
Abstract
The crustacean isopod Armadillidium vulgare is characterized by an unusual ∼42-kb-long mitochondrial genome consisting of two molecules co-occurring in mitochondria: a circular ∼28-kb dimer formed by two ∼14-kb monomers fused in opposite polarities and a linear ∼14-kb monomer. Here we determined the nucleotide sequence of the fundamental monomeric unit of A. vulgare mitochondrial genome, to gain new insight into its structure and evolution. Our results suggest that the junction zone between monomers of the dimer structure is located in or near the control region. Direct sequencing indicated that the nucleotide sequences of the different monomer units are virtually identical. This suggests that gene conversion and/or replication processes play an important role in shaping nucleotide sequence variation in this mitochondrial genome. The only heteroplasmic site we identified predicts an alloacceptor tRNA change from tRNAAla to tRNAVal. Therefore, in A. vulgare, tRNAAla and tRNAVal are found at the same locus in different monomers, ensuring that both tRNAs are present in mitochondria. The presence of this heteroplasmic site in all sequenced individuals suggests that the polymorphism is selectively maintained, probably because of the necessity of both tRNAs for maintaining proper mitochondrial functions. Thus, our results provide empirical evidence for the tRNA gene recruitment model of tRNA evolution. Moreover, interspecific comparisons showed that the A. vulgare mitochondrial gene order is highly derived compared to the putative ancestral arthropod type. By contrast, an overall high conservation of mitochondrial gene order is observed within crustacean isopods.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The role and function of mitochondrial products are well conserved across taxa but recent data from mitochondrial genomics research demonstrate that this conservation contrasts with an often-perplexing diversity in mitochondrial structure and gene order. In metazoans, the mitochondrial genome usually consists of a double-stranded circular DNA molecule that ranges in size from 15 to 20 kb and encodes a reduced number of essential mitochondrial products (Boore 1999). However, linear structures have been reported in various species, including unicellular eukaryotes, the liverwort Marchantia polymorpha, and several cnidarian species (Nosek and Tomaska 2003; Nosek et al. 1998; Oldenburg and Bendich 2001; Suyama and Miura 1968; Vahrenholz et al. 1993). Other structural exceptions include placozoan mitochondrial genomes that are 32–37 kb in length, owing to extensive length variation in intergenic spacer regions, open reading frames, and the presence of introns (Signorovitch et al. 2007). Although large animal mitochondrial genomes have been discovered, they are relatively rare and owe their large size to secondary expansions such as duplications, AT-rich regions, and multiple short tandem repeats (Boyce et al. 1989).
Most mitochondrial genomes have retained only 13 polypeptide genes, all of which encode essential components of the oxidative phosphorylation metabolic pathway (Boore 1999). Notable exceptions include the absence of Atp8 in nematodes and some mollusks (Boore 1999). The mitochondrial genome also encodes the 12S and 16S rRNA genes and the 22 tRNA genes required for mitochondrial protein synthesis (Boore 1999). In vertebrates, mitochondrial gene order remains practically invariant, but there is substantial variation in invertebrate phyla. In arthropods, complete or partial mitochondrial DNA (mtDNA) sequences have been determined for 69 hexapods, 23 chelicerates, 6 myriapods, and 34 crustaceans as of June 2007 (NCBI at http://www.ncbi.nlm.nih.gov/). Gene organization substantially varies both among (Boore and Brown 1998; Boore et al. 1995) and within major arthropod groups. Hence, many gene rearrangements relative to the putative arthropod primitive mitochondrial gene order have been reported within crustaceans (Hickerson and Cunningham 2000; Machida et al. 2002; Miller et al. 2004, 2005; Ogoh and Ohmiya 2004; Sun et al. 2005; Tjensvoll et al. 2005), including the two isopods Ligia oceanica and Idotea baltica (Kilpert and Podsiadlowski 2006; Podsiadlowski and Bartolomaeus 2006).
The isopod Armadillidium vulgare presents yet another kind of unusual mitochondrial genome, consisting of a remarkable length of ∼42 kb that is not consistent with amplification of sequence fragments or additional copy number of repeated sequences (Raimond et al. 1999). Instead, the A. vulgare mitochondrial genome consists of two molecules co-occurring in mitochondria: a circular ∼28-kb dimer formed by two ∼14-kb monomers fused in opposite polarities and a linear ∼14-kb monomer (Fig. 1) (Raimond et al. 1999). It has been hypothesized that the linear and circular molecules are not at equilibrium, and that the circular state might repeatedly be generated from the linear state, although the actual replication mechanism of this atypical mitochondrial genome currently is unknown (Raimond et al. 1999).

Schematic representation of the atypical structure of Armadillidium vulgare mitochondrial genome proposed by Raimond et al. (1999)
Another distinctive feature of A. vulgare is that mitochondria collocate within the cytoplasm with obligate intracellular Wolbachia bacteria that are responsible for the feminization of genetic males into functional females (Bouchon et al. 1998; Cordaux et al. 2004; Rigaud et al. 1997). There is evidence that mtDNA variability is influenced by the presence of Wolbachia in A. vulgare populations (Cordaux et al. 2004; Grandjean et al. 1993; Rigaud et al. 1999; Souty-Grosset et al. 1992), which raises the question about a possible link between the presence of Wolbachia and the atypical structure of the mitochondrial genome in A. vulgare.
To gain new insight into the structure and evolution of the atypical mitochondrial genome of A. vulgare and the extent of its association with Wolbachia, we sequenced the fundamental monomeric unit of the A. vulgare mitochondrial genome. We show that nucleotide sequences of the monomers constituting A. vulgare mitochondrial genome are virtually identical. This suggests that gene conversion and/or replication processes play an important role in shaping nucleotide sequence variation in this mitochondrial genome. We also identified an unusual dual tRNA locus in which tRNAAla and tRNAVal genes are found at the same locus in different monomers. Thus, our results provide empirical evidence for the tRNA gene recruitment model of tRNA evolution (Higgs et al. 2003; Lavrov and Lang 2005). Furthermore, our results highlight several gene rearrangements relative to the putative ancestral arthropod mitochondrial gene order. However, comparisons with I. baltica and L. oceanica (Kilpert and Podsiadlowski 2006; Podsiadlowski and Bartolomaeus 2006) suggest that the mitochondrial gene order found in A. vulgare is conserved among isopods.
Materials and Methods
Samples and mtDNA Extraction
A. vulgare individuals used for mitochondrial genome sequencing were obtained from an isofemale line (i.e., a lineage of individuals all descended from a single female) maintained in our laboratory (strain ZN 540; originally collected in Celles-sur-Belle, western France). Individuals used for restriction profile analyses were obtained from an isofemale line maintained in our laboratory (strain WX 1107; originally collected in Helsingor, Denmark). By using isofemale lines, we are able to pool mtDNA extracts from various individuals of each isofemale line to obtain sufficient amounts of mtDNA for analyses. The six individuals used for nucleotide sequence heteroplasmy analyses originated from four different sources: three individuals from lab strain ZN 540, one individual from lab strain ZU (originally collected in Nice, southeastern France), and two wild-caught individuals collected in Chizé (western France) and Porto Alegre (Brazil). Mitochondrial DNA was extracted from ovaries (for genome sequencing) and gonads, fat tissues, and nervous system (for restriction profile and heteroplasmy analyses) using an alkaline lysis protocol, as previously described (Souty-Grosset et al. 1992).
Mitochondrial Genome Sequencing
MtDNA was digested separately with the two restriction endonucleases, BglII and HincII. Restriction fragments were inserted in the dephosphoryled pUC 19 vector opened by an adequate enzyme: BamHI (complementary to BglII) and HincII (generating blunt ends). Next, we subcloned the large cloned fragment obtained from BglII digestion (∼9.6 kb) by digesting the insert with the restriction enzyme HindIII to create smaller fragments. All ligation products were then used to transform competent bacteria (Escherichia coli Xblue1 F’ Eurogentec) and colonies were grown overnight.
Plasmid DNA was extracted and purified with Wizard plus miniprep Kit (Promega) and then sequenced using the dideoxy terminator method. Inserts were sequenced using the universal sequencing primers M13F and M13R. For long restriction fragments, intermediate primers were designed by hand as part of a primer-walking strategy (Table 1). Sequencing products were precipitated with sodium acetate and absolute ethanol and resolved on an ABI Prism 310 automated DNA sequencer (Applied Biosystems).
Genome Annotation
Sequence assemblage and alignments were performed using the BioEdit software (Hall 1999). Gene identity was determined by BLAST searches of GenBank (Altschul et al. 1997). Start and stop codons of protein-coding genes were identified as previously described (Podsiadlowski and Bartolomaeus 2006). tRNAs were identified using the program tRNAscan-SE (Lowe and Eddy 1997) or by visual inspection of suspected regions after alignment with previously annotated isopod mitochondrial genomes. The partial DNA sequence (13,858 bp) of the monomer unit of A. vulgare mitochondrial genome was deposited in GenBank under accession number EF643519.
Sequence Conservation of the Monomer Unit
Based on the monomer sequence of A. vulgare mitochondrial genome, seven genomic regions were randomly selected and primer pairs were designed using the software Primer3 (Rozen and Skaletsky 2000). PCR reactions were performed on total DNA and the PCR products were directly sequenced to reveal heteroplasmic sites arising from nucleotide substitutions on different monomers of the mitochondrial genome.
mtDNA Digestions and Restriction Profiles
Total mtDNA from germinal and somatic tissues of both male and female individuals (1–3 μg in 12 μl) were digested with three endonucleases: BamHI, EcoRI, and XhoI. All digestions were carried out using 5 units of enzyme incubated at 37°C for 3 h. Samples were then mixed with a gel-laoding buffer and digested mtDNA was run on a 1.2% agarose gel in Tris EDTA phosphate buffer for 15 h at 30 V. Gels were stained with SYBR Green I and examined under UV light.
Results and Discussion
Atypical Genome Structure and Size
We determined 13,858 bp of the fundamental monomer sequence of the mitochondrial genome of A. vulgare. Given an expected global monomer size of ∼14 kb (Raimond et al. 1999), we estimate that our sequence covers ∼99% of the entire monomer sequence. We were not able to determine the nucleotide sequence between 12S rRNA and Cytb despite several attempts. We interpret this result as further evidence for the atypical structure of A. vulgare mitochondrial genome (Raimond et al. 1999). Indeed, this genome is thought to consist of a circular ∼28-kb dimer, formed by two ∼14-kb monomers fused in opposite polarities, and a linear ∼14-kb monomer (Raimond et al. 1999). Under this model, palindromic sequences are predicted to occur at the junction zones of monomers constituting the circular dimer structure (Fig. 1). Along with the unknown open ends of the linear monomer structure, it is not surprising under this model of mitochondrial structure that determination of the sequence located at the extremities of the monomer unit may be problematic. Correlatively, these results suggest that the junction zone between monomers of the dimer structure is located in or near the control region. This view is supported by two lines of evidence: (i) the control region is not included in the ∼13.9-kb mitochondrial sequence we obtained, and (ii) the control region is located between 12S rRNA and Cytb in the circular mitochondrial genome of the isopod L. oceanica (Kilpert and Podsiadlowski 2006).
With a fundamental unit ∼14 kb in length, A. vulgare mtDNA falls in the lower range of crustacean mitochondrial genomes that generally range between ∼14.6 and ∼18.4 kb (Lavrov et al. 2004; Machida et al. 2002), while the smallest-sized mitochondrial genome known in arthropods is that of the collembolan Onychiurus orientalis with ∼13.0 kb (Cook et al. 2005). Even within isopod crustaceans, there appears to be mitochondrial genome size variation, since the complete mitochondrial genome of L. oceanica is ∼15.3 kb long (Kilpert and Podsiadlowski 2006). Overall, these results suggest that the functional unit of A. vulgare mtDNA has experienced size reduction compared to other species.
Extreme Nucleotide Conservation Among Monomer Unit Sequences
The coexistence of various monomer units as constituents of the mitochondrial genome of A. vulgare raises the question of the extent of their relatedness. To address this issue, we directly sequenced seven randomly selected regions along the monomer unit, amplified by PCR from total DNA. This approach allowed us to simultaneously sequence homologous regions from both molecule types (i.e., linear monomers and circular dimers) and identify polymorphic sites among monomers. Overall, 2709 bp was sequenced using this strategy, which covers ∼20% of the monomer sequence. A single nucleotide position was found to be polymorphic, as shown by a double signal apparent at one position in a sequence trace file (Fig. 2). Therefore, the different monomers appear to exhibit extremely low nucleotide divergence. This conclusion is further supported by the fact that no nucleotide polymorphism among monomers was detected based on (i) an RFLP analysis of the mitochondrial genome of A. vulgare (Raimond et al. 1999) and (ii) direct sequencing of ∼400 bp of the 16S rRNA gene within each of several A. vulgare individuals (Cordaux et al. 2004; Michel-Salzat and Bouchon 2000).

Partial sequence trace file obtained after direct sequencing of a portion of Armadillidium vulgare mtDNA. Above the peaks are the deduced nucleotides. The heteroplasmic site (C/T) is highlighted by the yellow star
The extreme nucleotide conservation among monomer sequences suggests that recurrent gene conversion events may contribute to maintain sequence homogeneity among monomers. This process of concerted evolution has recently been suggested to represent an important evolutionary force in animal mtDNA (Tatarenkov and Avise 2007). Alternatively, it is possible that replication preferentially takes place on a particular structure (e.g., the monomer in the linear state) and that the other structure (e.g., the dimeric circular state) is repeatedly generated from the structure that replicates (Raimond et al. 1999). The two aforementioned explanations (gene conversion and structure-biased replication) are not mutually exclusive.
Interestingly, the heteroplasmic site we detected by direct sequencing of the monomer units displays two signals of roughly equal intensity, each of which is roughly half the intensity of the surrounding positions showing single signals (Fig. 2). Quantitatively, this observation suggests that monomer units exhibiting nucleotide C or T at the heteroplasmic site were in equal or nearly equal quantities in the sequencing reaction. This could be explained if the proportions of monomers and dimers are different, as previously suggested based on densitometry results following A. vulgare mtDNA digestion (Raimond et al. 1999). Alternatively, our sequencing results could be explained by two coexisting populations of mtDNA sequences, each with equal proportions of monomer and dimer structures in A. vulgare individuals. Also, analogous to the doubly uniparental mitochondrial inheritance in mussels in which males are normally heteroplasmic and females homoplasmic (Zouros 2000), it is conceivable that the monomer and dimer structures are characterized by different inheritance patterns, for example, in germinal vs. somatic tissues and/or in male vs. female individuals.
To gain new insight into tissue- and sex-specific inheritance patterns of mtDNA in A. vulgare, we extracted mtDNA separately from gonads and somatic tissues in both male and female individuals. Then we performed single digestions with three different restriction enzymes (EcoRI, BamHI, and XhoI) and compared the restriction profiles of the different samples. Two bands of expected ∼28- and ∼14-kb sizes were observed in all undigested mtDNA control samples, as well as in EcoRI-digested samples (suggesting that there is no EcoRI restriction site in these samples), whatever tissue and sex type (Fig. 3). BamHI and XhoI digestions confirmed these results since (i) identical profiles were obtained whatever tissue and sex type digested with each enzyme, and (ii) the profiles obtained with each enzyme were both consistent with the expected atypical mitochondrial structure (Fig. 3). These results indicated that the ∼28-kb dimer and ∼14-kb monomer are both present in germinal and somatic tissues, in both males and females, thus ruling out a mussel-like system of mitochondrial inheritance in A. vulgare.

Digestion profiles of Armadillidium vulgare mtDNA using female somatic (S) and germinal (G) tissues. Results for males were identical (data not shown). M: molecular ladder (sizes in kb shown on the left). Approximate band sizes (kb) are shown below profiles (see Raimond et al. 1999); values in brackets indicate incompletely digested bands. Schematics at the bottom: interpretations of the BamHI and XhoI digestion profiles, consistent with the atypical structure of the A. vulgare mitochondrial genome (see Fig. 1)
Dual tRNA Locus
Strikingly, the heteroplasmic site identified in Fig. 2 happens to correspond to the position that specifies a tRNA anticodon. Specifically, the C/T nucleotide polymorphism predicts an alloacceptor tRNA change involving tRNAVal and tRNAAla. In comparison, tRNAAla is present at the corresponding site in the two isopods I. baltica and L. oceanica (Kilpert and Podsiadlowski 2006; Podsiadlowski and Bartolomaeus 2006), suggesting that the ancestral tRNA at this locus in A. vulgare was tRNAAla. Subsequently, a mutation occurred at this site in an A. vulgare monomer, resulting in the presence of tRNAVal and tRNAAla at the same locus in different monomers, ensuring that both tRNAs are present in mitochondria.
To test whether the substitution is specific to the sequenced individual or shared with other A. vulgare individuals, we directly sequenced the tRNAVal/Ala locus in five additional A. vulgare individuals of different origins. All individuals were found to be heteroplasmic at the previously identified heteroplasmic position. Because it is highly unlikely that an identical mutation would have occurred independently at the same site in all individuals analyzed, we conclude that the mutation most likely occurred in the ancestor of these individuals. The simplest explanation for the sharing of an ancestral polymorphism in all these individuals drawn from different populations is that it is selectively maintained, probably because of the necessity of both tRNAs for maintaining proper mitochondrial functions.
Because no other tRNAVal locus was identified by the program tRNAscan-SE (Lowe and Eddy 1997), we visually inspected A. vulgare mitochondrial genome. No tRNAVal gene could be identified, suggesting that it has been lost or that it is degraded beyond recognition. Overall, our results provide empirical evidence for the tRNA gene recruitment model of tRNA evolution (Higgs et al. 2003; Lavrov and Lang 2005). Indeed, this model proposes that novel tRNA genes may evolve after duplication of pre-existing tRNA genes and point mutations subsequently occurring at anticodon positions that change tRNA amino acid identity. In A. vulgare, not only can duplications occur within monomers, but the presence of a three-monomer atypical mitochondrial structure actually triples the number of available tRNA genes in each mitochondrial genome, making A. vulgare an ideal system for gene recruitment. This is because a point mutation occurring in a tRNA gene of a given monomer and giving rise to a new tRNA can be generated without prior requirement for tRNA gene duplication, since other monomers that lack the mutation can continue to encode the ancestral tRNA gene. In A. vulgare, the mutation resulting in the dual tRNAVal/Ala was probably selectively advantageous since it has presumably been maintained for a long evolutionary period, at least long enough to lead to deletion or advanced degradation of the putative ancestral tRNAVal of A. vulgare. On the long term, such dual tRNA structure may in turn justify the evolutionary conservation of the two molecules types (i.e., linear monomer and circular dimer) at appropriate proportions in A. vulgare.
These results have interesting implications with respect to the processes that maintain extreme nucleotide conservation among monomer sequences, involving gene conversion and/or replication mechanisms (see above). Indeed, if replication was preferentially taking place on a particular structure (e.g., the monomer in the linear state) and that the other structure (e.g., the dimeric circular state) was repeatedly generated from the structure that replicates (Raimond et al. 1999), no ancestral polymorphism could be maintained in A. vulgare mitochondria. On the other hand, if gene conversion is responsible for the homogeneity of monomer sequences, then it is conceivable that strong selective constraints maintaining a polymorphism required for tRNA functionality could overcome the effects of gene conversion in this region of the mitochondrial monomer. In any event, further investigations are needed to elucidate the evolutionary processes governing A. vulgare mitochondrial nucleotide sequence evolution. For example, it would be desirable to sequence the control region, junction zones of the dimer structure, and ends of the linear structure of A. vulgare mitochondrial genome, as this could yield new insight into replication mechanisms. Other future directions include generation of additional mitochondrial sequence data to better understand patterns of nucleotide sequence evolution and inheritance in A. vulgare mtDNA.
Genome Base Composition
Analysis of nucleic acid composition indicated a strong bias in A. vulgare mitochondrial genome toward A+T nucleotides, which represent 71.2% of the bases, vs. 28.8% of G+C nucleotides. This high A+T value is at the upper range among crustaceans, where values for complete mtDNA sequence range from 60.8% in L. oceanica to 74.9% in Geothelphusa dehaani (Ivey and Santos 2007; Kilpert and Podsiadlowski 2006; Segawa and Aotsuka 2005). The most frequent nucleotide was the purine T (37.1%). This overall A+T richness in A. vulgare mitochondrial genome may also explain why we could not detect the mtDNA origin of replication, as it is known to be a single, large, A+T-rich noncoding region of the mtDNA genome (Wolstenholme 1992).
Mitochondrial Gene Rearrangements
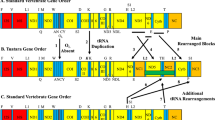
The gene order of A. vulgare mitochondrial monomer is shown in Fig. 4. Comparison with the putative ancestral arthropod mitochondrial genome (Shao et al. 2003) revealed several gene rearrangements in A. vulgare. The first rearrangement involved the 16S rRNA gene which in A. vulgare is translocated between Nad2 and Nad6, whereas it is located between 12S rRNA and Nad1 in the arthropod type (Fig. 4). The latter gene arrangement is considered ancestral for insects and crustaceans (Boore et al. 1998; Hwang et al. 2001). Similarly, a bloc of seven contiguous genes (Nad3, Cox3, Atp6, Atp8, Cox2, Cox1, and Nad2) has been translocated between Nad1 and 16S rRNA in A. vulgare (Fig. 4). Another rearrangement involved Cytb which has been translocated near Nad5 in A. vulgare (Fig. 4). Furthermore, a bloc including Nad5, Cytb, and tRNAPhe has been inverted to the opposite strand of the usual strand. Therefore, the mitochondrial gene order of A. vulgare appeared highly derived compared to the putative ancestral arthropod type.

Comparison of mitochondrial gene order in various species: putative ancestral arthropod type (A) and the isopods Armadillidium vulgare (B), Ligia oceanica (C), and Idotea baltica (D). Lines and arrows indicate the major gene order rearrangements between the different species. Gene names encoded on the (–) strand are shown in gray
By contrast, comparison of A. vulgare genome structure with that of L. oceanica and I. baltica (Kilpert and Podsiadlowski 2006; Podsiadlowski and Bartolomaeus 2006) indicated an overall high conservation of mitochondrial gene order within crustacean isopods (Fig. 4). In fact, all gene rearrangements noted above in A. vulgare relative to the arthropod type are shared with I. baltica and L. oceanica. However, A. vulgare apparently differs from the two other isopods in the position of tRNAVal, which is located between Nad1 and Nad3 in A. vulgare, whereas tRNAAla is present at the corresponding site in I. baltica and L. oceanica. The position of tRNAVal appears quite variable in isopods, as it turns out to be located on three different positions in the three isopod mitochondrial genomes from A. vulgare, I. baltica, and L. oceanica. Therefore, it may represent a character of interest for resolving phylogenetic questions related to isopod crustaceans.
Concluding Remarks
Mitochondrial genome sequence information for the three isopod species A. vulgare (this study), L. oceanica (Kilpert and Podsiadlowski 2006), and I. baltica (Podsiadlowski and Bartolomaeus 2006) indicates that they all share globally similar gene orders. This suggests a relatively well-conserved gene order in isopod mtDNA, distinct from the mitochondrial gene order reported in Decapoda and other Malacostraca. However, isopod mitochondrial genomes differ in their organization, since A. vulgare possesses an unusual mitochondrial genome consisting of a ∼14-kb linear monomer and a ∼28-kb circular dimer (Raimond et al. 1999). By contrast, L. oceanica is characterized by a typical ∼15.3-kb circular mitochondrial genome (Kilpert and Podsiadlowski 2006), although both A. vulgare and L. oceanica belong to the same suborder Oniscidea (Table 2). On the other hand, the mitochondrial genome of I. baltica (suborder Valvifera) is suspected to exhibit an atypical structure that may resemble that of A. vulgare (Podsiadlowski and Bartolomaeus 2006). These observations suggest that atypism of mitochondrial genome structure is not correlated with species phylogeny. If so, one might speculate that mitochondrial genome structure may have evolved multiple times independently toward atypical organizations in various isopod species. However, further studies of the taxonomic distribution of atypical mitochondrial structures among isopod species will be necessary before this question can be resolved.
Given that structural atypism of the mitochondrial genome such as observed in A. vulgare apparently so far seems restricted to isopod species, the question arises as to what factor(s) may cause this atypism to occur in unrelated isopod species such as A. vulgare and I. baltica. Interestingly, many isopod species are known to be infected by the intracellular endosymbiont Wolbachia which collocate with mitochondria in host cytoplasm (Bouchon et al. 1998; Cordaux et al. 2001; Rigaud et al. 1997). Based on these observations, it could be hypothesized that the presence of Wolbachia might somehow be linked to atypical mitochondrial genome organization. However, according to the three isopod species for which information is available on both mitochondrial genome organization and Wolbachia infection status, there apparently is no correlation between the two factors (Table 2). Moreover, other arthropods infected by Wolbachia do not exhibit atypical mitochondrial genomes. Thus, a contribution of Wolbachia to isopod mitochondrial atypism seems questionable. In any event, our results highlight the relevance of conducting further investigations of isopod mitochondrial evolution, with the aim of uncovering the evolutionary forces that make this crustacean group to be so prone to mitochondrial genome structure plasticity.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27:1767–1780
Boore JL, Brown WM (1998) Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev 8:668–674
Boore JL, Collins TM, Stanton D, Daehler LL, Brown WM (1995) Deducing the pattern of arthropod phylogeny from mitochondrial DNA rearrangements. Nature 376:163–165
Boore JL, Lavrov DV, Brown WM (1998) Gene translocation links insects and crustaceans. Nature 392:667–668
Bouchon D, Rigaud T, Juchault P (1998) Evidence for widespread Wolbachia infection in isopod crustaceans: molecular identification and host feminization. Proc Biol Sci 265:1081–1090
Boyce TM, Zwick ME, Aquadro CF (1989) Mitochondrial DNA in the bark weevils: size, structure and heteroplasmy. Genetics 123:825–836
Cook CE, Yue Q, Akam M (2005) Mitochondrial genomes suggest that hexapods and crustaceans are mutually paraphyletic. Proc Biol Sci 272:1295–1304
Cordaux R, Michel-Salzat A, Bouchon D (2001) Wolbachia infection in crustaceans: novel hosts and potential routes for horizontal transmission. J Evol Biol 14:237–243
Cordaux R, Michel-Salzat A, Frelon-Raimond M, Rigaud T, Bouchon D (2004) Evidence for a new feminizing Wolbachia strain in the isopod Armadillidium vulgare: evolutionary implications. Heredity 93:78–84
Grandjean F, Rigaud T, Raimond R, Juchault P, Souty-Grosset C (1993) Mitochondrial DNA polymorphism and feminizing sex factors dynamics in a natural population of Armadillidium vulgare (Crustacea, Isopoda). Genetica 92:55–60
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hickerson MJ, Cunningham CW (2000) Dramatic mitochondrial gene rearrangements in the hermit crab Pagurus longicarpus (Crustacea, anomura). Mol Biol Evol 17:639–644
Higgs PG, Jameson D, Jow H, Rattray M (2003) The evolution of tRNA-Leu genes in animal mitochondrial genomes. J Mol Evol 57:435–445
Hwang UW, Friedrich M, Tautz D, Park CJ, Kim W (2001) Mitochondrial protein phylogeny joins myriapods with chelicerates. Nature 413:154–157
Ivey JL, Santos SR (2007) The complete mitochondrial genome of the Hawaiian anchialine shrimp Halocaridina rubra Holthuis, 1963 (Crustacea: Decapoda: Atyidae). Gene 394:35–44
Kilpert F, Podsiadlowski L (2006) The complete mitochondrial genome of the common sea slater, Ligia oceanica (Crustacea, Isopoda) bears a novel gene order and unusual control region features. BMC Genomics 7:241
Lavrov DV, Lang BF (2005) Transfer RNA gene recruitment in mitochondrial DNA. Trends Genet 21:129–133
Lavrov DV, Brown WM, Boore JL (2004) Phylogenetic position of the Pentastomida and (pan)crustacean relationships. Proc Biol Sci 271:537–544
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Machida RJ, Miya MU, Nishida M, Nishida S (2002) Complete mitochondrial DNA sequence of Tigriopus japonicus (Crustacea: Copepoda). Mar Biotechnol (NY) 4:406–417
Michel-Salzat A, Bouchon D (2000) Phylogenetic analysis of mitochondrial LSU rRNA in oniscids. CR Acad Sci III 323:827–837
Miller AD, Nguyen TT, Burridge CP, Austin CM (2004) Complete mitochondrial DNA sequence of the Australian freshwater crayfish, Cherax destructor (Crustacea: Decapoda: Parastacidae): a novel gene order revealed. Gene 331:65–72
Miller AD, Murphy NP, Burridge CP, Austin CM (2005) Complete mitochondrial DNA sequences of the decapod crustaceans Pseudocarcinus gigas (Menippidae) and Macrobrachium rosenbergii (Palaemonidae). Mar Biotechnol (NY) 7:339–349
Nosek J, Tomaska L (2003) Mitochondrial genome diversity: evolution of the molecular architecture and replication strategy. Curr Genet 44:73–84
Nosek J, Tomaska L, Fukuhara H, Suyama Y, Kovac L (1998) Linear mitochondrial genomes: 30 years down the line. Trends Genet 14:184–188
Ogoh K, Ohmiya Y (2004) Complete mitochondrial DNA sequence of the sea-firefly, Vargula hilgendorfii (Crustacea, Ostracoda) with duplicate control regions. Gene 327:131–139
Oldenburg DJ, Bendich AJ (2001) Mitochondrial DNA from the liverwort Marchantia polymorpha: circularly permuted linear molecules, head-to-tail concatemers, and a 5′ protein. J Mol Biol 310:549–562
Podsiadlowski L, Bartolomaeus T (2006) Major rearrangements characterize the mitochondrial genome of the isopod Idotea baltica (Crustacea: Peracarida). Mol Phylogenet Evol 40:893–899
Raimond R, Marcade I, Bouchon D, Rigaud T, Bossy JP, Souty-Grosset C (1999) Organization of the large mitochondrial genome in the isopod Armadillidium vulgare. Genetics 151:203–210
Rigaud T, Juchault P, Mocquard JP (1997) The evolution of sex determination in isopod crustaceans. Bioessays 19:409–416
Rigaud T, Bouchon D, Souty-Grosset C, Raimond R (1999) Mitochondrial DNA polymorphism, sex ratio distorters and population genetics in the isopod Armadillidium vulgare. Genetics 152:1669–1677
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Segawa RD, Aotsuka T (2005) The mitochondrial genome of the Japanese freshwater crab, Geothelphusa dehaani (Crustacea: Brachyura): evidence for its evolution via gene duplication. Gene 355:28–39
Shao R, Dowton M, Murrell A, Barker SC (2003) Rates of gene rearrangement and nucleotide substitution are correlated in the mitochondrial genomes of insects. Mol Biol Evol 20:1612–1619
Signorovitch AY, Buss LW, Dellaporta SL (2007) Comparative genomics of large mitochondria in placozoans. PLoS Genet 3:e13
Souty-Grosset C, Raimond R, Tourte M (1992) Déterminisme épigénétique du sexe et divergence génétique de l’ADN mitochondrial chez Armadillidium vulgare Latr. (Crustacé Oniscoïde): variabilité inter et intrapopulations. CR Acad Sci Paris 314:119–125
Sun H, Zhou K, Song D (2005) Mitochondrial genome of the Chinese mitten crab Eriocheir japonica sinenesis (Brachyura: Thoracotremata: Grapsoidea) reveals a novel gene order and two target regions of gene rearrangements. Gene 349:207–217
Suyama Y, Miura K (1968) Size and Structural Variations of Mitochondrial DNA. Proc Natl Acad Sci USA 60:235–242
Tatarenkov A, Avise JC (2007) Rapid concerted evolution in animal mitochondrial DNA. Proc Biol Sci 274:1795–1798
Tjensvoll K, Hodneland K, Nilsen F, Nylund A (2005) Genetic characterization of the mitochondrial DNA from Lepeophtheirus salmonis (Crustacea; Copepoda). A new gene organization revealed. Gene 353:218–230
Vahrenholz C, Riemen G, Pratje E, Dujon B, Michaelis G (1993) Mitochondrial DNA of Chlamydomonas reinhardtii: the structure of the ends of the linear 15.8-kb genome suggests mechanisms for DNA replication. Curr Genet 24:241–247
Wolstenholme DR (1992) Animal mitochondrial DNA: structure and evolution. Int Rev Cytol 141:173–216
Zouros E (2000) The exceptional mitochondrial DNA system of the mussel family Mytilidae. Genes Genet Syst 75:313–318
Acknowledgments
We thank Nicolas Galtier and two anonymous reviewers for comments on an early version of the manuscript, Pierre Grève and Mathieu Sicard for constructive discussions, Yves Caubet and Sébastien Verne for providing samples, and Daniel Guyonnet for technical assistance. This research was funded by the Centre National de la Recherche Scientifique (CNRS) and the French Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Marcadé, I., Cordaux, R., Doublet, V. et al. Structure and Evolution of the Atypical Mitochondrial Genome of Armadillidium vulgare (Isopoda, Crustacea). J Mol Evol 65, 651–659 (2007). https://doi.org/10.1007/s00239-007-9037-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-007-9037-5