 min. Final elongation was at 72°C for 7 min. The 4.6-kb PCR product was extracted from the agarose gel and cloned into the pT7Blue vector using a TA cloning kit (Invitrogen, USA) according to the manufacturer’s instructions. DNA sequencing was carried out with the CEQ 2000XL DNA analysis system (Beckman Coulter, USA). A 4581-bp sequence encoding the full-length ORF of copia-like elements was characterized and referred to as PyRE10G.
The nucleotide sequence reported in this paper was entered into the DDBJ, EMBL, and GenBank nucleotide sequence databases with accession number AB286055.
Sequence Analysis
Isolated DNA sequences were analyzed by Genetyx-win software. Multiple alignments were created by ClustalW (Thompson et al. 1994) and the neighbor-joining phylogenetic tree of conserved domains was constructed using MEGA 3.1 software (Kumar et al. 2004). Bootstrap values of 1000 replicates above 50% only are indicated on the nodes.
Southern Hybridization
Total genomic DNA (1 μg) was digested with restriction enzymes (10 U). The digested DNA fragments were fractionated on 0.8 % (w/v) agarose gels in TAE buffer, hydrolyzed with 0.25 N HCl, renatured in 0.5 N NaOH and 1.5 M NaCl, and then transferred to Biodyne B membranes (PALL, USA). For hybridization, the digoxigenin (DIG)-labeled DNA probes complementary to the gag region (390 bp) and RT region (758 bp) were synthesized using a PCR DIG Probe Synthesis Kit (Roche Diagnostics, Mannheim, Germany). Prehybridization and hybridization were performed as described previously (Suzuki et al. 1998). The membranes were washed twice for 15 minutes in 2 × SSC, 0.1% (w/v) SDS at room temperature and twice for 15 min in 0.1 × SSC, 0.1% SDS at 65°C. The hybridized probes were immunodetected with an alkaline phosphatase-conjugated antidigoxigenin antibody and visualized with CSPD chemiluminescence substrate as per the supplier’s instructions (Roche Diagnostics).
Results
Gene Structure and Copy Number of the copia-like Element PyRE10G
The genomic DNA fragment (PyRE10G) isolated from Porphyra yezoensis contained a putative ORF with 1431 amino acids, although one stop codon was present within the frame (Fig. 1a). In our previous report, a reverse transcriptase-encoding gene from P. yezoensis (PyRE2A) lacked protein genes other than RT and RNase H (Zhang et al. 2006). Therefore, candidate proteins encoded by PyRE10G were sought using BlastX (Altschul et al. 1997). The results showed that PyRE10G gag and protease regions were weakly related to those of viruses (data not shown). Then amino acid sequences of PyRE10G gag and protease were compared with those of viruses and retrotransposons. The CCHC motif conserved in the nucleic acid-binding gag proteins of viruses and retrotransposons (Mount and Rubin 1985) was also identified in PyRE10G (Fig. 1b). The three conserved domains of viral protease sequences recognized by McClure (1991) were partially preserved in PyRE10G as in other LTR retrotransposons (Fig. 1c). These results suggest that PyRE10G encodes gag- and protease-like sequences. Further, a BlastX search suggested that PyRE10G possesses integrase, RT, and RNase H sequences in the order of the arrangement shown in Fig. 1a. Based on the order of polyprotein gene arrangement, PyRE10G can be considered a copia-like retrotransposon. Southern hybridization of P. yezoensis total DNA with probes against gag and RT indicated that PyRE10G exists as a single copy in the genome (Fig. 1d).

Schematic representation of gene structure and Southern blot analysis of PyRE10G. a A schematic representation of the gene structure of PyRE10G. The positions of the translation start codon and stop codon are indicated as base pairs in parentheses. The asterisk at the integrase region indicates the stop codon. DNA probes (P1 and P2) used for Southern blot are indicated as bold bars. b Amino acid sequences conserved in gag proteins. c Amino acid sequences conserved in proteases of viruses and retrotransposons. Conserved signatures among retroelements are indicated above the alignment. Accession numbers are PyRE10G (this study; AB286055), HIV-1 (M93258), HTLV-I (AF033817), Copia (M11240), RIRE1 (D85597), Tto1 (D83003), Tnt1 (X13777), Osser (X69552), Tal3 (X13291), Ty3 (M23367), and Gypsy (M12927). d Southern blot analysis of total DNA. Total DNA (1 μg) was digested with restriction enzymes PvuII (P) and KpnI (K). Their recognition sites are indicated in a. The undigested (U) and digested DNAs were transferred onto the membrane and hybridized with the P1 or P2 probe
Since the phylogenetic relationship of retrotransposons has been evaluated through similarities between the conserved domains of RT (Xiong and Eickbush 1990), RNase H (Malik and Eickbush 2001; Malik 2005), and integrase (Capy et al. 1996), we compared the amino acid sequences of the respective conserved domains of PyRE10G to those of the red alga-derived PyRE2A (Zhang et al. 2006) and other retroelements.
copia-like Sequences of RT and RNase H
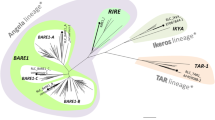
RT is found in a wide variety of organisms from bacteria to retroviruses. Most RT sequences have seven conserved domains, but copia retrotransposons lack the sixth domain (Xiong and Eickbush 1990). Like the RTs of PyRE2A and copia-like retrotransposons, the RT of PyRE10G lacked the sixth domain (Fig. 2a). Further, the amino acid sequences of PyRE10G RT were more related to those of copia-like retrotransposons. A neighbor-joining tree of 208 amino acids of conserved RT domains also supports this result in that PyRE10G is located in the clade of copia elements and grouped with PyRE2A (Fig. 2b).

Multiple alignment and phylogenetic tree of the RT domains of PyRE10G and other elements. a Alignment of the RT region. The seven domains are indicated by bars and roman numbers and the conserved amino acids are highlighted. b Phylogenetic tree. Accession numbers of the retrotransposons used are BARE1 (Z17327), PyRE2A (AB248913), Hopscotch (U12626), TED (M32662), Sushi (AF030881), MAGGY (L35053), Skippy (L34658), Osvaldo (AJ133521), Cin4 (Y00086), R1Dm (P16425), R1Bm (AB182560), Jockey (P21328), and T1 (B34751). Accession numbers of other elements are described in the legend to Fig. 1
RNase H is involved in the cleaving of the RNA strand in RNA-DNA hybrids during cDNA synthesis. RNase H domains have been found in all Eukarya, one Archaea, many Eubacteria, a few non-LTR retrotransposons, and all LTR retrotransposons (Malik and Eickbush 2001). Determination of phylogenetic relationships of retrotransposons through similarities between conserved domains of RNase H proteins has been reported (Malik and Eickbush 2001; Malik 2005). Based on these similarity studies, we aligned the RNase H catalytic domains of PyRE10G along with those of the red alga-derived PyRE2A and LTR retrotransposons (Fig. 3a). The residues believed to be important for the catalytic activity of RNase H (D10, E45, D70, and D134) were entirely conserved in PyRE10G as in other major LTR retrotransposons. A neighbor-joining tree of 158 amino acid sequences from PyRE10G RNase H domains was constructed in comparison with Eukarya, Eubacteria, and selected non-LTR and LTR retrotransposons to analyze the evolutionary relationship of PyRE10G (Fig. 3b). The phylogenetic tree suggests that PyRE10G is located within the clade of copia elements.

Multiple alignment and phylogenetic tree of RNase H. a Alignment of catalytic domains of RNase H was constructed according to Malik and Eickbush (2001). The two elements PyRE10G and PyRE2A are aligned with other retrotransposons and conserved sequences are highlighted. Catalytically important amino acids are indicated above the alignment. b A neighbor-joining tree of RNase H domains of elements from Eubacteria, Eukarya, non-LTR, and LTR retrotransposons. Accession numbers of the elements used here are 412 (X04132), Lian (U87543), MGL (AF018033), Schizosaccharomyces (AF048992), Giardia (AACB01000112), Caenorhabditis (U41994), Thermus (X60507), and Haemonphilus (AE017143); those mentioned in the legends to Figs. 1 and 2 are not repeated
gypsy-like Sequences of PyRE10G Integrase
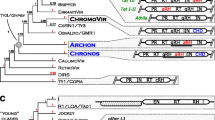
Integrase is required for insertion of the DNA form of the retrotransposon into a new chromosome location. The HHCC signature is conserved in retrotransposons and retroviruses, whereas the DDE signature is conserved in the integrases of LTR retrotransposons, retroviruses, and the transposase of DNA transposons (Khan et al. 1991). This signature is crucial for the integration of these elements (Kulkosky et al. 1992). We aligned PyRE10G integrase with integrases of copia- and gypsy-like retrotransposons (Fig. 4a). The alignment showed a perfect conservation of HHCC and DDE signatures in PyRE10G integrase (Fig. 4a; only DDE alignment shown). Although it was difficult to evaluate whether PyRE10G integrase is more related to copia or gypsy integrases, a BlastX search suggested that PyRE10G integrase was most related to integrases of gypsy-like retrotransposons (data not shown). In a phylogenetic tree using the region of the DDE signature, copia- and gypsy-like retrotransposons were reported to stand in different clades (Capy et al. 1996). We therefore constructed a DDE region phylogenetic tree using nucleotide sequences because the number of amino acids in this region was relatively small compared to that in RT and RNase H coding regions. The tree revealed that PyRE10G integrase is located within the same clade as gypsy-like retrotransposons, whereas all integrases from the copia-like elements stand within the copia clade (Fig. 4b).

Multiple alignment and phylogenetic tree of catalytic domains of integrases. a Alignment of integrase catalytic domains. The DDE signatures are indicated above the alignment. b Phylogenetic tree of DDE signatures. A neighbor-joining tree of nucleotide sequences of the DDE region was constructed by deleting sequences of 399 to 404 amino acids (gap) in PyRE10G and the corresponding region from the aligned elements. c Alignment of PyRE10G C-terminal putative chromodomain sequences with Drosophila Polycomb chromoprotein and other chromodomain-containing gypsy retrotransposons. The conserved amino acids are highlighted. Accession numbers of the elements used here are Tst1 (X52387), Polycomb (P26017), Grh (M77661), and Del (X13886); the rest of the accession numbers are given in the legends to Figs. 1– 3
Integrases of many gypsy elements contain a C-terminal chromodomain-like module, downstream of the catalytic DDE motif (Malik and Eickbush 1999). This module was found to be functionally involved in transposition of the MAGGY retrotransposon (Nakayashiki et al. 2005). We therefore aligned the C-terminal sequences of PyRE10G integrase with Drosophila chromoprotein, Polycomb, and chromodomain-containing gypsy elements (Fig. 4c). We found partially conserved chromodomain sequences in PyRE10G integrase. On the other hand, PyRE10G integrase did not show a GKGY motif at about 60 residues downstream of the DDE motif, a universal feature of copia-elements integrases (Peterson-Burch and Voytas 2002).
Discussion
This is the first example of a full-length LTR retrotransposon in red algae. The most prominent property of the protein structure of this element was that PyRE10G RT and RNase H were related to those of copia-like retrotransposons (Figs. 2 and 3), whereas its integrase was related to gypsy-like elements (Fig. 4). Based on the gene order of the polyproteins, PyRE10G belongs to the family of copia-like retrotransposons. Phylogenetic analysis revealed that PyRE10G and PyRE2A RTs are primitive copia-like elements (Fig. 2b). This is consistent with the phylogenetic placement of red algae as a primitive plant lineage. It was unexpected that the PyRE10G integrase would be more related to gypsy-retrotransposons than copia-elements. So far, Volvox Osser is the only single example of a full-length copia retrotransposon other than those found in seed plants (Lindauer et al. 1993). Based on the gene order and similarity of RT and RNase H sequences, Osser is defined as a copia-like element. In the present study, the integrase sequence of Osser was related to those of copia-like elements (Fig. 4b), contrasting with the unique property of PyRE10G integrase.
Based on the gene structure, PyRE10G seems to be a chimera of copia and gypsy elements. A hybrid element of copia retrotransposons, Ty1/Ty2, is observed in Saccharomyces cerevisiae and its generation has been explained by recombination due to two RT-mediated template switches between Ty1 and Ty2 families (Jordan and McDonald 1999). Template switching is recognized as a mechanism for generating chimeric (hybrid) elements between different family elements. However, this process cannot explain the origin of PyRE10G, because the RT/RNase H and integrase genes are arranged in different orders between the two elements.
Retroelements are known to capture host genome sequences through a process known as transduction (Bureau et al. 1994; Palmgren 1994). Some LTR retrotransposons have been found to contain a retroviral envelope-like gene (Britten 1995; Laten et al. 1998; Wright and Voytas 1998). Therefore, retroviruses are speculated to have evolved from retrotransposons by capturing an envelope gene (Malik and Eickbush 2001). On the other hand, LTR retrotransposons are also believed to be derived from common segments including RTs gene by acquisition of retroviral-like poly protein genes (McClure 1991; Malik and Eickbush 2001). However, direct evidence for this concept has not been demonstrated. The unusual structure of PyRE10G evokes the idea that copia-like and gypsy-like polyprotein genes might have fused during the evolution of PyRE10G.
Several models for the evolution of retrotransposons have been provided. Non-LTR retrotransposons are believed to be more primitive than LTR retrotransposons in that the RT sequences (Xiong and Eickbush 1990) and RNase H (Malik and Eickbush 2001) of non-LTR retrotransposons are more related to those of bacterial elements. Since non-LTR elements do not encode integrase genes, the acquisition of integrase in LTR retrotransposons seems to be a key step in the evolution of LTR retrotransposons. From the sequence similarities among integrases of LTR retrotransposons and DNA transposases, Capy et al. (1996) postulated that the integrase of LTR retrotransposons originated from bacterial insertional elements. According to a model proposed by Malik and Eickbush (2001) and Malik (2005), at the early stage of LTR retrotransposon evolution, the transposase gene was fused with an ancestor of non-LTR retrotransposons. The order of arrangement of integrase and RT/RNase H genes is different between copia and gypsy elements. In copia elements the integrase gene is located upstream of the RT/RNase H region, while the inverse occurs in gypsy elements. It is therefore speculated that the transposase captured upstream RT/RNase H gene evolved into an extant copia retrotransposon, while downstream insertion of RT/RNase H evolved into a gypsy retrotransposon. These models are essential for explaining how copia and gypsy elements have diverged. However, no evidence supporting this model has been found for extant LTR retrotransposons, until the present study, which shows that red algal retrotransposons possess copia-like RT/RNase H and gypsy-like integrase. We therefore propose that at the early stage of PyRE10G generation, a transposase gene that evolved into extant gypsy-like retrotransposons was fused upstream of the RT/RNase H gene. However, this explanation includes an additional assumption that the integrases of copia and gypsy element are derived from different transposase genes. Based on the sequence similarities, retrotransposon integrase genes are usually grouped as either copia and gypsy elements. Therefore, it has not been discussed whether the origin of the integrases of copia and gypsy elements is monophyletic or diphyletic. The contradictory finding that the copia-like PyRE10G possesses gypsy-like integrase highlights this problem and supports the diphyletic model described above. To further evaluate this idea, we must isolate typical gypsy-like elements. However, we cannot abandon the monophyletic model stating that all LTR retrotransposons from P. yezoensis might have originated from only one transposase gene, until we can isolate other copia elements from P. yezoensis.
We previously speculated that PyRE2A isolated from P. yezoensis was a single RT/RNase H gene and that it was a progenitor of extant copia-like retrotransposons (Zhang et al. 2006). As shown in Fig. 2b, the RT of PyRE10G was most related to that of PyRE2A. This finding evokes the possibility that PyRE10G has evolved from PyRE2A. However, the phylogenetic tree of RNase H does not support this assumption in that the position of PyRE2A was far from that of PyRE10G (Fig. 3b). These findings further evoke the possibility that at the early stage of evolution of copia-like retrotransposons, fusion of the RNase H gene with the RT gene also occurred. Considering that PyRE10G and PyRE2A contain a stop codon(s) in the ORF and exist as single copies in the genome, it is likely that they represent different stages of the evolution of copia-like retrotransposons. Characterization of red algal retrotransposon sequences other than PyRE2A and PyRE10G will provide valuable information about the evolution of plant retrotransposons.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Apt KE, Grossman AR (1993) Characterization and transcript analysis of the major phycobiliprotein subunit genes from Aglaothamnion neglectum (Rhodophyta). Plant Mol Biol 21:27–38
Bennetzen JL (2000) Transposable element contributions to plant gene and genome evolution. Plant Mol Biol 42:251–269
Britten RJ (1995) Active gypsy/Ty3 retrotransposons or retroviruses in Caenorhabditis elegans. Proc Natl Acad Sci USA 92:599–601
Bureau TE, White SE, Wessler SR (1994) Transduction of a cellular gene by a plant retroelement. Cell 77:479–480
Capy P, Vitalis R, Langin T, Higuet D, Bazin C (1996) Relationships between transposable elements based upon the integrase-transposase domains: Is there a common ancestor? J Mol Evol 42:359–368
Jordan IK, McDonald JF (1999) Tempo and mode of Ty element evolution in Saccharomyces cerevisiae. Genetics 151:1341–1351
Khan E, Mack JPG, Katz RA, Kulkosky J, Skalka AM (1991) Retroviral integrase domains: DNA binding and the recognition of LTR sequences. Nucleic Acids Res 19:851–860
Kulkosky J, Jones KS, Katz RA, Mack JPG, Skalka AM (1992) Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol Cell Biol 12:2331–2338
Kumar A, Bennetzen JL (1999) Plant Retrotransposons. Annu Rev Genet 33:479–532
Kumar S, Tamura K, Nei M (2004) MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Kuwano K, Aruga Y, Saga N (1996) Cryopreservation of clonal gametophytic thalli of Porphyra (Rhodohyta). Plant Sci 116:117–124
Laten HM, Majumdar A, Gaucher EA (1998) SIRE-1, a copia/Ty1–like retroelement from soybean, encodes a retroviral envelope-like protein. Proc Natl Acad Sci USA 95:6897–6902
Lindauer A, Fraser D, Bruderlein M, Schmitt R (1993) Reverse transcriptase families and a copia-like retrotransposon, Osser, in the green alga Volvox carteri. FEBS Lett 319:261–266
Malik HS (2005) Ribonuclease H evolution in retrotransposable elements. Cytogenet Genome Res 110:392–401
Malik HS, Eickbush TH (1999) Modular evolution of the integrase domain in the Ty3/Gypsy class of LTR retrotransposons. J Virol 73:5186–5190
Malik HS, Eickbush TH (2001) Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res 11:1187–1197
Matsuzaki M, Misumi O, Shin IT, Maruyama S, Takahara M, Miyagishima SY, Mori T, Nishida K, Yagisawa F, Nishida K, Yoshida Y, Nishimura Y, Nakao S, Kobayashi T, Momoyama Y, Higashiyama T, Minoda A, Sano M, Nomoto H, Oishi K, Hayashi H, Ohta F, Nishizaka S, Haga S, Miura S, Morishita T, Kabeya Y, Terasawa K, Suzuki Y, Ishii Y, Asakawa S, Takano H, Ohta N, Kuroiwa H, Tanaka K, Shimizu N, Sugano S, Sato N, Nozaki H, Ogasawara N, Kohara Y, Kuroiwa T (2004) Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature 428:653–657
McClure MA (1991) Evolution of retroposons by acquisition or deletion of retrovirus-like genes. Mol Biol Evol 8:835–856
Mount SM, Rubin GM (1985) Complete nucleotide sequence of the Drosophila transposable element copia: Homology between copia and retroviral proteins. Mol Cell Biol 5:1630–1638
Nakayashiki H, Awa T, Tosa Y, Mayama S (2005) The C-terminal chromodomain-like module in the integrase domain is crucial for high transposition efficiency of the retrotransposon MAGGY. FEBS Lett 579:488–492
Nikaido I, Asamizu E, Nakajima M, Nakamura Y, Saga N, Tabata S (2000) Generation of 10,154 expressed sequence tags from a leafy gametophyte of a marine red alga, Porphyra yezoensis. DNA Res 7:223–227
Nozaki H, Takano H, Misumi O, Terasawa K, Matsuzaki M, Maruyama S, Nishida K, Yagisawa F, Yoshida Y, Fujiwara T, Takio S, Tamura K, Chung SJ, Nakamura S, Kuroiwa H, Tanaka K, Sato N, Kuroiwa T (2007) A 100%-complete sequence reveals unusually simple genomic features in the hot-spring red alga Cyanidioschyzon merolae. BMC Biol 5:28
Palmgren MG (1994) Capturing of host DNA by a plant retroelement:Bs1 encodes plasma membrane H+-ATPase domains. Plant Mol Biol 25:137–140
Peterson-Burch BD, Voytas DF (2002) Genes of the Pseudoviridae (Ty1/copia retrotransposons). Mol Biol Evol 19:1832–1845
Suzuki T, Takio S, Satoh T (1998) Light-dependent expression in liverwort cells of chlL/N and chlB identified as chloroplast genes involved in chlorophyll synthesis in the dark. J Plant Physiol 152:31–37
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTALW. Nucleic Acids Res 22:4673–4680
Wright DA, Voytas DE (1998) Potential retroviruses in plants:Tat1 is related to a group of Arabidopsis thaliana Ty3/gypsy retrotransposons that encode envelope-like proteins. Genetics 149:703–715
Xiong Y, Eickbush TH (1990) Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J 9:3353–3362
Zhang W, Sakai M, Lin X, Takechi K, Takano H, Takio S (2006) Reverse transcriptase like sequences related to retrotransposon in a red alga, Porphyra yezoensis. Biosci Biotechnol Biochem 70:1999–2003
Acknowledgments
We would like to thank Dr. Naotune Saga of Hokkaido University, Japan, for providing the gametophyte of P. yezoensis. This study was supported by Grants-in-Aid for Scientific Research on Priority Areas (to H. T.) from the Ministry of Education, Science, Sports and Culture of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peddigari, S., Zhang, W., Sakai, M. et al. A Copia-like Retrotransposon Gene Encoding Gypsy-like Integrase in a Red Alga, Porphyra yezoensis . J Mol Evol 66, 72–79 (2008). https://doi.org/10.1007/s00239-007-9057-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-007-9057-1