
Abstract
Dengue virus (DENV) is the agent of the most widespread vector-borne viral disease of humans. To infer the timescale of DENV evolution with as much accuracy as possible, we compared, within a Bayesian Markov Chain Monte Carlo (MCMC) framework, estimates of phylogenetic tree length using both covarion and noncovarion models of molecular evolution, the latter also incorporating lineage-specific rate variation through a “relaxed” molecular clock. Using a data set of 32 complete genome sequences representing all four viral serotypes, we found evidence for covarion-like evolution at second codon positions in specific DENV genes, although rarely at the level of complete gene or genomes. Further, the covarion model had little effect on estimates of tree length and hence time to the Most Recent Common Ancestor (MRCA). We conclude that although covarion models can improve descriptions of the dynamics of amino acid substitution, they have little effect on estimates of the timescale of viral evolution, which in the case of DENV covers a period of no more than 2000 years.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dengue has imposed a serious disease burden on human populations for centuries. The first clear description of epidemics of dengue illness occurred in the Americas during the 17th and 18th centuries, suggesting that the causative dengue virus (DENV) was imported from Africa during the slave trade. Dengue is manifest in a variety of disease syndromes ranging from asymptomatic dengue fever (DF), to severe manifestations, often classified as dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS). It is estimated that between 50 million and 100 million cases of DF occur globally each year (Gubler 2002), with an estimated 500,000 individuals hospitalized with severe dengue disease.
DENV is an arthropod-borne RNA virus (family Flaviviridae, genus Flavivirus) that probably arose via cross-species transmission from closely related viruses that infect nonhuman primates in Africa and Asia (Gubler and Kuno 1997; Wang et al. 2000). The genome of DENV is comprised of a single, positive-sense RNA molecule of approximately 11 kb that is translated as a single polyprotein, and that exists as four antigenically distinct serotypes (denoted DENV 1–4). The virus is transmitted between humans by mosquito vectors, principally the anthropophilic species Aedes aegypti, and has a sylvatic or “jungle” cycle involving monkeys and sylvatic mosquito species, again primarily of the genus Aedes.
There have been several attempts to estimate the time to the Most Recent Common Ancestor (MRCA) of sampled DENV isolates. In the first such study, Zanotto et al. (1996) used patristic (branch length) distances of nonsynonymous substitutions to infer that the common ancestor of the four serotypes existed approximately 1500 to 2000 years ago. A more robust time to the MRCA of approximately 1000 years was obtained using a maximum likelihood (ML) method, and accounted for the sampling date of the isolates in question (Twiddy et al. 2003). This study also provided the first comprehensive estimate of rates of nucleotide substitution in all serotypes of DENV—ranging from 4.55 × 10−4 to 1.16 × 10−3 substitutions per site, per year (subs/site/year) and, therefore, broadly similar to those seen in other RNA viruses (Jenkins et al. 2002). Hence, although some variation is apparent, those studies undertaken to date suggest that DENV originated within the last few thousand years. However, despite the similarity of the inferred evolutionary dynamics, none have accounted for the full complexities of viral evolution so that the timescale of DENV evolution is still in doubt.
Most previous estimates of the rates and dates of DENV evolution, and of RNA viruses in general, have made two simplifying assumptions; (i) that although viral sequences have been sampled at different times, evolutionary rates are constant across lineages—the assumption of a “molecular clock”; and (ii) that the distribution of variable and invariable nucleotide sites is the same across all lineages. However, both these assumptions are highly questionable for DENV, a virus that exists as four lineages that are as genetically different as some “species” of Flavivirus (Kuno et al. 1998). It is therefore possible that the use of incorrect evolutionary models has resulted in erroneous estimates of divergence times (Holmes 2003).
Recently, there have been major theoretical and logistic improvements in the study of gene sequence evolution, which could have a major impact on estimates of the timescale of viral evolution. First, analytical methods have been developed that explicitly account for lineage-specific rate through the use of a “relaxed” molecular clock (Drummond et al. 2006). Second, changes in the distribution of variable and invariable sites across a phylogeny can now be accommodated through the use of the covarion model of molecular evolution (Galtier 2001; Huelsenbeck 2002; Wang et al. 2007). The covarion model, first proposed by Fitch and Markowitz (1970), considers differential patterns of nucleotide or amino acid evolution across the phylogenetic tree. In essence, this model determines the proportion of sites that are invariable due to functional and structural constraints, and how the epistatic effect of fixed mutations can lead to an invariable site becoming variable, and vice versa; hence, changing the proportion of sites that are either switched “on” or switched “off” over time. The main prediction of the covarion model is therefore that lineages will differ in the proportion of sites that are variable over time. As such, it has been proposed, although not explicitly tested, that the covarion model could have a major impact on estimates of viral divergence times (Holmes 2003).
To assess whether the current estimates of the timescale of DENV evolution are accurate, and how covarion-like evolution might affect these estimates, we calculated divergence times in a data set of complete viral genomes under both the relaxed molecular clock and the covarion models of DNA substitution.
Materials and Methods
Analyses were conducted on a data set of 32 complete genome sequences representative of the genetic diversity in human DENV from all four serotypes. All sequences were taken from patients diagnosed with dengue at the Queen Sirikit National Institute of Child Health, Bangkok, from 1973 to 2002 (previously published and available at GenBank; Klungthong et al. 2004; Zhang et al. 2005, 2006). More information on the epidemiological background of the patients is given by Nisalak et al. (2003). All sequences were aligned manually using the SE-AL program (Rambaut 1996). Within these data, we studied complete genomes (coding regions only), as well as component genes (C, PrM/M, E, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5), and second and third position codon data sets separately (as these roughly correspond to nonsynonymous and synonymous sites, respectively)
To estimate rates of evolutionary change and divergence times (time to the MRCA) under a noncovarion substitution model, we employed the Bayesian Markov Chain Monte Carlo (MCMC) approach available in the BEAST package (Drummond and Rambaut 2003). For all data sets we employed both a relaxed (uncorrelated exponential) and a constant (strict) molecular clock (Drummond et al. 2006). For each data set we also utilized the demographic models of constant population size and exponential population growth, assuming the GTR+I+Γ4 model of nucleotide substitution. Uncertainty in parameter estimates is reflected in the 95% highest probability density (HPD) values.
We used the covarion model with gamma-distributed rate variation of Huelsenbeck (2002) that was originally adopted from the model of Tuffley and Steel (1998). This model has two additional parameters—s01 (rate of switching from off to on) and s10 (rate of switching from on to off)—and the substitution rate follows a general reversible (GTR) distribution when the switch rate is on (Huelsenbeck 2002). The on and off processes and the substitutions that occur when the switch process is “on” are independent events (for more details see Huelsenbeck 2002). Two different substitution models were therefore run on each data set in the MrBayes 2.01 program (Huelsenbeck and Ronquist 2001): (i) the “standard” noncovarion model of sequence evolution and (ii) the covarion model assuming a constant rate of nucleotide substitution and a constant distribution of switching rates. These models were compared using Akaike’s Information Criterion (AIC). In all cases, we employed the GTR+I+Γ model of nucleotide substitution with the convergence of parameter values confirmed using Tracer v1.3 (http://www.evolve.zoo.ox.ac.uk/software.html?id=tracer). Finally, to assess how each model affects estimates of the time to the MRCA we calculated the ratio of the tree length (TL; the total number of substitutions from the root to the tip of the tree) obtained under both the noncovarion and the covarion models.
Results and Discussion
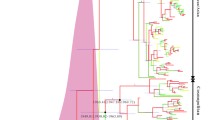
In all cases the relaxed molecular clock (noncovarion) model provided a better fit to our data set of 32 complete DENV genomes than the strict molecular clock (data not shown; available from the authors upon request). Parameter estimates and likelihood values from the relaxed molecular clock analysis are given in Table 1, while a phylogeny of the four DENV serotypes inferred using the covarion model in MrBayes (see below) is shown in Fig. 1. Our focus is on the time to the MRCA of the four DENV serotypes. A model of exponential population growth applied to complete genome sequences gave a mean time to the MRCA of 600 years (95% HPD of 193–1308 years), while at second codon positions this demographic model produced a mean estimate of 684 years (95% HPD of 199–1428 years). Under the constant population size model for complete genome sequences we estimated the mean time to the MRCA as 828 years (95% HPD of 269–1836 years), with similar estimates again obtained for second codon positions—858 years (95% HPD of 201–1739 years). Overall, the upper range of times of origin for DENV under a relaxed molecular clock were between 1300 and 1900 years ago. This is in line with previous estimates of the age of the MRCA of DENV (Zanotto et al. 1996; Lanciotti et al. 1997; Wang et al. 2000; Twiddy et al. 2003) and indicates that lineage-specific rate variation is not having a substantial impact on rates of nucleotide substitution and consequently divergence times. Further, that the constant population size and exponential growth models gave similar estimates for the time to the MRCA suggests that the underlying demographic model similarly has a relatively minor effect on estimates of divergence time.

Consensus Bayesian phylogenetic tree of 32 complete genomes of DENV estimated using the covarion model in MrBayes. All horizontal branch lengths are drawn to a scale of nucleotide substitutions per site, and posterior probability values are shown for key nodes. A topologically identical phylogeny was obtained under the noncovarion model.
Next, we assessed whether the evolution of DENV is better described by a covarion model of molecular evolution, considering whole viral genomes, individual genes, and specific codon positions. At the whole-genome level (either all sites or second and third codon positions concatenated), the noncovarion model was always a better fit to the DENV sequence data than the covarion model (Tables 2–4). However, more complex results were obtained when genes were considered individually. An analysis of whole genes (all codon sites) provided similar results to those of whole genomes, with the covarion model only providing a significantly better fit to the data in the case of the short prM/M gene (Table 2). However, very different results were obtained when the second codon positions of genes were considered in isolation. Here, the covarion model provided a better description of sequence evolution than the noncovarion model in 7 of the 10 genes, the exceptions being the E, NS2A, and long NS5 gene (Table 3). The preference for the covarion model in these cases suggests that DENV lineages (most likely the four serotypes) often differ in selective pressure at nonsynonymous sites, although the underlying causes are unknown and the effect is dissipated when whole genomes are considered. Finally, only the E gene was found to support the covarion model in an analysis of gene-specific third codon positions (Table 4). Although the E gene has previously been proposed as the site of positive selection, presumably due to immune pressure (Bennett et al. 2006; Twiddy et al. 2002), whether equivalent selection pressures operate on synonymous substitutions, which may affect both RNA secondary structure and codon usage bias, is unknown. Indeed, it is likely that the occurrence of multiple synonymous substitutions at third codon positions among the four serotypes will dilute lineage-specific differences. In this context it is also important to note that there are important differences in the parameter estimates for the second and third codon position data sets, mostly likely reflecting differences in overall substitution rate; the analysis of switch rates indicates that s01 is lower than s10 for second codon positions compared to third codon positions. This indicates that second codon positions are more constrained and implies that estimates of divergence times from third codon positions alone (and at synonymous sites in general) may not be accurate, even under the most sophisticated substitution models.
Finally, to determine how covarion-like evolution might affect estimates of divergence time, we compared the values of total tree length of the covarion and noncovarion models (Tables 2–4). Strikingly, the TL values under both models do not vary significantly at either the genic or the genomic levels—the TL ratios are usually close to 1.0—indicating that the covarion process does not significantly alter estimates of DENV divergence times. Indeed, in the case of whole genes and third codon positions the covarion model sometimes produced shorter tree lengths. Unusually high TL ratios were only apparent for the E, NS3, and NS5 genes in the second position codon analysis (range, 7.21–8.97), indicating that the noncovarion model may have underestimated the true number of nonsynonymous substitutions in these cases (although the covarion model was not supported in NS5). Hence, although the covarion model may better describe some aspects of DENV evolution than other models of DNA substitution, particularly at nonsynonymous sites, that it has relatively little effect on tree lengths indicates that it will similarly have a small effect on estimates of the time to common ancestry. In more general terms, application of the covarion model is therefore unlikely to explain the paradox that, although they are ubiquitous, most RNA viruses have inferred times of origin dating back only a few thousand years at most (Holmes 2003).
Although there has been some controversy over the estimated recent origin of DENV, and of flaviviruses in general, our results are in accord with previous studies which suggest that the evolutionary history of DENV is a relatively recent one. Hence, modern human dispersal is the most likely factor shaping the genetic structure of DENV populations; it is likely that sylvatic DENV could not establish itself in humans until a sufficient number and density of susceptible human hosts were available, corresponding to the age of urbanization and global travel (Zanotto et al. 1996).
References
Bennett SN, Holmes EC, Chirivella M, Rodriguez DM, Beltran M, Vorndam V, Gubler DJ, McMillan WO (2006) Molecular evolution of dengue 2 virus in Puerto Rico: positive selection in the viral envelope accompanies clade reintroduction. J Gen Virol 87:885–893
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88
Drummond AJ, Rambaut A (2003) BEAST version 1.3. Available at: http://www.evolve.zoo.ox.ac.uk/beast/
Fitch WM, Markowitz E (1970) An improved method for determining codon variability in a gene and its application to the rate of fixation of mutations in evolution. Biochem Genet 4:579–593
Galtier N (2001) Maximum-likelihood phylogenetic analysis under a covarion-like model. Mol Biol Evol 18:866–873
Holmes EC (2003) Molecular clocks and the puzzle of RNA virus origins. J Virol 77:3893–3897
Huelsenbeck JP, Ronquist F (2001) MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Huelsenbeck JP (2002) Testing a covariotide model of DNA substitution. Mol Biol Evol 19:698–707
Jenkins GM, Rambaut A, Pybus OG, Holmes EC (2002) Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J Mol Evol 54:152–161
Klungthong C, Zhang C, Mammen Jr. MP, Ubol S, Holmes EC (2004) The molecular epidemiology of dengue virus serotype 4 in Bangkok, Thailand. Virology 329:168–179
Kuno G, Chang G-J, Tsuchiya KR, Karabatsos N, Cropp CB (1998) Phylogeny of the genus Flavivirus. J Virol 72:73–83
Lanciotti RS, Gubler DJ, Trent DW (1997) Molecular evolution and phylogeny of dengue-4 viruses. J Gen Virol 78:2279–2286
Nisalak A, Endy TP, Nimmannitya S, Kalayanarooj S, Thisayakorn U, Scott RM, Burke DS, Hoke CH, Innis BL, Vaughn DW (2003) Serotype-specific dengue virus circulation and dengue disease in Bangkok, Thailand from 1973–1999. Am J Trop Med Hyg 68:191–202
Rambaut A (1996) Se-Al: Sequence Alignment Editor. Available at: http://www.evolve.zoo.ox.ac.uk/
Twiddy SS, Woelk CH, Holmes EC (2002) Phylogenetic evidence for adaptive evolution of dengue viruses in nature. J Gen Virol 83:1679–1689
Twiddy SS, Holmes EC, Rambaut A (2003) Inferring the rate and time-scale of dengue virus evolution. Mol Biol Evol 20:122–129
Tuffley C, Steel M (1998) Modeling the covarion hypothesis of nucleotide substitution. Math Biosci 147:63–91
Wang E, Ni H, Xu R, Barrett ADT, Watowich SJ, Gubler DJ, Weaver SC (2000) Evolutionary relationships of endemic/epidemic and sylvatic dengue viruses. J Virol 74:3227–3234
Wang HC, Spencer M, Susko E, Roger AJ (2007) Testing for covarion-like evolution in protein sequences. Mol Biol Evol 24:294–305
Zanotto PM de A, Gould EA, Gao GF, Harvey PH, Holmes EC (1996) Population dynamics of flaviviruses revealed by molecular phylogenies. Proc Natl Acad Sci USA 93:548–553
Zhang C, Mammen MP Jr, Chinnawirotpisan P, Klungthong C, Rodpradit P, Monkongdee P, Nimmannitya S, Kalayanarooj S, Holmes EC (2005) Clade replacements in dengue virus serotypes 1 and 3 are associated with changing serotype prevalence. J Virol 79:15123–15130
Zhang C, Mammen MP Jr, Chinnawirotpisan P, Klungthong C, Rodpradit P, Nisalak A, Nimmannitya S, Kalayanarooj S, Vaughn DW, Holmes EC (2006) Structure and age of genetic diversity of dengue virus type 2 in Thailand. J Gen Virol 87:873–883
Acknowledgment
This work was supported by the Alfred P. Sloan Foundation Graduate Scholarship.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Nicolas Galtier]
Rights and permissions
About this article
Cite this article
Dunham, E.J., Holmes, E.C. Inferring the Timescale of Dengue Virus Evolution Under Realistic Models of DNA Substitution. J Mol Evol 64, 656–661 (2007). https://doi.org/10.1007/s00239-006-0278-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-006-0278-5