
Abstract
During the last decade, the major histocompatibility complex (MHC) has received much attention in the fields of evolutionary and conservation biology because of its potential implications in many biological processes. New insights into the gene structure and evolution of MHC genes can be gained through study of additional lineages of birds not yet investigated at the genomic level. In this study, we characterized MHC class II B genes in five families of birds of prey (Accipitridae, Pandionidae, Strigidae, Tytonidae, and Falconidae). Using PCR approaches, we isolated genomic MHC sequences up to 1300 bp spanning exons 1 to 3 in 26 representatives of each raptor lineage, finding no stop codons or frameshift mutations in any coding region. A survey of diversity across the entirety of exon 2 in the lesser kestrel Falco naumanni reported 26 alleles in 21 individuals. Bayesian analysis revealed 21 positively selected amino acid sites, which suggests that the MHC genes described here are functional and probably expressed. Finally, through interlocus comparisons and phylogenetic analysis, we also discuss genetic evidence for concerted and transspecies evolution in the raptor MHC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The major histocompatibility complex (MHC) is a multigene family that plays a central role in the vertebrate immune system. MHC genes encode protein receptors that recognise and bind foreign peptides for presentation to specialised immune cells and subsequent initiation of an immune response (Klein 1986). MHC genes are the most highly polymorphic genes described in vertebrates with several hundred of different alleles at some loci, for instance, in humans (Robinson et al. 2000). Two main types of balancing selection, heterozygote advantage and frequency-dependent selection, have been suggested to be important in maintaining the high levels of MHC polymorphism needed to counteract the selection pressures imposed by pathogens (Bernatchez and Landry 2003; Hedrick 1999). Patterns of polymorphism in the MHC have been the focus in studies of evolutionary ecology and conservation as a consequence of their suggested implication in many relevant biological processes, including self versus non-self recognition, susceptibility to infectious diseases, individual odours, mating preferences, kin recognition or pregnancy outcome (Brown and Eklund 1994; Grimholt et al. 2003; Westerdahl et al. 2005; Zelano and Edwards 2002; Singh et al. 1987; Tregenza and Wedell 2000; Knapp et al. 1996). This widespread relevance for ecological processes has made MHC genes excellent models for the investigation of adaptive variation in vertebrates (see the recent reviews by Sommer 2005; Piertney and Oliver 2006).
There are two major classes of MHC molecules (class I and class II), which act in different ways. Class I molecules are heterodimers expressed in all nucleated cells that play an essential role in immune defence against intracellular pathogens by binding peptides mainly derived from viral proteins or cancer-infected cells. Class II molecules are primarily expressed on antigen-presenting cells of the immune system and bind peptides derived from the processing of extracellular pathogens such as bacteria or parasites. The MHC class II protein consists of two amino acid chains, called α and β, encoded by MHC class II A and MHC class II B genes, respectively. While both amino acid chains shape the peptide-binding region (PBR), the second exon of the B gene is known to hold the majority of the polymorphism. Several studies have characterized the second exon of MHC class II B genes in a wide variety of non-model vertebrates including mammals (Otting et al. 2002; Musolf et al. 2004), reptiles (Miller et al. 2005; Shi et al. 2004), amphibians (Bos and DeWoody 2005), and fishes (Consuegra et al. 2005; Wegner et al. 2006).
Studies of the MHC in birds have been restricted mainly to galliform species or passerines (i.e. Wittzell et al. 1999a; Ye X et al. 1999; Edwards et al. 1998; Miller and Lambert 2004; Jarvi et al. 2004; Bonneaud et al. 2004), with few examples of other avian groups (Ekblom et al. 2003; Tsuda et al. 2001). These studies have shown that there is substantial variation in MHC gene structure and number between different species. Thus, this emerging picture encourages gathering of information from a wide array of taxa to broaden our understanding of the evolution of MHC genes (Edwards et al. 2000). By far the best-studied bird MHC is the B-complex of the chicken (Gallus gallus), although integration of the updated chicken genome and of the less-well-characterised Rfp-Y complex may update even this picture (Hunt et al. 2006; Miller et al. 2003). Early studies soon revealed striking differences between the genomic organization of the MHC in chickens and mammals (Trowsdale 1995). The chicken MHC appears to be much smaller and compact, with shorter introns, a lower number of genes and rare occurrence of pseudogenes (Bourlet et al. 1988; Guillemot et al. 1989; Kauffman 2000). For example, the mammalian MHC encodes multiple loci for both class I and class II genes whereas in the chicken the B-complex codes for only two class I and two class II genes. These findings led to the formulation of the minimal essential MHC hypothesis (Kaufman and Salomonsen 1997), which highlights that the chicken MHC is selected to be as small and compact as possible, containing only enough expressed genes to ensure resistance to common pathogens. While most passerines have many copies of both class I and class II genes and pseudogenes are abundant (i.e. Sato et al. 2000; Hess et al. 2000), the genomic complexity of other non-passerine birds such as the great snipe Gallinago media seems to be intermediate between chicken and passerines, with at least two class II genes and intermediate gene lengths (Ekblom et al. 2003).
In this study, we have developed the molecular tools for the characterization of MHC class II B genes in birds of prey, a group of vertebrates including species of high conservation concern. We investigated 26 different species from the major raptor families (Aves: Accipitridae, Pandionidae, Strigidae, Tytonidae and Falconidae, Brooke and Birkhead 1991), making ours one of the largest phylogenetic surveys of MHC diversity in any avian group. We also conducted a wide survey of exon 2 diversity for the lesser kestrel Falco naumani. These data permit a preliminary investigation and testing of different mechanisms of molecular evolution already documented in the avian MHC, such as balancing selection (Hedrick 1999; Ekblom et al. 2003), concerted evolution (Edwards et al. 1995a; Wittzell et al. 1999a), and transspecies polymorphism (Klein 1987; Richardson and Westerdahl 2003).
Materials and Methods
Study Species and DNA Isolation
The species we investigated and the numbers of individuals analysed per species are shown in Table 1. Blood or tissue samples were collected from different individuals in the field or at rehabilitation centres in Spain, Argentina and Namibia. The extraction protocol we used is a modification of that described by Gemmell and Akiyama (1996). Blood or tissues were digested by incubating with proteinase K for at least 3 hours. DNA purification was carried out using 5M LiCl, organic extraction with chloroform-isoamylic alcohol (24:1) and DNA precipitation with absolute ethanol. Pellets hence obtained were dried and washed twice with 70% ethanol, and later stored at −20°C in 0.1–0.2 ml TE buffer.
Amplification, Sequencing and Alignment of MHC Fragments
Amplification strategies relying on the polymerase chain reaction (PCR) were performed over genomic DNA in a PTC-100 programmable thermal controller (MJ Research Inc.). The basic PCR profile for all amplifications was composed of 4 min at 94°C following 35 cycles of 40 s at 94°C, 40 s at 56–58°C and 40–80 s at 72°C, and finally 4 min at 72°C. Each 25 μl reaction contained 0.2 units Taq polymerase (Bioline), 1× PCR buffer, 1–1.5 mM MgCl2, 0.02% gelatine, 5% DMSO, 0.12 mM of each dNTP, 10–20 pmol of each primer and approximately 10 ng of genomic DNA. Sequencing reactions were carried out using Big Dye 1.1 Terminator technology and labelled fragments were subsequently resolved in a 3100 automated sequencer (Applied Biosystems). DNA sequences were aligned and edited using the software BioEdit (Hall 1999).
Amplification of Short and Long MHC Fragments
The degenerate primers 326 and 325 (Table 2; Edwards et al. 1995b) were employed to perform partial amplification of exon 2. We designed new degenerate primers (AlEx3F, AlEx3R; Table 2) across conserved regions emerging from an alignment of exon 3 sequences of different vertebrate taxa including birds (species names and GenBank accession number: Homo sapiens NM 002124, Gallus gallus DQ008586, Coturnix japonica AB110479, Agelaius phoeniceus U23971, Gallinago media AF485406, Sphenodon punctatus DQ124234). One individual of the following raptor species were sequenced: lesser kestrel Falco naumanni, peregrine falcon Falco peregrinus, Eurasian black vulture Aegypius monachus, booted eagle Hieraaetus pennatus, northern goshawk Accipiter gentilis, barn owl Tyto alba, little owl Athene noctua and eagle owl Bubo bubo. PCR reactions at this stage contained 20 pmol of each primer and 1 mM MgCl2. The annealing temperature was 56°C. Uncloned PCR products were directly sequenced in order to confirm appropriate amplification of MHC genes and to detect conserved regions among species.
Once obtaining partial exon 2 and exon 3 sequences, new primers will be designed across conserved regions in order to amplify intron 1 and intron 2. Primer design will be tested using Oligo 6.0 (Molecular Biology Insights). An additional forward primer in exon 1 needed to amplify intron 1 (MHC05 and 34F; Miller and Lambert 2004, Ekblom et al. 2003) (Fig. 1) was also used. At this stage, our aim was obtaining the intron sequences flanking the highly polymorphic exon 2 to design new primers for the amplification of the whole exon in a single PCR. Furthermore, there are no available primers able to amplify long MHC class II fragments including exon 1, exon 2, introns 1 and 2, and a large part of exon 3 in one single PCR from diverse species of birds. We utilized the primers targeted to exon 1 (MHC05 and 34F; Fig. 1, Table 2) and a newly designed reverse primer annealing to a distal conserved region of exon 3 that we identified after the alignment of different bird sequences deposited in the data bases (species names and GenBank accession number: Gallus gallus DQ008586, Coturnix japonica AB110479, Agelaius phoeniceus U23971, Gallinago media AF485406). The amplification of long MHC fragments was checked in all raptor species investigated here (Table 1) using the basic PCR profile described above but extending the extension time for the Taq to 80 s.

Schematic illustration of MHC class II B genes in various families of birds of prey. The positions of the primers used in this study are indicated by arrows. Boxes represent exons and the shaded box, the highly polymorphic exon 2 that codes for the antigen binding sites. LP indicates the peptide leader sequence located in exon 1
Molecular Cloning
Investigation of variation at single MHC loci requires separating the different PCR amplification products because of the possibility of amplifying more than one locus, and because individuals are likely to be heterozygous for these loci. We cloned PCR products resulting from the amplification of the complete second exon in all the species investigated here as well as long MHC fragments in which cloning was the only alternative to obtain unambiguous and complete sequences of the introns linked to both exons. After PCR clean-up in Microcon centrifuge tubes (Millipore), PCR products were cloned into bacterial plasmid using the PGEM-T easy vector system II (Promega). Clones were screened for the expected insert size in 1.5% agarose gels by running a second PCR with M13 primers. Positive clones (8–10 per individual) were selected for sequencing analysis when investigating polymorphisms in exon 2. Following Edwards and co-workers (1995b), rare exon 2 sequences found only once and differing by less than 3 bp from a redundant sequence of the same PCR product were considered artefacts of PCR errors and were discarded. Since recombination of cloned PCR products is an additional source of artefacts (Bradley and Hillis 1996; Meyerhans et al. 1990), direct sequencing of uncloned PCR products was used to get agreement for polymorphic sites.
Analyses of Intraspecific Polymorphism
A wide survey of intraspecific polymorphism in exon 2 was conducted for 21 lesser kestrels hatched in Spain. Three white-backed vultures, Gyps africanus, and three cape vultures, Gyps coprotheres, from Namibia were also analysed. Polymorphism statistics were generated using the software DNAsp 4.0 (Rozas et al. 2003).
Test for Positive Selection in Exon 2 Sequences Using Maximum Likelihood Analyses
An excess of nonsynonymous substitutions (d N) over synonymous substitutions (d S) in functionally important amino acid sites indicates that positive selection is occurring. Then, ω = d N/d S > 1. We used the programme CODEML in the PAML package, version 3.15 (Yang 2000) to test for the presence of codon sites affected by positive selection and to identify those sites in exon 2 sequences of the lesser kestrel. This fact precludes assuming that codons comprising the PBR in birds are the same as in the human MHC class IIB genes (see Brown et al. 1993). The models considered in this study were M7 (β) and M8 (β and ω). Under the model M7 (β), the ω ratio varies according to the β distribution and does not allow for positively selected sites (0 < ω < 1). Model M8 provides an additional site class to account for sites under positive selection (ω > 1). Models M7 and M8 were compared using likelihood ratio tests (LRT) (Nielsen and Yang 1998). The LRT statistics calculates twice the log-like-likelihood difference compared with a χ 2 distribution with degrees of freedom equal to the difference in the number of parameters between the two compared models. The best tree by maximum-likelihood search was in accordance with the one-ratio model (M0) used to provide phylogenetic information. Finally, we used a Bayesian approach implemented in CODEML to identify residues under positive selection in the kestrel MHC class II sequences.
Phylogenetic Relationships of MHC Class II B Genes in Birds of Prey
The phylogenetic relationships of MHC class II B sequences were visualized through Neighbour-Net networks based on Kimura’s two parameter model that were built in the software Splits Tree 4 using maximum likelihood distances (Huson and Bryant 2006). Under complex models of evolution involving gene loss and duplication, hybridization, horizontal gene transfer or recombination, phylogenetic networks can provide a useful representation of the genetic relationships among sequences as compared to traditional phylogenetic trees. In this regard, gene loss and duplication in addition to recombination have been widely described in the MHC (i.e. Nei et al. 1997; Miller and Lambert 2004; Hess and Edwards 2002; Schaschl et al. 2006). Raptor exon 2 sequences jointly with exon 2 sequences obtained from galliform species (GenBank accession numbers: AM489776, AB282651, AJ224352, AY928104), passerines (GenBank accession numbers: L42335, AJ404376, AY437913, AF328737, U24411, U24426) and a tuatara Sphenodon punctatus sequence (GenBank accession number: DQ124237) as an outgroup were analysed. In addition, we built another network containing only intron 2 and exon 3 sequences from different species within the Accipitridae family. This network included 10 different sequences, from at least two different loci, that were isolated in three white-backed vultures and three different sequences from one cape vulture, respectively. Our aim at this point was to look for specific clusters that may reflect different loci within the same species or orthologus relationships among loci from different species.
Finally, the occurrence of gene conversion was assessed using the software GENECONV version 1.81 (Sawyer 1999). GENECONV analyses the distribution of nucleotide differences to detect gene conversion events by looking for stretches of nucleotides in a pair of sequences that are more similar to each other than would be expected by chance (Drouin et al. 1999). Putative gene conversion events were considered significant when the simulated global P value < 0.05. Such simulations were based on 10,000 permutations of the original data. The analysis was performed on a 1274 bp alignment of MHC class II B sequences from three white-backed vultures (n = 10), one cape vulture (n = 3) and one Eurasian black vulture Aegypius monachus (n = 1). Gscale values of 0, 1 and 2 were used, allowing for varying levels of mismatches (i.e. subsequent mutation) within the gene conversion event to take into account.
Results
Amplification of Conserved MHC, Complete Exon 2 and Intronic Regions
A 159 bp fragment of exon 2 and an 81 bp fragment of exon 3, excluding primers sequences, were successfully obtained in multiple species using degenerate primers. GenBank accession numbers are not given here since PCR products were not cloned and because these sequences will overlap with longer MHC sequences we describe. Primers 34F and MHC05 (Miller and Lambert 2004; Ekblom et al. 2003), in combination with new primers designed across conserved regions of exon 2, successfully amplified intron 1 in all raptor species tested so far. A novel battery of primers designed across conserved regions at the family level of exons 2 and 3 successfully amplified intron 2 (Fig. 1, Table 2).
Newly designed primers targeting conserved regions at the familial level, which were located at the end of intron 1 and at the beginning of intron 2, cleanly amplified the entire exon 2 in all species of birds of prey we investigated (Table 2, Fig. 1). These regions were preferentially chosen because of the high G + C content of both introns in the remainder sequence (Fig. 4). In addition, using primers 34F and RapEx3CR, we successfully amplified a long MHC class II fragment (> 1100 bp) in hawks and allies as well as in owls. Long MHC sequences for each species are deposited in GenBank (EF370953–EF370990). Whereas some species yielded a clean band of the expected size, other species required the excision of the band from the gel before cloning. Nonspecific bands exhibited low sizes (< 500 bp), and therefore were unlikely to include the entire region. The length of intron 1 (> 1–1.5 kb), as estimated in 1.5% agarose gels, precluded the amplification of this fragment in falcons and kestrels in a single PCR. For these species we utilized two different PCR reactions, one for the amplification of intron 1 using primers MHC05 and Fal2R, and the other for the amplification of the remaining downstream sequence using primers Fal2FC and RapEx3CR (Fig. 1, Table 2).
MHC Class II Polymorphism and Gene Duplications
The number of alleles per individual ranged from 1 to 5 and the number of potential MHC class II B loci per species ranged from 1 to 3 (Table 1). Analyses of intraspecific polymorphism in 21 lesser kestrels revealed 26 exon 2 alleles. (GenBank accession numbers: EF370767–370788; see Fig. 2 and Table 3 for polymorphism statistics). No more than two alleles were found in any lesser kestrel we investigated. The double peaks observed in the sequencing chromatograms of uncloned PCR products are congruent with the two cloned alleles obtained in each bird. The specific amplification of one single locus in this species is also supported by an ongoing study in which the segregation of the alleles in lesser kestrel families with at least four nestlings adjusts to a single model of biparental inheritance (Alcaide et al. unpublished data). On the other hand, we also found high intraspecific polymorphism in the white-backed vulture and in the cape vulture. Through the genotyping of three individuals from each species, we found 12 and 11 exon 2 alleles, respectively (Fig. 3). The finding of up to four alleles per individual in both species suggests the existence of at least two different MHC class II B loci; this also applies to other birds of prey. All exon 2 alleles differed in at least one nonsynonymous nucleotide substitution. A survey of interspecific polymorphism is shown in Fig 3. No identically shared alleles among any of the species we investigated were found.

Alignment of putative amino acid sequences of 26 MHC class II B exon 2 alleles of the lesser kestrel and other Falco species. Dots indicate identity with the top sequence. Asterisks on top indicate codons comprising the peptide-binding region in humans (Brown et al. 1993). Crosses indicate PBRs under strong positive selection in lesser kestrels (> 0.95 posterior probabilities)

Complete amino acid sequences of exon 2 in 25 raptor species. Two alleles per species are shown, as well as the intraspecific polymorphism found in the white-backed vulture and the cape vulture. Dots indicate identity with the top sequence. See Table 1 for species codes
Tests of Selection
The LRT statistic comparing M7 and M8 model indicates that M8 fitted the data significantly better than M7 (P < 0.001). The estimates from M8 suggested that about 23% of the exon 2 amino acid sites were under strong positive selection in the lesser kestrel (ω = 8.216; Table 4). Bayesian identification of sites under positive selection is listed in Table 4. As can be noticed in Fig. 2, there are slight differences regarding the human PBR-sites (HLA-DRB1 gene; Brown et al. 1993).
Gene Structure and Evolution of MHC Class II B Genes in Birds of Prey
At about 1.1 kb, the most compact MHC class II B regions spanning exon 1 to exon 3 was found in the barn owl (Tyto alba) and other strigiformes. The length and sequence of introns were generally quite conserved within the same raptor family but not between families (Fig. 4). The length of intron 2 appears to be smaller in birds of prey (250–280 bp) than in passerines (380–950 bp or longer) (Edwards et al. 1998; Hess et al. 2000; Gasper et al. 2001). Nonetheless, the length of intron 1 in passerines (about 440 bp) is similar to the ones detected in the majority of raptors, except for the Falco species, where extremely long introns (1–1.5 kb) were documented (Fig. 1).

Alignment of intron 2 sequences isolated in different raptorial genera. See Table 1 for species codes
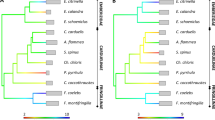
The network relationships between different avian exon 2 sequences are presented in Fig. 5. This phylogeny shows that the major families of birds of prey are upheld but provides low resolution at the family level. Some sequences that were isolated in different species of the same family appeared to be more similar to each other than to sequences within species. These results are consistent with the transspecies evolution of the polymorphism typically found in MHC class II exon 2 alleles within particular loci (reviewed in Hedrick 2001). In contrast, the phylogenetic relationships among intron 2 and exon 3 sequences from different Accipitridae species show that MHC sequences cluster together within species. Even though this network includes ten different sequences from three white-backed vultures and three different sequences from one cape vulture, there is no specific clustering of sequences that might suggest the presence of different loci within species or orthologous loci between species (Fig. 6). Overall, the extent of homology in the whole sequences, excluding exon 2, from the same white-backed vulture and the same cape vulture is about 97%. The similarity still remains quite high (about 94%) when comparing these species. Recent gene duplication events or concerted evolution could explain this finding. In this regard, a total of three significant gene conversion events across long stretches of MHC sequences were detected using GENECONV in the white-backed vulture (Table 5).

A Neighbour-Net constructed from 102 MHC class II B exon 2 sequences isolated from 37 avian species. The major clusters reflecting different avian groups are indicated. Evidence for transspecies allelism at the familial level is indicated by arrows. See Table 1 for species codes

Phylogenetic network among intron 2 and exon 3 sequences from different Accipitridae species. Ten sequences from three white-backed vultures and three sequences from one cape vulture are included. Sequences that were isolated in the same individual are named as Gyaf1.1, 1.2, and so on. Notice that there is no evidence of clusters within species reflecting different loci. See Table 1 for species codes
Discussion
Genomic Architecture and Polymorphism of Raptor MHC Genes
Based on our PCR survey, the genomic structure of raptor MHC genes resembles nonpasserine species in displaying the comparably lower genomic complexity documented in the chicken MHC versus passerines (Zoorob 1990; Ekblom et al. 2003). We have found evidence for a small number of MHC class II loci (1–3 genes) in comparison to passerine species, where up to six different loci have been reported (Sato et al. 2000). Whereas we have commonly detected only two alleles per individual in some species, such as the goshawk Accipiter gentilis or Falco species, up to four alleles per individual were frequently found in the majority of hawks and allies. Sequence evidence for three loci came from the finding of five exon 2 alleles in the little owl Athene noctua (Table 1). Nonetheless, our PCR survey is likely to be biased downward since PCR might selectively amplify particular genes in multigene families (Wagner et al. 1994), and Southern blots would help resolve this issue further (Edwards et al. 2000; Westerdahl et al. 1999; Wittzell et al. 1999b). The lack of stop codons or frameshift mutations in any coding region reported here also suggests a low incidence of pseudogenes in the MHC of birds of prey. Pseudogenes have been commonly documented in passerines for both class I and II (Hess et al. 2000; Edwards et al. 2000; Westerdahl et al. 1999) but appear to be almost absent in other avian groups (Kauffman et al.1999; Ekblom et al. 2003).
Our survey of intraspecific polymorphism reveals high genetic diversity at the MHC genes investigated here (Table 1). In addition, positive selection at several amino acid sites comprising the PBR indicates that balancing selection is in operation. Although we have not performed gene expression analyses in this study, research in this topic has found high expression levels at the same loci we have characterized in the barn owl (R. Burri et al., personal communication), and in general, studies have observed a correlation between signatures for balancing selection and level of expression (Zoorob 1990).
Concerted Evolution Leads to Sequence Homogenization at Multiple Loci in the MHC of Birds of Prey
Concerted evolution is the tendency of different genes within multigene families to undergo genetic exchange. Without concerted evolution, genes are expected to evolve independently and therefore differences in the length of introns and the sequence of exons might be expected. The non-mutually exclusive birth-and-death model of molecular evolution typically documented in the mammalian MHC (Nei et al. 1997) allows the recognition of orthologous loci in such distant species as humans and mice (Trowsdale 1995). In contrast, revealing orthologous loci in birds has sometimes required the sequencing of the 3′ untranslated (UT) region of the cDNA (Wittzell et al. 1999a; Miller & Lambert 2004) because high homology among MHC sequences derived from putative different loci has typically been found (Edwards et al. 1995a). Gene conversion events across long regions of avian MHC genes cannot only explain the high diversity levels documented in the second exon of class II B genes but might also be responsible for the sequence homogenization of other parts of the gene within species, such as introns and exon 3 (Martinsohn et al. 1999). Indeed, our GENECONV analyses revealed significant gene conversion events in the white-backed vulture involving stretches of sequences of up to 574 bp (Table 5). Nonetheless, postspeciation duplication is a process that can produce patterns that mimic recent concerted evolution in birds (Edwards et al. 1999). In this regard, our phylogenetic analyses have revealed high homology in intron and exon 3 sequences not only within species but also among species belonging to the same family (Fig. 3). Furthermore, we only found two fixed nucleotide differences out of 238 bp sequenced among 14 different exon 3 sequences from three vulture species. Genetic data therefore suggest that gene duplications have taken place before a relatively recent split of the evolutionary lineages and not after.
Conclusions
The sequence data from this avian group presented in this paper, should contribute to a better understanding of the evolutionary significance and conservation implications of the MHC. In addition, our results suggest the occurrence of non-mutually exclusive concerted and transspecies evolutionary processes in the raptor MHC, and provide new insights into the structure and diversity of genetic processes in a diverse but phylogenetically problematic avian order. Because of their importance for conservation genetics (Edwards and Potts 1996; Hedrick 2001), our sequences may also aid in the conservation of genetic diversity in this globally threatened clade.
References
Bernatchez L, Landry C (2003) MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol 16:363–377
Bonneaud C, Sorci G, Morin V, Westerdahl H, Zoorob R, Wittzell H (2004) Diversity of Mhc class I and IIB genes in house sparrows (Passer domesticus). Immunogenetics 55:855–865
Bos DH, DeWoody JA (2005) Molecular characterization of major histocompatibility complex class II alleles in wild tiger salamanders (Ambystoma tigrinum). Immunogenetics 57:775–781
Bourlet Y, Behar G, Guillemot F, Frechin N, Billault A, Chausse A-M, Zoorob R, Auffrey C (1988) Isolation of major histocompatibility complex class II (B-L) b chain sequences: comparison with mammalian a chains and expression in lymphoid organs. EMBO J 7:1031
Bradley RD, Hillis DM (1996) Recombinant DNA sequences generated by PCR amplification. Mol Biol Evol 14:592–593
Brooke M, Birkhead M (1991) The Cambridge encyclopedia of ornithology. Cambridge University Press, Cambridge
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39
Brown JL, Eklund A (1994) Kin recognition and the major histocompatibility complex: An integrative review. Am Nat 143:435–461
Consuegra S, Megens HJ, Leon K, Stet RJM, Jordan WC (2005) Patterns of variability at the major histocompatibility class II alpha locus in Atlantic salmon contrast with those at the class I locus. Immunogenetics 57:16–24
Drouin G, Prat F, Ell M, Clarke GDP (1999) Detecting and characterizing gene conversion events in multigene family members. Mol Biol Evol 16:1369–1390
Edwards SV, Wakeland EK, Potts WK (1995a) Contrasting histories of avian and mammalian Mhc genes revealed by class II B sequences from songbirds. Proc Natl Acad Sci USA 92: 12200–12204
Edwards SV, Grahn M, Potts WK (1995b) Dynamics of Mhc evolution in birds and crocodilians: amplification of class II genes with degenerate primers. Mol Ecol 4: 719–729
Edwards SV, Potts WK (1996) Polymorphism of genes in the major histocompatiblility complex: implications for conservation genetics of vertebrates. In: Smith TB, Wayne RK (eds) Molecular genetic approaches in conservation. Oxford University Press, Oxford, pp 214–237
Edwards SV, Chesnut K, Satta Y, Wakeland EK (1997) Ancestral polymorphism of MHC class II genes in mice: implications for balancing selection and the mammalian molecular clock. Genetics 146:655–668
Edwards SV, Gasper J, Stone M (1998) Genomics and polymorphism of Agph-DAB1, an Mhc class II B gene in red-winged blackbirds (Agelaius phoenicus). Mol Biol Evol 15:236–250
Edwards SV, Hess CM, Gaspar J, Garrigan D (1999) Toward and evolutionary genomics of the avian MHC. Immunol Rev 167:119–132
Edwards SV, Nusser J, Gasper J (2000) Characterization and evolution of major histocompatibility complex (MHC) genes in non-model organisms, with examples from birds. In: Baker AJ (ed) Molecular methods in ecology. Blackwell, Oxford, pp 168–207
Ekblom R, Grahn M, Höglund J (2003) Patterns of polymorphism in the MHC class II of a non-passerine bird, the great snipe (Gallinago media). Immunogenetics 54:734–741
Gasper JS, Shiina T, Inoko H, Edwards SV (2001) Songbird genomics: Analysis of 45 kb upstream of a polymorphic MHC class II gene in red-winged blackbirds (Agelaius phoeniceus). Genomics 75:26–34
Grimholt U, Larsen S, Nordmo R, et al. (2003) MHC polymorphism and disease resistance in Atlantic salmon (Salmo salar): facing pathogens with single expressed major histocompatibility class I and class II loci. Immunogenetics 55:210–219
Gemmell NJ, Akiyama S (1996) An efficient method for the extraction of DNA from vertebrate tissues. Trends Genet 12:338–339
Guillemot F, Kaufman JF, Skjoedt K, Auffray C (1989) The major histocompatibility complex in the chicken. Trends Genet 5:300–304
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hedrick PW (1999) Balancing selection and MHC. Genetica 104:207–214
Hedrick PW (2001) Conservation genetics: where are we now? Tree 16:629–636
Hess CM, Gasper J, Hoekstra HE, Hill CE, Edwards SV (2000) MHC class II pseudogene and genomic signature of a 32-kb cosmid in the house finch (Carpodacus mexicanus). Genome Res 10:613–623
Hess CM, Edwards SV (2002) The evolution of major histocompatibility genes in birds. Bioscience 52:423–431
Hunt HD, Goto RM, Foster DN, Bacon LD, Miller MM (2006) At least one YMHCI molecule in the chicken is alloimmunogenic and dynamically expressed on spleen cells during development. Immunogenetics 58:297–307
Huson H, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Jarvi SI, Tarr CL, McIntosh CE, Atkinson CT, Fleischer RC (2004) Natural selection of the major histocompatibility complex (Mhc) in Hawaiian honeycreepers (Drepanidinae). Mol Ecol 13:2157–2168
Kaufman J, Salomonsen J (1997) The “minimal essential MHC” revisited: Both peptide-binding and cell surface expression level of MHC molecules are polymorphisms selected by pathogens in chickens. Hereditas 127:67–73
Kaufman J, Milne S, Göbel T, Walker BA, Jacob JP, Auffrey C, Zoorob R, Beck S (1999) The chicken B locus is a minimal-essential major histocompatibility complex. Nature 401:923–925
Kaufman J (2000) The simple chicken major histocompatibility complex: life and death in the face of pathogens and vaccines. Philos Trans R Soc Lond Biol Sci 355:1077–1084
Klein J (1987) Origin of major histocompatibility complex polymorphism – the transspecies hypothesis. Human Immunol 19:155–162
Klein J (1986) Natural history of the major histocompatibility complex. Wiley, New York
Klein J, Bontrop RE, Dawkins RL, Erlich HA, Gyllensten UB, Heise ER, Jones PP, Parham P, Wakeland EK, Watkins DI (1990) Nomenclature for the major histocompatibility complexes of different species: a proposal. Immunogenetics 31:217–219
Knapp LA, Ha JC, Sackett GP (1996) Parental MHC antigen sharing and pregnancy wastage in captive pigtailed macaques. J Reprod Immunol 32:73–78
Martinshon JT, Sousa AB, Guethlein LA, Howard JC (1999) The gene conversion hypothesis of MHC evolution: A review. Immunogenetics 50:168–200
Meyerhans A, Vartanian JP, Wain-Hobson S (1990) DNA recombination during PCR. Nucleic Acids Res 18:1687–1690
Miller MM, Shiina T, Goto R, Inoko H (2003) Rfp-Y, the second cluster of MHC loci in the chicken, contains large families of distinctive NK lectin-like and class Ib loci. Faseb J 17:C161–C161
Miller HC, Lambert DM (2004) Gene duplication and gene conversion in class II MHC genes of New Zealand robins (Petroicidae). Immunogenetics 56:178–191
Miller HC, Belov K, Daugherty CH (2005) Characterisation of MHC class II genes from an ancient reptile lineage, Sphenodon (tuatara). Immunogenetics 57:883–891
Musolf K, Meyer-Lucht Y, Sommer S (2004) Evolution of MHC-DRB class II polymorphism in the genus Apodemus and a comparison of DRB sequences within the family Muridae (Mammalia: Rodentia). Immunogenetics 56:420–426
Nei M, Gu X, Sitnikova T (1997) Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc Natl Acad Sci USA 94:7799–7806
Nielsen R, Yang A (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Otting N, de Groot NG, Doxiadis GG, Bontrop RE (2002) Extensive Mhc-DQB variation in humans and non-human primate species. Immunogenetics 54:230–239
Piertney SB, Oliver MK (2006) The evolutionary ecology of the major histocompatibility complex. Heredity 96:7–21
Richardson D, Westerdahl H (2003) A comparison of MHC diversity in two Acrocephalus species: the outbred great reed warbler and the inbred Seyshelles warbler. Mol Ecol 12:3523–3529
Richman A, Herrera LG, Nash D, Schierup MH (2003) Relative roles of mutation and recombination in generating allelic polymorphism at MHC class II locus in Peromyscus maniculatus. Genet Res 82:89–99
Robinson J, Malik A, Parham P, Bodme JG, Marsh SGE (2000) IMGT/HLA database — A sequence database for the human major histocompatibility complex. Tissue Antigens 55:280–287
Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Sato A, Figueroa F, Mayer WE, Grant PR, Grant BR, Klein J (2000) Mhc class II genes of Darwin’s Finches: divergence by point mutations and reciprocal recombination. In: Kasahara M (ed) Major histocompatibility complex: evolution, structure and function. Springer, Berlin
Sawyer SA (1999) GENECONV: a computer package for the statistical detection of gene conversion. Distributed by the author, Department of Mathematics, Washington University in St. Louis. http://www.math.wustl.edu/∼sawyer
Schaschl H, Wandeler P, Suchentrunk F, Obexer-Ruff G, Goodman SJ (2006) Selection and recombination drive the evolution of MHC class II DRB diversity in ungulates. Heredity 97:427–437
Shi Y, Xiao-bing W, Peng Y, Bi-hui C (2004) Cloning and sequences analysis of the second exon of MHC class II B genes in Chinese alligator, Alligator sinensis. Zool Res 25:415–421
Singh PB, Brown RE, Roser B (1987) MHC antigens in urine as olfactory recognition cues. Nature 327:161–164
Sommer S (2005) The importance of immune gene variability in evolutionary ecology and evolution. Front Zool 2:16
Tregenza T, Wedell N (2000) Genetic compatibility, mate choice and patterns of parentage: Invited review. Mol Ecol 9:1013–1027
Trowsdale J (1995) ‘Both bird and man and beast’: comparative organization of MHC genes. Immunogenetics 41:1–17
Tsuda TT, Tsuda M, Naruse T, Kawata H, Ando A, Shiina T, Fukuda M, Kurita M, LeMaho I, Kulski JK, Inoko H (2001) Phylogenetic analysis of penguin (Spheniscidae) species based on sequence variation in MHC class II genes. Immunogenetics 53:712–716
Wagner A, Blackstone N, Cartwright P, Dick M, Misof B, Snow P, Wagner G, Bartels J, Murtha M, Pendleton J (1994) Surveys of gene families using polymerase chain reaction: PCR selection and PCR drift. Syst Biol 43:250–261
Wegner KM, Kalbe M, Rauch G, Kurtz J, Schaschl H, Reusch TBH (2006) Genetic variation in MHC class II expression and interactions with MHC sequence polymporphism in three-spined sticklebacks. Mol Ecol 15:1153–1164
Westerdahl H, Wittzell H, von Schantz T (1999) Polymorphism and transcription of Mhc class I genes in a passerine bird, the great reed warbler. Immunogenetics 49:158–170
Westerdahl H, Waldenstrom J, Hansson B, Hasselquist D, von Schantz T, Bensch S (2005) Associations between malaria and MHC genes in a migratory songbird. Proc Biol Sci 272:1511–1518
Wittzell H, Bernot A, Auffrey C, Zoorob R (1999a) Concerted evolution of two Mhc class II B loci in pheasants and domestic chickens. Mol Biol Evol 16:479–490
Wittzell H, Madsen T, Westerdahl H, Shine R, von Schantz T (1999b) MHC variation in birds and reptiles. Genetica 104:301–309
Yang Z (2000) Phylogenetic analysis by maximum likelihood (PAML), version 3.0. University College, London
Ye X, Zhu J, Velleman SG, Bacon WL, Nestor KE (1999) Analysis of genetic polymorphism in the major histocompatibility complex of Japanese quail. Poult Sci 78:8–14
Zelano B, Edwards SV (2002). An Mhc component to kin recognition and mate choice in birds: predictions, progress, and prospects. Am Nat 160:S225–S237
Zoorob R, Behar G, Kroemer G, Auffrey C (1990) Organization of a functional chicken class II B gene. Immunogenetics 31:179–187
Acknowledgments
We thank Christopher N. Balakrishnan for his comments and suggestions during the preparation of this manuscript. This study and a research visit for MA to Harvard University was supported by a grant from the MCyT (project REN2001-2310) to MA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Alcaide, M., Edwards, S.V. & Negro, J.J. Characterization, Polymorphism, and Evolution of MHC Class II B Genes in Birds of Prey. J Mol Evol 65, 541–554 (2007). https://doi.org/10.1007/s00239-007-9033-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-007-9033-9